РАДИОХИМИЯ, 2023, том 65, № 2, с. 188-193
УДК 546.799.5
ВЫДЕЛЕНИЕ АМЕРИЦИЯ ИЗ
АКТИНИД-ЛАНТАНИДНОЙ ФРАКЦИИ
ВЫСОКОАКТИВНЫХ ОТХОДОВ ПРИ ЕГО
ПЕРЕВОДЕ В СОСТОЯНИЕ ОКИСЛЕНИЯ Am(V)
© 2023 г. П. А. Осин*, Т. И. Трофимов, К. С. Пилюшенко, Ю. М. Куляко,
С. Е. Винокуров, Б. Ф. Мясоедов
Институт геохимии и аналитической химии им. В. И. Вернадского РАН,
119991, Москва, ул. Косыгина, д. 19
* e-mail: Posingeohi@rambler.ru
Поступила в редакцию 02.12.2022, после доработки 28.12.2022, принята к публикации 30.12.2022
Показаны условия окисления Am(III) до Am(VI), его восстановления до Am(V) и определена
устойчивость этих форм в 0.1 моль/л азотнокислом модельном растворе актинид-лантанидной фракции
высокоактивных отходов (ВАО), содержащем изотопы америция и кюрия, а также празеодим как
имитатор поведения осколочных лантанидов. Установлено, что около 30% америция в состоянии
окисления Am(V) от его исходного содержания в модельном растворе, содержащем высаливатель -
нитрат аммония - в количестве 8 моль/л, переходит в органическую фазу за одну стадию экстракции
30%-ным раствором трибутилфосфата в разбавителе Изопар М. При этом Cm и Pr экстрагируются на
80-85%. Коэффициент распределения Am составил около 0.1, Cm и Pr - около 1, а фактор отделения Am
от Cm и Pr составил около 10. Таким образом, показана возможность селективного выделения америция
из ВАО для его последующей трансмутации в быстрых реакторах.
Ключевые слова: америций, кюрий, лантаниды, празеодим, экстракция, трибутилфосфат, выделение,
разделение, высаливатель
DOI: 10.31857/S0033831123020089, EDN: XLTBWF
ВВЕДЕНИЕ
глубинного захоронения, что может быть достиг-
нуто при глубоком фракционировании ВАО, позво-
Технология переработки отработавшего ядер-
ляющем выделенный в самостоятельную фракцию
ного топлива (ОЯТ) основана на экстракционном
америций затем трансмутировать в реакторах на
Пурекс-процессе, в котором в качестве экстраген-
быстрых нейтронах [1].
та используется трибутилфосфат (ТБФ) в легком
Ранее нами был предложен способ выделения
разбавителе. После извлечения урана и плутония
актинид-лантанидной фракции из высокоактивного
из раствора ОЯТ в рафинате остаются трансплуто-
рафината при экстракции ТБФ в изопарафиновом
ниевые элементы (ТПЭ) - долгоживущие изотопы
разбавителе в присутствии нитрата железа как вы-
241,243Am (T1/2 = 433 и 7370 лет соответственно) и от-
саливателя [2]. При этом одной из важнейших ради-
носительно короткоживущий 244Cm (T1/2 = 18 лет),
охимических задач является последующее отделе-
а также группа осколочных лантанидов. При этом
ние америция от кюрия и лантанидов. Такое разде-
через несколько сотен лет основной вклад в радио-
ление необходимо по ряду причин. Во-первых, лан-
активность ВАО будут вносить долгоживущие изо-
таниды являются нейтронными ядами; некоторые
топы америция. Современная стратегия развития
изотопы имеют высокое сечение захвата нейтронов,
атомной энергетики России предусматривает замы-
и их присутствие в топливе будет ингибировать
кание ядерного топливного цикла (ЯТЦ), включая
трансмутацию америция [3, 4]. Во-вторых, вовле-
минимизацию объемов радиоактивных отходов для
чение изотопов кюрия в замкнутый ЯТЦ пробле-
188
ВЫДЕЛЕНИЕ АМЕРИЦИЯ ИЗ АКТИНИД-ЛАНТАНИДНОЙ ФР
АКЦИИ
189
матично, поскольку он обладает высоким уровнем
На данный момент известно лишь несколь-
нейтронного и теплового излучения, что усложнит
ко других работ [14-16] по использованию ТБФ в
изготовление и эксплуатацию регенерированного
качестве экстрагента для отделения Am от других
гомогенного ядерного топлива или мишеней амери-
трансурановых элементов или лантанидов. Напри-
ция для его трансмутации.
