РАДИОХИМИЯ, 2023, том 65, № 3, с. 214-225
УДК 66.061.16:546.79
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ
В КАРБОНАТНЫХ РАСТВОРАХ1
© 2023 г. Н. М. Червякова, б, А. В. Бояринцева, б, *,
Г. В. Костиковав, С. И. Степанова, б
а Российский химико-технологический университет им. Д.И. Менделеева,
125047, Москва, Миусская пл., д. 9
б Озерский технологический институт Национального ядерного университета «МИФИ»,
456783, Озерск Челябинской обл., пр. Победы, д. 48
в Институт физической химии и электрохимии им. А.Н. Фрумкина РАН,
119071, Москва, Ленинский пр., д. 31, корп. 4
* e-mail: aboyarincev@muctr.ru
Поступила в редакцию 08.12.2022, после доработки 17.01.2023, принята к публикации 18.01.2023
Стадия растворения уранового и смешанного уран-плутониевого оксидного отработавшего ядерного
топлива является ключевой начальной стадией новой альтернативной гидрометаллургической
технологии - КАРБЭКС-процесса. В работе рассмотрены карбонатные окислительные системы
NaHCO3/Na2CO3-H2O2/2Na2CO3·3H2O2/М2S2O8, где M = Na+, K+ или NH+, для растворения порошков
оксидов актиноидов. Определены химические и физические факторы, определяющие скорость
окислительного растворения порошков индивидуальных оксидов UO2, U3O8, PuO2 и NpO2 в карбонатных
средах. Полученные результаты являются важными для разработки вариантов окислительного и
сонохимического растворения высокопрокаленных кристаллических образцов оксидов урана, плутония
и нептуния, а также отработавшего ядерного топлива в карбонатных средах.
Ключевые слова: диоксид урана, октаоксид триурана, диоксид плутония, диоксид нептуния,
окислительное растворение, карбонатные среды, пероксид водорода, персульфат
DOI: 10.31857/S0033831123030024, EDN: ENKEJB
ВВЕДЕНИЕ
11]. В условиях электрохимического анодного или
реагентного окислительного растворения в присут-
Принципиальная возможность извлечения урана
ствии пероксида водорода оксиды UO2 и U3O8 бы-
из техногенных и минеральных источников в кар-
стро и полностью растворяются в водных растворах
бонатных окислительных системах определила их
Na2CO3 или (NH4)2CO3, а продукты деления (ПД),
применение в новых подходах некислотной перера-
такие как редкоземельные элементы РЗЭ(III), Ru,
ботки ОЯТ и РАО в Японии, Южной Корее, США и
Rh, Pd, Sr, Ba, остаются в нерастворимом остатке.
Российской Федерации [1-6]. Карбонатные окисли-
Таким образом, уже на стадии растворения ОЯТ в
тельные среды рассматриваются в качестве нетра-
карбонатных средах происходит фракционирование
диционных альтернативных систем для растворе-
ПД. В тоже время Cs, Mo, частично Zr и Tc пере-
ния диоксида урана UO2 как основного компонента
ходят в карбонатный раствор совместно с ураном,
ОЯТ и октаоксида триурана U3O8 как основного
компонента волоксидированного ОЯТ, а также для
поэтому возникает необходимость в проведении
выщелачивания урана из различных материалов [7-
дополнительных аффинажных операций с целью
-------------
получения очищенных соединений урана для фа-
1По материалам доклада на Х Российской конференции с меж-
брикации новых партий оксидного уранового или
дународным участием «Радиохимия-2022» (Санкт-Петербург,
26-30 сентября 2022 г.)
смешанного уран-плутониевого топлива.
214
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
215
Важным требованием к современным радиохи-
топлива ((U,Gd)O2 [16, 17], твердых отходов про-
мическим технологиям является возможность пере-
изводства изотопа 99Mo [18-20], урансодержащего
рабатывать ОЯТ с большой глубиной выгорания, ха-
шлама и известкового осадка [21, 22], а также при
рактеризующееся высоким содержанием плутония,
растворении металлического урана [23].
минорных актиноидов и других ПД. Выделение и
Скорость окислительного растворения оксидов
фракционирование плутония, нептуния и америция
урана в карбонатных растворах в присутствии H2O2
расширяет возможности радиохимической техноло-
возрастает с увеличением доли U(VI) в оксиде в
гии, позволяет повысить глубину и комплексность
следующем порядке: UO2 < U3O8 < UO3·H2O [24-
переработки ОЯТ, вовлечь делящиеся материалы в
26]. Растворимость UO2 и U3O8 в карбонатных сре-
ядерный топливный цикл и снизить количество вы-
дах повышается с ростом концентрации карбоната,
сокотоксичных радиоактивных отходов, подлежа-
окислителя и температуры среды [7-11, 16]. Ско-
щих захоронению.
рость растворения UO2 или U3O8 в карбонатно-пе-
Выбор карбонатных сред для переработки ок-
роксидных растворах в зависимости от природы
сидного уранового и уран-плутониевого ОЯТ свя-
катиона карбонатной соли возрастает в следующем
зан с высокой растворимостью и стабильностью
ряду: Li2СО3 > Na2СО3 > K2СО3 > (NH4)2СО3 [7].
карбонатных комплексов актиноидов [12], а также
Значительное влияние на скорость окислитель-
высокой селективностью и широкой вариативно-
ного растворения оксидов урана оказывают такие
стью по составу карбонатных сред, обусловленных
свойства исходных порошков, как удельная поверх-
уникальностью координационной химии карбонат-
ность, распределение частиц по размерам и их мор-
ных и смешанных карбонатно-пероксидных, карбо-
фология [27].