мер, в работах [14, 16] предложено проводить экс-
тракцию Am(VI) напрямую из азотнокислых рас-
Известная проблема разделения Am, Cm и лан-
творов без добавления высаливателя. Достижения
танидов определяется сходством в их химических
высоких коэффициентов распределения удается
свойствах [5, 6]. ТПЭ и лантаниды являются 5f- и
добиться путем корректировки кислотности раство-
4f-элементами соответственно, они характеризуют-
ся заполнением внутренних f-орбиталей с увеличе-
ра. На первоначальном этапе предлагается снижать
кислотность для эффективного окисления Am(III)
нием атомного номера. Это оказывает небольшое
до Am(VI), затем увеличивать до 1 M по HNO3. Ко-
влияние на химические свойства, которые в основ-
эффициент разделения для пары Am-Nd составил
ном зависят от процессов во внешних электронных
около 50; в то же время разделение пары Am-Np
оболочках. Ввиду этого эти элементы преимуще-
не происходило. В работе [15] предложено исполь-
ственно существуют в водных растворах в состоя-
зовать дигидрофосфат для связывания Nd в более
нии окисления (III). Ионные радиусы этих катионов
также сопоставимы. Ионы америция и кюрия име-
устойчивый комплекс по сравнению с Am(VI), и та-
ким образом удается добиться коэффициента разде-
ют изоструктурные аналоги в серии лантанидов:
ления около 120.
r(Nd3+) ≈ r(Am3+), r(Sm3+) ≈ r(Cm3+). В результате
затруднено отделение трехвалентных Am и Cm от
Настоящая работа посвящена изучению экстрак-
лантанидов путем корректировки только размера
ции Am в высших состояниях окисления в слабокис-
координационной сферы органических лигандов.
лых растворах - имитаторах актинид-лантанидной
фракции высокоактивного рафината, содержащих
Существует несколько подходов к разделению
Am от Cm и лантанидов, и большинство из них ос-
изотопы америция и кюрия и весовые количества
лантанидов (на примере празеодима) - при исполь-
новано на экстракционных методах [7, 8]. Ключе-
зовании ТБФ и в присутствии нитрата аммония, как
вым различием между этими катионами является то,
высаливателя, не образующего нерастворимых со-
что америций может окисляться до высших состоя-
лей с актинидами и продуктами деления, в том чис-
ний окисления Am(V,VI), что меняет его поведение
ле с лантанидами.
при экстракции [9, 10]. Ранее в наших работах [11,
12] показано, что, получив Am(VI) в слабокислых
растворах, можно извлечь Cm(III) из их смеси, ис-
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
пользуя экстрагенты 1-фенил-3-метил-4-бензоил-
пирозолон-5 и триоктилфосфиноксид, тогда как
Элементный состав ОЯТ реакторов типа
Am(VI), восстанавливаясь органической фазой до
ВВЭР-1000 с выгоранием 60 ГВт·сут/(т U) и вы-
Am(V), остается в водном растворе. При этом при-
держкой 8 лет рассчитан в работе [17]. Показано,
менение этих экстрагентов в производственных
что на долю осколочных лантанидов приходится
условиях проблематично. По этой причине предло-
17.2 мг на 1 г исходного урана. При условии, что
жено [13] для решения задачи по разделению Am
при растворении ОЯТ планируется достижение кон-
от Cm использовать ТБФ как надежный и хорошо
центрации урана до 600 г/л, на долю актинид-лан-
опробованный в технологии экстрагент. Так, кюрий
танидной фракции будет приходиться до 10.5 г/л
извлекали из растворов, содержащих ≤0.1 M HNO3
осколочных лантанидов. В качестве имитатора лан-
и ~8 M NH4NO3, с помощью 30%-ного ТБФ в Изо-
танидов в данной работе использовали Pr, полосы
паре M практически количественно, тогда как аме-
поглощения которого не совпадают с полосами по-
риций только частично (≤30%) переходил в органи-
глощения Am(III), Am(V) и Am(VI), что позволяет
ческую фазу в виде Am(III). При этом достигалась
определять концентрации в растворе как Pr, так и
высокая степень отделения Cm от Am(V), остающе-
Am в различных состояниях окисления методом
гося в водной фазе (≥99.9%).
спектрофотометрии.
РАДИОХИМИЯ том 65 № 2 2023
190
ОСИН и др.
Для приготовления модельного раствора акти-
Перед экстракцией в полученные растворы, со-
нид-лантанидной фракции использовали изото-
держащие Am(VI) или Am(V), а также трехвалент-
пы 241Am, 243Am, 244Сm, концентрации которых
ные Cm и Pr, вносили навеску NH4NO3 для дости-
в исходном растворе соответственно были равны
жения его концентрации в растворе 8 моль/л, как и
2.48 × 10-3, 1.28 × 10-1, 1.38 × 10-4 г/л. Концентра-
в работе [13]. Экстракцию актинидов и Pr проводи-
ция Pr составляла 10.5 г/л.
ли в течение 1 мин при соотношении водной (ВФ) и
органической фаз (ОФ) 1 : 5.
Перевод америция в высшие состояния окисле-
ния Am(VI) и затем в Am(V) осуществляли соглас-
Как было определено нами ранее [13], для уста-
но нижеприведенных реакций (1) и (2) соответ-
новления термодинамического равновесия при
ственно:
распределении Am(III) и Cm между азотнокислым
раствором, содержащим NH4NO3, в качестве выса-
2Am3+ + 3S2O2- + 4Н2О = 2AmO22+ + 6SO42- + 8H+, (1)
ливателя, и 30%-ным ТБФ в изопаре М достаточно
2AmO22+ + 2H2O2 = 2AmO+ + О2 + 2H2O.
(2)
1 мин. Фазы приводили в контакт с друг другом,
встряхивая пробирки вручную в течение 1 мин. За-
Окисляли Am(III) до Am(VI) в растворе
тем фазы центрифугировали для их расслаивания
0.1 моль/л HNO3, содержащем в качестве окислите-
в течение примерно 1 мин при скорости вращения
ля 0.2 моль/л (NH4)2S2O8 при нагревании раствора
2000 об/мин. Далее разделяли фазы и проводили
при 90°С в течение 20 мин в соответствии с методи-
анализ.
кой, описанной в работе [11]. Эффективность дан-
ного способа обычно ограничивается концентраци-
Общую концентрацию Am и Cm в использован-
ных растворах определяли методом альфа-спектро-
ями азотной кислоты не более 0.5 М [18]. При более
метрии (Alpha Analyst, Canberra, США).
высоких концентрациях кислоты персульфат аммо-
ния разлагается с образованием перекиси водоро-
Используемые в работе нитрат аммония, гекса-
да - эффективного восстановителя для AmO2+ [19].
гидрат нитрата празеодима, персульфат аммония,
Однако этот эффект, судя по всему, можно нивели-