натно-фторидных и других комплексов актиноидов.
В зависимости от состава среды и других ус-
Высоким потенциалом для применения в процессах
ловий, H2O2 может проявлять как окислительные,
переработки ОЯТ и РАО обладают карбонатно-пе-
так и восстановительные свойства. Пероксид водо-
роксидные системы. Роль H2O2 в таких системах не
рода обладает подходящим окислительно-восста-
ограничивается только окислением U(IV). В отли-
новительным потенциалом (ОВП) для окисления
чие от кислотных сред, добавка пероксидного ли-
в порошках UO2 или U3O8 до UO3, в то время как
ганда в карбонатный раствор не приводит к осажде-
порошки PuO2 и NpO2 H2O2 практически не окисля-
нию урана. В растворах Na2CO3 образуются устой-
ются [8]. Для перевода Pu(IV) и Np(IV) из оксидных
чивые смешанные карбонатно-пероксидные моно-
фаз (MeO2-х) в составе ОЯТ [28] в карбонатный рас-
ядерные {Na4[UO2(O2)x(CO3)3-х]} и полиядерные
твор требуется их окисление до высших валентных
{Na4[(UO2)2(O2)2(CO3)2], Na6[(UO2)3(O2)2(CO3)4]}
состояний. В случае анодного растворения ОЯТ в
анионные комплексы, обладающие более высокой
карбонатных средах Np(IV) и Pu(IV) в составе ин-
растворимостью - 150-180 г/л U(VI) - по сравне-
дивидуальных оксидных фаз могут быть окислены
нию с комплексом Na4[UO2(CO3)3] [1-11]. Pu(IV),
до Np(VI) и Pu(VI) и стабилизированы в этом ва-
Np(IV) и Np(VI) также склонны к образованию
лентном состоянии озоном при его барботаже через
карбонатно-пероксидных комплексов
[7, 13, 14],
слой суспензии [29]. Однако поведение индивиду-
однако химия и свойства таких соединений пока
альных оксидов трансурановых элементов (ТУЭ)
мало изучены. Регулирование параметров процес-
и в составе ОЯТ в процессе окислительного рас-
са окислительного растворения, таких как концен-
творения в карбонатно-пероксидных средах будет
трация и тип окислителя, карбонатного реагента и
отличаться. Во-первых, при воздействии высоких
рН раствора, позволяет селективно или совместно
радиационных полей при переработке ОЯТ, про-
с минорными актиноидами переводить U(VI) в кар-
исходит радиолиз воды с образованием H2O2, O2 и
бонатный раствор.
различных радикальных продуктов. Радикалы O• и
Эффективность использования растворов сме-
OH• обладают чрезвычайно сильными окислитель-
сей Na2CO3-H2O2, (NH4)2CO3-H2O2 в качестве рас-
ными свойствами, в том числе и в щелочных сре-
творителя была продемонстрирована при извлече-
дах, что повышает окислительно-восстановитель-
нии U(VI) из образцов коммерческого ОЯТ [15], от-
ный потенциал (ОВП) растворительной системы.
ходов производства уран-гадолиниевого оксидного
Несмотря на низкий выход, в присутствии продук-
РАДИОХИМИЯ том 65 № 3 2023
216
ЧЕРВЯКОВ и др.
тов радиолиза может происходить окислительное
рения оксидов актиноидов в карбонатных средах.
(радиолитическое) растворение NpO2 и PuO2 в во-
Изучение химического поведения оксидов актинои-
дных растворах даже в отсутствие комплексообра-
дов в карбонатных окислительных системах важно
зующих лигандов СO2- и HCO– [30, 31]. При этом
с точки зрения как фундаментальной науки, так и
скорость радиолитического растворения уменьша-
практического применения при обращении с ради-
ется в ряду UO2 > NpO2 > PuO2. Во-вторых, ТУЭ в
оактивными отходами в ядерном топливном цикле,
ОЯТ обычно находятся не только в виде интегри-
включая хранение и переработку ОЯТ и РАО.
рованных в основную фазу UO2 самостоятельных
В настоящей работе исследовано окислительное
фаз отдельных соединений, но и в большей степени
растворение порошков диоксидов урана, плутония,
в виде твердого раствора в фазе UO2. В результа-
нептуния и октаоксида триурана в водных карбо-
те воздействия высоких температур (до 1700°С) и
натных растворах.
чрезвычайно интенсивных радиационных полей
при работе ядерного реактора может протекать об-
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
разование твердых растворов включения оксидов
ТУЭ в матрице UO2 [32-34]. Возможность количе-
В работе использовали кристаллические соедине-
ственного извлечения урана и ТУЭ из ОЯТ на осно-
ве UO2 c глубиной выгорания 30-45 ГВт·сут/(т U)
ния Na2CO3, перкарбонат натрия (2Na2CO3·3H2O2),
после 27-28-летней выдержки в водных растворах
NH4HCO3, (NH4)2S2O8, Na2S2O8, K2S2O8, 8-оксихи-
(NH4)2CO3-H2O2 была продемонстрирована в рабо-
нолин, динатриевую соль этилендиаминтетрауксус-
те [15].
ной кислоты (Трилон Б) и 30%-ный раствор H2O2
квалификации х.ч. Для приготовления водных рас-
Из-за недостаточной изученности поведения
творов использовали дистиллированную воду.