азотная кислота и пероксид водорода имели квали-
ровать путем увеличения концентрации окислителя
фикацию ч.д.а.
до 1.6 M [14]; тогда удавалось окислить Am(III) до
Am(VI) в 1 M HNO3 на 90 ± 5%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Раствор, содержащий Am(V), получали добавле-
нием 1 мкл раствора 37%-ного H2O2 к 1 мл раствора
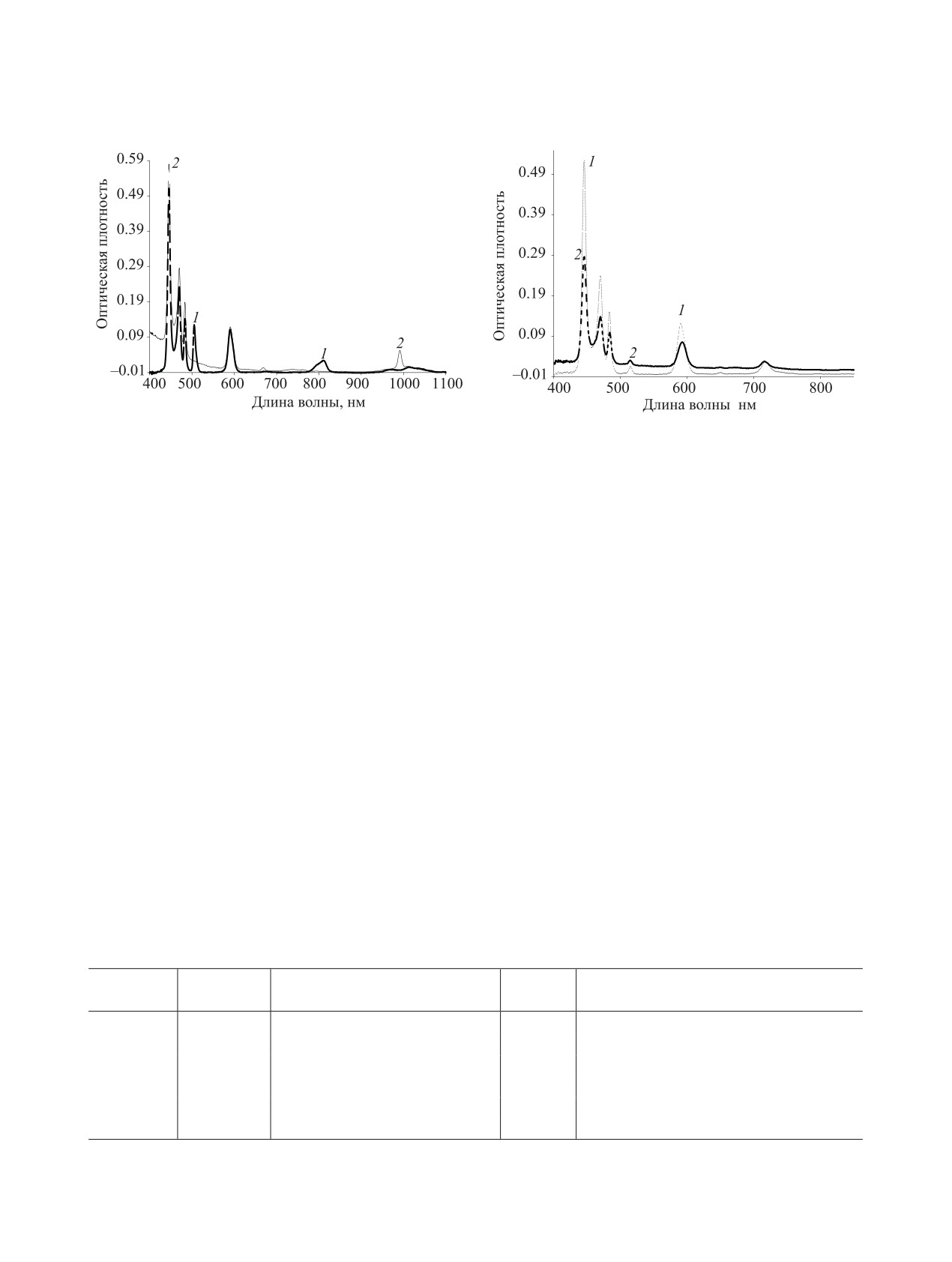
На рис. 1 показан спектр (1) исходного раствора
Am(VI) в 0.1 моль/л HNO3, предварительно полу-
0.1 моль/л HNO3, содержащего трехвалентные Am,
ченного, как описано выше. Авторы работы [20] со-
Cm и Pr с концентрациями, приведенными в табл. 1,
общают о возможности прямого получения Am(V)
а также спектр (2) того же раствора после количе-
в 0.1 M HNO3 при нагревании до 80-100°С и кон-
ственного окисления Am(III) до Am(VI) по мето-
центрации персульфата аммония 1 моль/л.
дике, описанной в экспериментальной части. Как
Полноту окисления Am(III) до Am(VI) и его по-
видно из рис. 1, присутствие в растворе даже почти
следующего восстановления до Am(V), их поведе-
100-кратного избытка Pr по сравнению с Am (10.5 и
ние при экстракции, а также концентрацию Pr(III)
0.13 г/л соответственно) не влияет на процесс окис-
в растворе контролировали спектрофотометриче-
ления Am(III).
ским методом (Unicam UV-300, Unicam Instruments,
Предварительно исследовали экстракцию
Великобритания) по полосам поглощения при 503 и
Am(VI) и трехвалентных Cm и Pr из 0.1 моль/л
814 нм для Am(III), 513 и 717 нм для Am(V), 666 и
HNO3 в присутствии 8 моль/л NH4NO3 30%-ным
996 нм для Am(VI) и 445 и 592 нм для Pr.
ТБФ в Изопаре М (табл. 1). Из приведенных в
В экстракционных экспериментах использова-
табл. 1 данных видно, что в исследуемых условиях
ли 30%-ный раствор ТБФ в Изопаре М, который
с высокой степенью экстрагируются (до 95 мас%)
предварительно контактировали в течение 5 мин с
как трехвалентные Cm и Pr, так и Am(VI). Коэффи-
равным объемом раствора 0.1 моль/л HNO3, содер-
циенты распределения (D) Am(VI), Cm(III) и Pr(III)
жащего 0.2 моль/л (NH4)2S2O8.