оксидов PuO2 и NpO2 в процессах окислительно-
го растворения в карбонатных средах нет полного
В работе использовали диоксид плутония-239,
понимания, как наиболее эффективно растворять
полученный из оксалата плутония-239 при терми-
оксидные фазы этих элементов в карбонатных сре-
ческой выдержке в течении 4 ч при 1200°С в воз-
дах. Поэтому возникает необходимость проведения
душной атмосфере, представляющий кристалли-
более детальных систематических исследований,
ческий порошок оливково-зеленого цвета. Также
результаты которых позволят определить наиболее
использовали порошки диоксида нептуния-237 и
эффективные альтернативные системы и оценить
оксидов природного урана: UO2.25, U3O8. Значения
их потенциал для применения в процессах раство-
величины удельной поверхности (Sуд) используе-
рения оксидного уранового и смешанного уран-плу-
мых в работе образцов порошков оксидов актино-
тониевого ОЯТ в новом водно-химическом процес-
идов представлены в табл. 1. Образцы порошков
се - КАРБЭКС-процессе. Использование персуль-
U3O8 получали при термической обработке порош-
фатов щелочных металлов или аммония в качестве
ка UO2.25 в атмосфере воздуха в режиме 120 мин
окислителей Pu(IV) и Np(IV), привлечение методов
нагрев-120 мин изотермическая выдержка-мед-
химической и механической интенсификации мо-
ленное охлаждение вместе с печью. С повышением
гут стать определяющими факторами повышения
температуры обработки UO2.25 происходит сниже-
скорости растворения кристаллических порошков
ние Sуд образцов U3O8, что обусловлено увеличе-
оксидов актиноидов, прокаленных при высокой
нием среднего размера частиц (табл. 1). Средний
температуре, в том числе продуктов высокотем-
размер частиц (D50) для порошков U3O8 возрастает
пературной волоксидации, обладающих прочной
кристаллической решеткой, в карбонатных окисли-
Таблица 1. Величины Sуд и D50 образцов порошков PuO2,
тельных средах. Решение сложной задачи полного
UO2.25 и U3O8
окислительного растворения PuO2 и NpO2 совмест-
Образец
PuO2
UO2.25
U3O8
но с UO2 или U3O8 в карбонатных растворах позво-
tпрок, °C
1200
-
480
600
800
1000
1200
лит в значительной степени расширить возможно-
Sуд, м2/г
0.2
3.3
3.8
3.7
1.8
0.8
0.1
сти КАРБЭКС-процесса в технологии переработки
D50
23.5
2.6
5.8
5.9
8.6
9.5
32.2
ОЯТ и РАО, а также обосновать варианты раство-
РАДИОХИМИЯ том 65 № 3 2023
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
217
от 2.6 до 32.2 мкм при увеличении температуры об-
агентов в реактор включали подачу газообразного
работки (tпрок) UO2.25 от 480 до 1200°С.
СО2, расход которого 0.2 л/мин устанавливали при
помощи газового расходомера. Процесс проводили
Содержание U(VI) в растворах с концентрацией
выше 1.0 г/л определяли титрованием с раствором
в режиме 5 мин перемешивания магнитной мешал-
0.0084 моль/л ванадата аммония в присутствии ин-
кой - 5 мин УЗО.
дикатора - натриевой соли дифениламин-4-суль-
Окислительное растворение порошков PuO2 и
фокислоты [35]. Содержание U(VI) в растворах с
NpO2 проводили в пластиковых пробирках. Наве-
концентрацией ниже 1.0 г/л определяли спектро-
ску образца порошка оксида массой 2-5 мг взвеши-
фотометрическим методом с арсеназо III по погло-
вали c точностью ±0.1 мг на аналитических весах
щению зелено-голубого комплексного соединения
HR-250AZG (AND, Китай). Перемешивание про-
уранил-арсеназо (λmax = 653 нм, предел обнаруже-
водили при помощи магнитной мешалки со скоро-
ния ~0.5 мкг/л) [36].
стью 500 об/мин. Для определения концентраций
Концентрации плутония и нептуния в водных
металлов из пробирки отбирали 0.5 мл жидкой
растворах определяли радиометрическим мето-
фазы. Перед анализом жидкие образцы центрифу-
дом по α-счету с использованием гамма-бета-
гировали при 5000 об/мин в течение 5-10 мин на
альфа-спектрометра-радиометра марки МКГБ-01
микроцентрифуге СМ-50 (ELMI, Латвия) для отде-
(НТЦ «РАДЭК», Россия) и интегрального аль-
ления остатков твердой фазы. Растворы после цен-
фа-спектрометра Alpha Analyst (Canberra, США).
трифугирования направляли на α-спектрометрию.
Концентрацию H2O2 в растворах определяли ти-
Для ультразвуковой интенсификации растворения
порошка NpO2 использовали ультразвуковую ванну
трованием аликвоты раствором 0.1 моль/л перман-
ганата калия. Водные растворы H2O2 использовали
Elmasonic xtra TT 30H (W = 130 Вт, ν = 37 кГц).
незамедлительно после приготовления.
Концентрацию CO2- и HCO– в растворе опреде-
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
ляли потенциометрическим титрованием аликвоты
раствором 0.1 моль/л HCl со стеклянным электро-
Окислительное растворение диоксида урана и
дом P13/BNC при использовании цифрового ионо-
октаоксида триурана в карбонатных средах
мера Elite 3320.