за одну экстракцию составили около 2.5, 3.6 и 1.8
РАДИОХИМИЯ том 65 № 2 2023
ВЫДЕЛЕНИЕ АМЕРИЦИЯ ИЗ АКТИНИД-ЛАНТАНИДНОЙ ФР
АКЦИИ
191
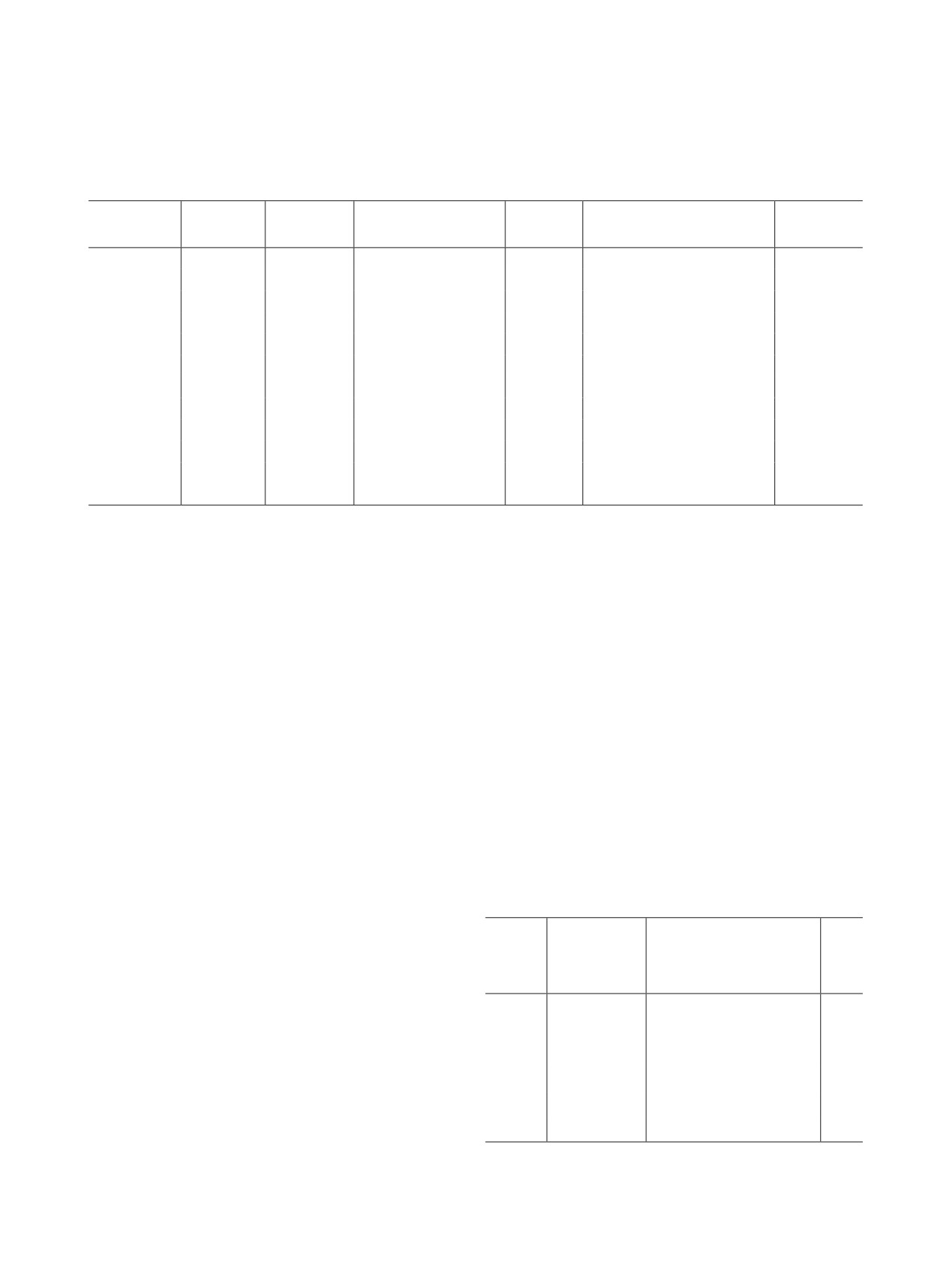
Рис. 1. Спектры поглощения Pr(III) и Am(III) в 0.1 моль/л
Рис. 2. Спектры раствора Am(V) и Pr(III) в 0.1 моль/л
HNO3 (1) и того же раствора после количественного
HNO3 до (1) и после (2) внесения в него высаливателя
окисления Am(III) до Am(VI) (2).