Удельную поверхность рассчитывали по изо-
Необходимым условием полноты растворения
терме адсорбции газообразного аргона на испыту-
UO2 и U3O8 в растворах M2CO3, где M - катион
емых образцах порошков, полученную на приборе
щелочного металла или аммония, является дости-
Quadrasorb Kr/SI (Quantachrome Instruments, США)
жение и поддержание требуемого значения ОВП в
с использованием программного обеспечения
системе. Пероксид водорода является бессолевым
QuadraWin (версия 5.02).
реагентом и имеет подходящий ОВП (200-250 мВ),
Окислительное растворение порошков UO2.25
который может окислять уран и демонстрирует вы-
и U3O8 проводили в стеклянном термостатиру-
сокую скорость окисления U(IV) по сравнению с
емом реакторе емкостью 100 мл при перемеши-
другими окислителями [7]. Пероксид ион является
вании суспензии магнитной мешалкой со скоро-
лигандом для UO22+-иона (пероксидная группа O22-
стью 500 об/мин в выбранном интервале темпера-
встраивается во внутреннюю координационную
тур (25-75) ± 0.1°С.
сферу карбонатных комплексов уранила, образуя
В случае ультразвуковой интенсификации про-
высоко растворимые в карбонатных растворах кар-
цесса в реактор помещали волновод, соединенный
бонатно-пероксидные комплексы), а протон сла-
с генератором ультразвуковых колебаний и пультом
бой кислоты, которой является H2O2, нейтрализу-
управления установки Булава-П УЗАП-3/22-ОП
ет NaOH, образующийся при растворении оксида.
(Центр ультразвуковых технологий, Россия). Уль-
Вариант однократного добавления стехиометриче-
тразвуковую обработку (УЗО) в процессе раство-
ского количества H2O2 в карбонатный раствор в на-
рения проводили при частоте (ν) 22 ± 1.65 кГц и
чале процесса, как правило, не позволяет добиться
интенсивности 10 Вт/см2. После загрузки всех ре-
полного растворения UO2 или U3O8. Избыток H2O2
РАДИОХИМИЯ том 65 № 3 2023
218
ЧЕРВЯКОВ и др.
полностью разлагается в растворе через 10-20 мин
влияющими на скорость растворения в карбонат-
перемешивания в зависимости от концентрации
ных средах.
карбоната и температуры [37], в результате чего
Химические и механические методы могут быть
карбонатная система быстро теряет окислительную
эффективными для интенсификации окислитель-
способность. Такого короткого интервала времени
ного растворения UO2 и U3O8 [38, 39]. Химическая
недостаточно для полноты окисления и взаимодей-
интенсификация заключается в добавлении ком-
ствия H2O2 c прокаленными и хорошо кристалли-
плексообразующих лигандов, которые образуют с
зованными порошками UO2 и U3O8. Для поддер-
уранилом хорошо растворимые стабильные ком-
жания постоянного значения ОВП в карбонатной
плексные анионы. В качестве таких реагентов, на-
окислительной системе необходимо осуществлять
пример, могут выступать Трилон Б и 8-оксихинолин
дробную подачу концентрированного раствора
[40]. Добавка 0.1 моль/л 8-оксихинолина или Три-
Н2О2 [27].
лона Б к смеси 1.0 моль/л Na2CO3-0.1 моль/л H2O2
Кристаллический
перкарбонат
натрия
в условиях одностадийного растворения при 75°С,
2Na2CO3·3H2O2 рассматривается в качестве аль-
Т : Ж = 1 : 50 и дробной подаче окислителя позво-
тернативного раствору H2O2 окислителя в карбо-
лила повысить степень растворения порошка U3O8
натных системах в отличие от пероксида натрия,
полученного при 1200°C, с 42.7 до 90% в случае
который характеризуется низкой эффективностью
раствора 0.1 моль/л 8-оксихинолина и более 99.9%
в процессах окислительного растворения порошков
в случае раствора 0.1 моль/л Трилона Б (табл. 2).
UO2 и U3O8. В процессе разложения пероксида на-
Ультразвуковая обработка суспензии порошка
трия происходит образование и накопление NaOH в
U3O8, выдержанного при 800°C, в смеси 1.0 моль/л
карбонатном растворе, что приводит к повышению
Na2CO3-0.1 моль/л H2O2 позволяет значительно
рН и выделению урана(VI) из раствора в виде мало-
увеличить скорость окислительного растворения и
растворимых полиуранатов.
получить карбонатные растворы, содержащие бо-
Порошкообразные образцы UO2 и образцы U3O8,
лее 150 г/л по U(VI) при снижении Т : Ж от 1 : 50
полученные в диапазоне температур 480-800°C,
до 1 : (3-5) [38]. Однако в этом случае требуется
могут быть быстро (в течение 90-180 мин) и коли-
регулировка рН карбонатного раствора для предот-
чественно (>99.9%) растворены в растворах, содер-
вращения образования малорастворимых соедине-
жащих
1.0 моль/л NaHCO3/Na2CO3-x моль/л H2O2
ний U(VI) из-за накопления щелочи, образующейся
где x = 0.1-0.5, в условиях дробной подачи H2O2
при растворении UO2 и U3O8. Такая корректировка
или
2Na2CO3·3H2O2. Повышение концентрации
рН может быть осуществлена, например, ограни-
H2O2, карбонатного реагента и температуры среды,
приводит к повышению скорости растворения UO2
ченной подачей газообразного CO2 в объем суспен-
и U3O8 (табл. 2), что согласуется с литературными
зии [38]. При низком содержании U(VI) в карбонат-
данными [7, 27].