(8 моль/л NH4NO3).
соответственно. DAm(VI) = 2.5 соотносится с данны-
успешно экстрагируется 30%-ным ТБФ в Изопаре
ми работы [21], где максимальный коэффициент
М из 0.1 моль/л HNO3 в присутствии высаливателя
распределения был достигнут при 4 М HNO3 и ми-
NH4NO3, поэтому этот подход к отделению Am(VI)
нимальном времени контакта фаз, а также несколько
от Cm и лантанидов в данных условиях технологи-
ниже, чем в статье, где для экстракции использовал-
чески нереализуем.
ся 100%-ный ТБФ [16]. Спектрофотометрическим
Исследовали экстракцию Am(V) и трехвалент-
методом подтвердили также, что в ОФ Am(VI) вос-
ных Cm и Pr из 0.1 моль/л HNO3 в присутствии
станавливается в основном до Am(V) и частично до
8 моль/л NH4NO3 30%-ным ТБФ в Изопаре М.
Am(III), а после 1 сут хранения Am(V) полностью
Предварительно подтвердили, что добавление вы-
переходит в Am(III). Однако в отдельных работах
саливателя NH4NO3 к раствору не приводит к из-
отмечены противоречия по вопросу устойчивости
менению состояния окисления Am(V) (рис.
2).
Am(VI) в присутствии ТБФ. Так, авторы работ [14,
Выполнили эксперименты, в которых содержание
16] указывают на относительную устойчивость
Pr составляло 1.05 или 10.5 г/л при постоянном со-
Am в присутствии ОФ, в тоже время в работе [21]
держании Am(V) и Cm; экстракцию проводили при
заявлено, что контакт с ОФ приводил к быстрому
соотношении ВФ : ОФ = 1 : 5.
восстановлению америция, в результате чего полу-
чался более низкий D(Am), чем ожидалось. Таким
Из представленных в табл. 2 результатов видно,
образом, из данных табл. 1 можно сделать вывод,
что различное содержание Pr не приводит к изме-
что Am(VI), так же как трехвалентные Cm и Pr,
нению степени извлечения Am(V) за одну стадию
Таблица 1. Экстракция Am(VI), Cm(III) и Pr(III) из 0.1 моль/л HNO3 в присутствии высаливателя (8.0 моль/л NH4NO3)
30%-ным ТБФ в Изопаре М. Содержание Am, Cm и Pr в исходном растворе 0.13, 1.38 × 10-4 и 10.5 г/л соответственно;
соотношение объемов ВФ : ОФ = 1 : 5
Содержание элемента в растворе, мас%
Раствор
Элемент
Концентрация элемента, г/л
D
от исходного
Экстракт
Am
2.42 × 10-2
2.5
92.7
Cm
2.62 × 10-5
3.6
94.7
Pr
1.89
1.8
90.0
Рафинат
Am
9.49 × 10-3
-
7.3
Cm
7.31 × 10-6
-
5.3
Pr
1.05
-
10
РАДИОХИМИЯ том 65 № 2 2023
192
ОСИН и др.
Таблица 2. Экстракция Am(V), Cm(III) и Pr(III) из 0.1 моль/л HNO3 в присутствии высаливателя (8.0 моль/л
NH4NO3) 30%-ным ТБФ в Изопаре М. Концентрации Am, Cm и Pr в исходном растворе 0.13, 1.38 × 10-4 и 10.5 мг/мл
соответственно; соотношение объемов ВФ : ОФ = 1 : 5
Концентрация
Содержание элемента в
№ опыта
Раствор
Элемент
D
SF
элемента, мг/мл
растворе, мас%
1
Рафинат
Am
8.64 × 10-2
67
Cm
2.35 × 10-5
17
Pr
1.58
15
Экстракт
Am
8.72 × 10-3
0.10
33
Cm
2.29 × 10-5
0.99
83
9.9а
Pr
1.78
1.15
85
11.5б
2
Рафинат
Am
8.77 × 10-2
68
Cm
2.21 × 10-5
16
Pr
0.21
20
Экстракт
Am
8.46 × 10-3
0.10
32
Cm
2.32 × 10-5
1.05
84
10.5а
Pr
0.17
0.81
80
8.1б
а Фактор разделения SFСm/Am.