ном растворе, когда степень превращения мала или
отношение Т : Ж повышено в пользу жидкой фазы,
Скорость растворения порошков U3O8, получен-
изменение рН при окислительном растворении не-
ных при температурах выше 1000°C, значительно
значительно вследствие буферных свойств карбо-
снижается, в результате не достигается полнота их
натных и/или бикарбонатных солей. Кроме того,
растворения даже при повышенном расходе окис-
механическое воздействие ультразвуковой кавита-
лителя и при непрерывном длительном агитаци-
онном перемешивании. Это вызвано уменьшением
ции на твердую фазу позволяет устранять образова-
удельной поверхности, увеличением размера ча-
ние сплошных пленок малорастворимых продуктов
стиц и кристалличности порошков U3O8 при повы-
окисления на поверхности оксида, приводящих к
шении температуры прокаливания исходного по-
повышению диффузионного сопротивления в гете-
рошка UO2. Таким образом, физические свойства -
рогенной системе и замедляющих скорость раство-
морфология, удельная поверхность, размер частиц
рения порошков оксидов урана вплоть до его пол-
порошков U3O8 - являются важными факторами,
ного прекращения.
РАДИОХИМИЯ том 65 № 3 2023
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
219
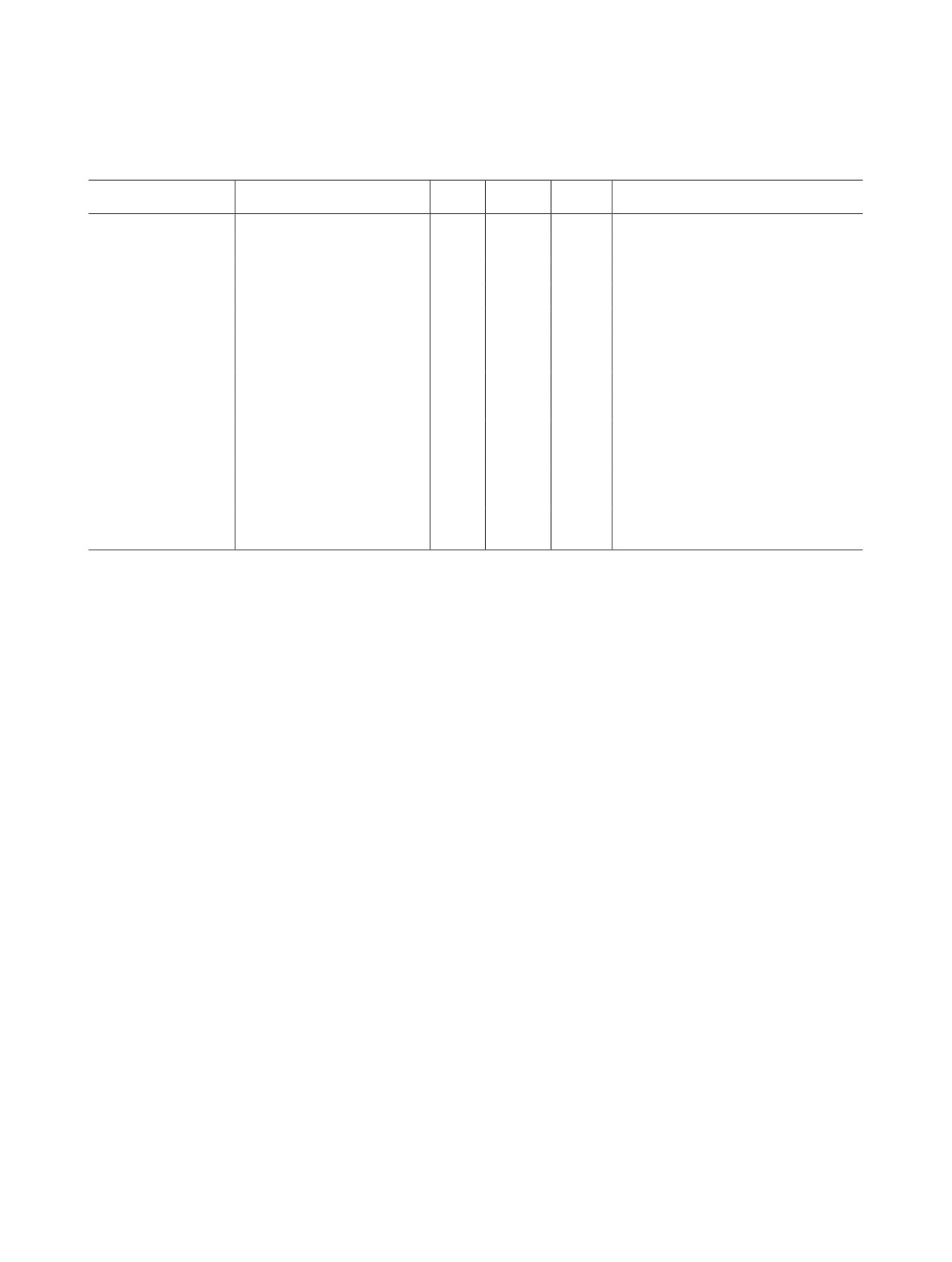
Таблица 2. Окислительное растворение порошков оксидов UO2 и U3O8 в карбонатных и бикарбонатных средах при
дробной подаче окислителя. t - температура среды, τ - время растворения, α - степень перевода порошка оксида в
раствор
Образец, tпрок, °C
Sуд, м2/г
Среда
Окислитель
t, °C
τ, мин
α, %
UO2.25
3.3
1.0 моль/л Na2CO3
0.1 моль/л H2O2
75
60
33.2
0.9 моль/л H2O2
60
45.0
1.0 моль/л NaHCO3
0.1 моль/л H2O2
25
90
62.6
75
60
80.0
1.0 моль/л Na2CO3а
0.1 моль/л 2Na2CO3-3H2O2
25
210
77.9
75
180
96.4
U3O8, 480°C
3.8
1.0 моль/л Na2CO3
0.1 моль/л H2O2
75
90
>99.9
U3O8, 800°C
1.8
120
89.0
U3O8, 1000°C
0.8
300
67.2
U3O8, 600оС
3.7
1.0 моль/л Na2CO3
0.1 моль/л H2O2
25
300
34.5
75
90
>99.9
U3O8, 1200°C
0.1
25
300
21.7
75
240
42.7
0.2 моль/л H2O2
62.3
0.5 моль/л H2O2
86.0
U3O8, 480°C
3.8
1.0 моль/л Na2CO3а
0.1 моль/л 2Na2CO3-3H2O2
25
210
96.4
75
60
>99.9
U3O8, 1200°C
0.1
25
270
38.0
75
150
99.7
U3O8, 480°C
3.8
1.0 моль/л NaHCO3
0.1 моль/л H2O2
25
210
47.2
75
30
>99.9
U3O8, 600оС
3.7
25
180
34.2
75
90
>99.9
U3O8, 800оС
1.8
25
240
31.9
75
180
>99.9
U3O8, 1200°C
0.1
25
210
8.0
75
66.9
1.0 моль/л Na2CO3
0.1 моль/л H2O2
50
240
54.3
0.1 моль/л
75
89.9
8-оксихинолин
1.0 моль/л Na2CO3
50
95.8
0.1 моль/л Трилон Б
75
>99.9
а Повышение концентрации Na2CO3 в карбонатном растворе до 1.0 моль/л происходит в процессе дробной подачи всех порции
кристаллического 2Na2CO3-3H2O2 в течение времени проведения процесса.
Окислительное растворение диоксида плутония
морфология, особенно температура и время про-
в карбонатных средах
каливания при получении порошка), так и условий
растворения. В области рН 7.5-9.6 при низких кон-
Порошки оксида PuO2 плохо растворяются не
центрациях карбоната растворимость кристалличе-
только в карбонатных, но и в азотнокислых раство-
рах. Растворимость PuO2 в этих средах зависит как
ского PuO2 и аморфных гидратированных порошков
от физических свойств порошков (размер частиц,
оксида PuO2∙xH2O не превышает 10-8 моль/л. По-
РАДИОХИМИЯ том 65 № 3 2023
220
ЧЕРВЯКОВ и др.
вышение концентрации карбоната в растворе со-
6
4
провождается увеличением растворимости оксида
Pu(IV) благодаря образованию устойчивых раство-
римых комплексных карбонатных анионов [41, 42].
4
Ключевым условием растворения порошков PuO2 в
карбонатных средах является окисление Pu(IV) до
3
Pu(VI), представляющее собой сложную задачу. В
2
2
присутствии H2O2 скорость растворения порошков
PuO2 в растворах Na2CO3 очень низкая. Раствори-
1
мость порошка PuO2 в карбонатном растворе изме-
0
няется в интервале 0.01-0.07 г/л Pu в зависимости
от способа его получения (температура и время
IJ ɦɢɧ
прокаливания) и условий растворения. Прокален-
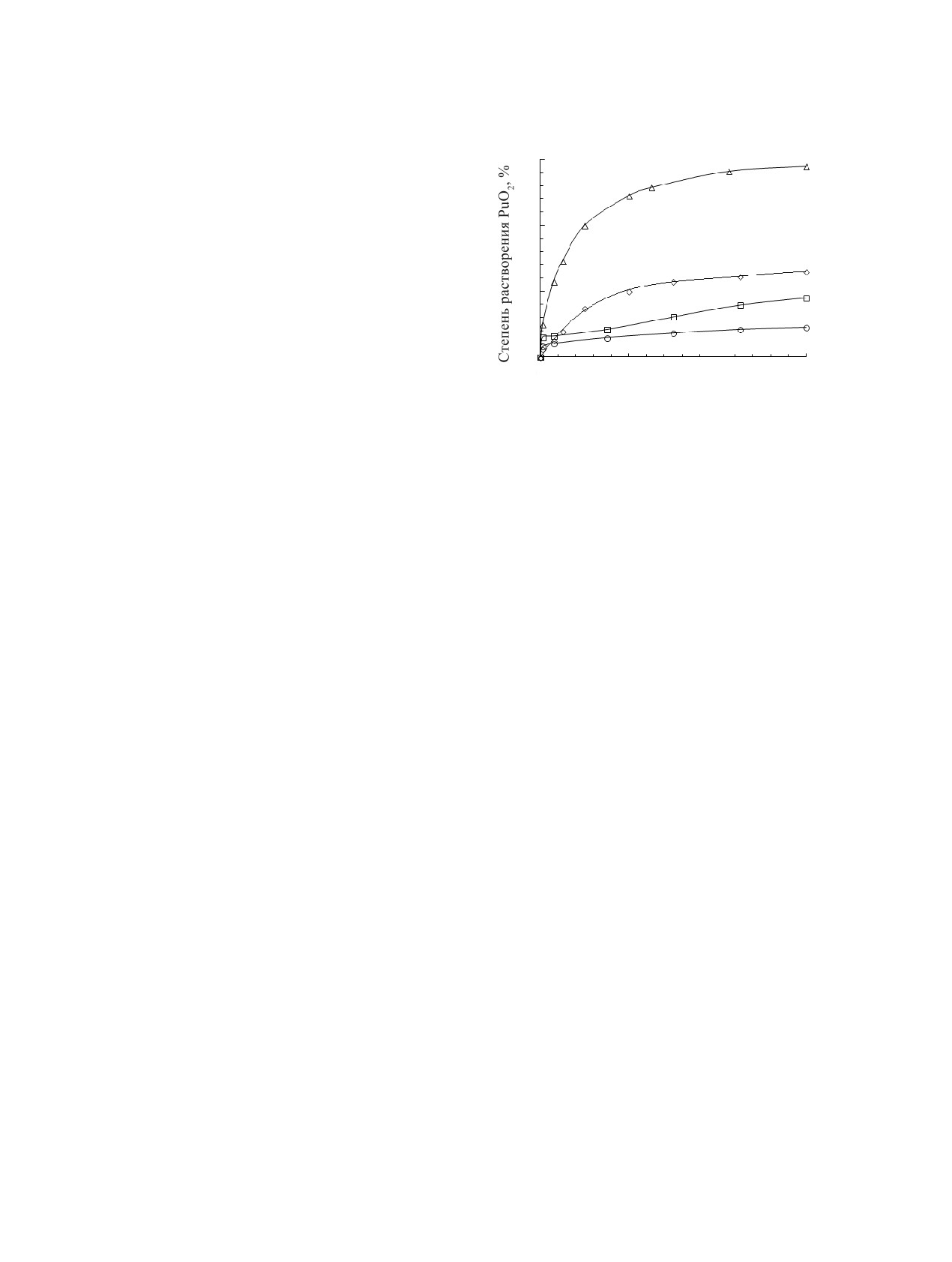
Рис. 1. Кинетические кривые растворения порошка PuO2
ный при высокой температуре порошок PuO2
прак-
в 1.0 моль/л Na2CO3 растворе в присутствии 0.2 моль/л
(NH4)2S2O8 при 20°С (1) и 2.0 моль/л (NH4)2S2O8 при
тически не растворяется в карбонатных растворах,
20 (2), 50 (3) и 85°С (4).
в то время как свежеосажденный Pu(OH)4 достаточ-
но хорошо растворяется в карбонатных и щелочных
мых карбонатных окислительных системах перок-
растворах [43].
сид-ион не проявляет окислительных свойств по
В качестве эффективных окислителей Pu(IV)
отношению к PuO2, но может принимать участие
в карбонатных средах хорошо зарекомендовали