б SFPr/Am.
экстракции. Коэффициент распределения Am(V)
ЗАКЛЮЧЕНИЕ
оказался равным около 0.1; америций извлекается
в ОФ не более, чем на 30% от его содержания в ис-
Установлено, что около 30% Am(V) извлекается
ходном растворе, что согласуется с данными рабо-
30%-ным раствором ТБФ в Изопаре М из 0.1 моль/л
ты [13]. При этом коэффициенты распределения Pr
HNO3 в присутствии 8 моль/л NH4NO3 за одно-
и Cm оказались весьма близки - около 1 (табл. 2), а
стадийную экстракцию при отношением объемов
степень их извлечения составляла 80-85% за один
цикл экстракции.
ВФ : ОФ = 1 : 5, в то время как Cm и Pr извлека-
При этом отмечено, что фактор разделения
ются более, чем на 80%. При этом на примере Pr
SFСm/Am оказался существенно ниже, чем в рабо-
показано, что замена одностадийной экстракции с
те [13], когда удавалось извлекать Cm практически
отношением ВФ : ОФ = 1 : 5 на трехстадийную с
нацело в ОФ и SFСm/Am составлял 65. Очевидно,
это можно объяснить влиянием присутствующего в
Таблица 3. Экстракция Pr(III) из 0.1 моль/л HNO3
растворе более чем стократного количества Pr отно-
в присутствии высаливателя
(8.0 моль/л NH4NO3)
сительно исходного содержания Am, а также следу-
30%-ным ТБФ в Изопаре М при использовании
трехстадийной экстракции при соотношении объемов
ет учитывать, что концентрации Am, Cm в исходном
ВФ : ОФ = 1 : 1
растворе были на несколько порядков выше (0.13 и
Содержание РЗЭ
1.38 × 10-4 соответственно), чем в работе [13].
№
[Pr(III)],
в растворе от его
D
В то же время показано, что фактор разделения
стадии
0.046 M
исходного содержания,
может быть повышен при использовании трех-
мас%
стадийной экстракции при соотношении объемов
1-ая
Рафинат-1
51.2
1.0
ВФ : ОФ = 1 : 1, как это было продемонстрировано
Экстракт-1
48.8
на примере Pr в отсутствие ТПЭ (табл. 3). В этом
2-ая
Рафинат-2
10.5
8.5
случае степень его извлечения достигала 98%, даже
Экстракт-2
89.5
несмотря на то, что суммарный объем экстрагента
3-я
Рафинат-3
1.2
82.3
был в 1.6 раза меньше, чем в случае одностадийной
Экстракт-3
98.8
экстракции с отношением ВФ : ОФ = 1 : 5.
РАДИОХИМИЯ том 65 № 2 2023
ВЫДЕЛЕНИЕ АМЕРИЦИЯ ИЗ АКТИНИД-ЛАНТАНИДНОЙ ФР
АКЦИИ
193
отношением объемов ВФ : ОФ = 1 : 1 позволяет ко-
5.
Morss L.R., Edelstein N.M., Fuger J. The Chemistry
личественно извлекать Cm и Pr.
of Actinide and Transactinide Elements. New York:
Springer, 2006. Vol. 2.
Основной проблемой использования Am(V) для
6.
Morss L.R., Edelstein N.M., Fuger J. The Chemistry
его отделения от Cm и РЗЭ является его восстанов-
of Actinide and Transactinide Elements. New York:
ление до хорошо экстрагируемого в этих условиях
Springer, 2006. Vol. 3.
Am(III), происходящее под действием продуктов ра-
7.
Matveev P.I., Mohapatra P.K., Kalmykov S.N.,
диолиза [22], диспропорционирования Am(V) [23]
Petrov V.G. // Solvent. Extr. Ion Exch. 2021. Vol. 39,
и вследствие контакта с ОФ. Нахождение условий,
N 7. P. 679.
8.
Hudson M.J., Harwood L.M., Laventine D.M.,
при которых извлечение Am(V) в ТБФ будет мини-
Lewis F.W. // Inorg. Chem. 2013. Vol. 52, N 7. P. 3414.
мальным без замены ТБФ на экстрагент TODGA,
9.
Runde W.H., Mincher B.J. // Chem. Rev. 2011. Vol. 111,
пока недоступный для промышленной технологии,
N 9. P. 5723.
как это сделано в работах [24, 25], позволит достичь
10. Mincher B.J.; Martin L.R.; Schmitt N.C. // Solvent Extr.
количественного отделения америция от кюрия, а
Ion Exch. 2012. Vol. 30, N 5. P. 445.