в процессах образования комплексных смешанных
себя персульфаты щелочных металлов и аммо-
пероксо-карбонатных соединений Pu(IV) [2, 13],
ния. Применение персульфата калия в процессе
растворимость и устойчивость которых в карбо-
натных средах на данный момент мало изучены.
окислительного растворения ограничено его от-
носительно низкой растворимостью в водных кар-
В отсутствие H2O2 в системе Na2CO3-(NH4)2S2O8,
повышение температуры с 20 до 90°С приводит к
бонатных растворах. Добавка Na2S2O8 в раствор
повышению α(PuO2) с 6 до 24% (табл. 3).
Na2CO3 способствует окислению Pu(IV) до Pu(VI)
с образованием устойчивого растворимого соеди-
Одним из подходов к растворению PuO2 в кар-
нения Na4[PuO2(CO3)3], при этом растворимость
бонатных средах является предварительный пе-
окисленного плутония повышается до 0.5-1.0 г/л.
ревод диоксида в другую форму, которая легче
Растворимость «состаренного» в течение двух не-
переходит в раствор, чем исходное соединение.
дель Pu(OH)4 в растворе Na2CO3 в присутствии
В качестве возможного варианта такого подхода
0.1 моль/л K2S2O8 составляет 0.25 г/л Pu [43].
является конверсия диоксида в пероксидное со-
единение при вымачивании в концентрирован-
Режим дробной подачи окислителя в карбо-
ном водном растворе H2O2, при которой прочная
натный раствор является важным условием для
кристаллическая решетка диоксида разрушается
повышения степени растворения порошков PuO2
и перестраивается в решетку другого, более рас-
в растворах Na2CO3 - (NH4)2S2O8/Na2S2O8. Повы-
творимого соединения. Например, при обработке
шение температуры карбонатного раствора и кон-
гидроксида Pu(IV) концентрированным раствором
центрации окислителя, приводит к повышению
H2O2 происходит его трансформация в пероксид-
растворимости порошка PuO2 в карбонатной систе-
ное соединение Pu(IV), которое после окисления в
ме (рис. 1).
присутствии персульфат-иона легко растворяется в
В смеси (NH4)2S2O8 и Н2О2 скорость растворе-
0.5 моль/л Na2CO3 [43]. Без окисления персульфа-
ния порошка PuO2 в растворе 1.0 моль/л Na2CO3
том продукты пероксидной конверсии малораство-
повышается, а значение α(PuO2) за 180 мин пере-
римы в растворе 0.5-1.0 моль/л Na2CO3 [44], тогда
мешивания при дробной подаче H2O2 достигает
как в присутствии 0.5 моль/л (NH4)2S2O8 протека-
~25% (табл. 3).