также лантанидов.
11. Мясоедов Б.Ф., Молочникова Н.П., Лебедев И.А //
Таким образом, важной задачей будущих иссле-
ЖАХ. 1971. Т. 26, № 10. С. 1984.
12. Каралова З.В., Букина Т.И., Лавринович Е.А., Трофи-
дований является стабилизация америция в его ма-
мов Т.И., Куляко Ю.М., Мясоедов Б.Ф. // Радиохимия.
лоэкстрагируемой форме Am(V).
1988. Т. 30, № 2. С. 203.
13. Куляко Ю.М., Маликов Д.А., Трофимов Т.И., Вино-
ФОНДОВАЯ ПОДДЕРЖКА
куров С.Е., Пилюшенко К.С., Зевакин Е.А., Мясое-
дов Б.Ф. // Радиохимия. 2018. Т. 60, № 1. С. 19.
Работа выполнена при финансовой поддержке
14. Kamoshida M., Fukasawa T., Kawamura P. // J. Nucl.
Минобрнауки России (соглашение о предоставле-
Sci. Technol. 1999. Vol. 36, N 6. P. 535.
15. Koma Y., Aoshima A., Kamoshida M,; Sasahira A. // J.
нии гранта № 075-15-2020-782).
Nucl. Sci. Technol. 2002. Vol. 39, N 3. P. 317.
16. Kamoshida M., Fukasawa T. // J. Nucl. Sci. Technol.
КОНФЛИКТ ИНТЕРЕСОВ
1996. Vol. 33, N 5. P. 403.
17. Фёдоров Ю.С., Куляко Ю.М., Блажева И.В., Го-
Авторы заявляют об отсутствии конфликта ин-
лецкий Н.Д., Зильберман Б.Я., Металиди М.М., Пе-
тересов.
тров Ю.Ю., Рябкова Н.В., Винокуров С.Е., Трофи-
мов Т.И., Мясоедов Б.Ф. // Радиохимия. 2016. Т. 58,
№ 3. С. 229.
СПИСОК ЛИТЕРАТУРЫ
18. Ермаков В.А., Рыков А.Г., Тимофеев Г.А. // Радиохи-
мия. 1971. T. 13. C. 826.
1.
OECD-NEA Potential Benefits and Impacts of Advanced
19. Moore F.L. // Anal. Chem. 1963. Vol. 35. P. 715.
Nuclear Fuel Cycles with Actinide Partitioning and
20. Mincher B.J., Schmitt N.C., Schuetz B.K., Shehee T.C.,
Transmutation; Nuclear Energy Agency (NEA). Paris:
Hobbs D.T. // RSC Adv. 2015. Vol. 5. P. 27205.
OECD, 2011. P. 78.
21. Mincher B.J., Martin L.R., Schmitt N.C. // Inorg. Chem.
2.
Винокуров С.Е., Куляко Ю.М., Маликов Д.А., Пере-
2008. Vol. 47. P. 6984.
валов С.А., Пилюшенко К.С., Савельев Б.В., Трофи-
22. Hall G.R., Herniman P.D. // J. Chem. Soc. 1954. P. 2214.
мов Т.И., Федоров Ю.С. // Патент RU 2774155. При-
23. Horne G.P., Grimes T.S., Bauer W.F., Dares C.J.,
оритет от 27.07.2021
Pimblott S.M., Mezyk S.P. Mincher B.J. // Inorg. Chem.
3.
Modolo B.G., Wilden A., Geist A., Magnusson D.,
2019. Vol. 58, N 13. P. 8551.
Malmbeck R.A. // Radiochim. Acta. 2012. Vol. 100.
24. Wang Lu J.B., Dong X., Yan Q., Feng X.G., Hu H.S.,
P. 715.
Wang S.A., Chen J., Li J., Xu C. // J. Am. Chem. Soc.
4.
Salvatores M., Slessarev I., Ritter G., Fougeras P.,
2022. Vol. 144, N 14. P. 6383.
Tchistiakov A., Youinou G., Zaetta A. Nucl. Instrum.
25. Wang Z.P., Dong X., Yan Q., Chen J., Xu C. // Anal.
Meth. Phys. Res. A. 1998. Vol. 414, N 1. P. 5.
Chem. 2022. Vol. 94, N 22. P. 7743.
РАДИОХИМИЯ том 65 № 2 2023