ет окисление Pu(IV) и растворимость продуктов
Повышение температуры с 20 до 85°С позво-
пероксидной конверсии значительно повышается.
ляет повысить α(PuO2) на ~15%. В рассматривае-
Помимо малорастворимых в карбонатных средах
РАДИОХИМИЯ том 65 № 3 2023
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
221
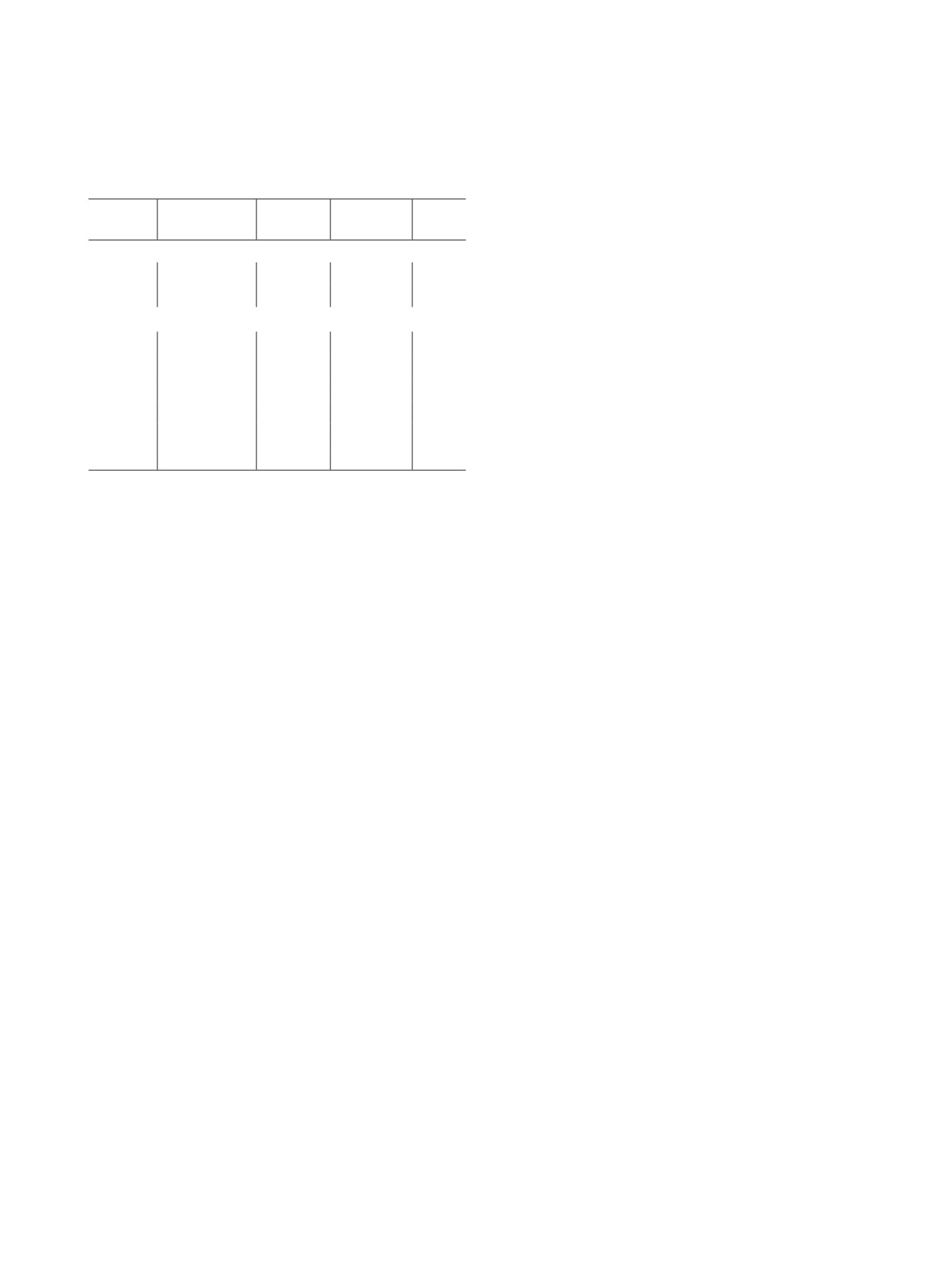
Таблица 3. Растворение порошка PuO2 в карбонатных окислительных средах при Т : Ж = 1 : 2000, различных темпе-
ратурах и режимах подачи окислителя
Среда
Окислитель
t, °С
τ, мин
α, %
Режим подачи окислителя
1.0 моль/л Na2CO3
0.2 моль/л (NH4)2S2O8
25
90
1.7
Разовая подача кристаллического
(NH4)2S2O8
2.0 моль/л (NH4)2S2O8
25
120
3.6
50
40
2.0
85
85
5.6
4.0 моль/л (NH4)2S2O8
95
120
18.4
2.0 моль/л Na2CO3
4.0 моль/л (NH4)2S2O8
25
60
6.0
Дробная подача кристаллического
(NH4)2S2O8
2.0 моль/л Na2CO3
4.0 моль/л (NH4)2S2O8
25
60
6.1
Разовая подача кристаллического
(NH4)2S2O8
1.0 моль/л Na2CO3
1.0 моль/л Na2O2
95
60
4.4
1.0 моль/л H2O2+
25
60
7.7
Разовая подача (NH4)2S2O8, дробная
1.0 моль/л (NH4)2S2O8
подача H2O2
1.0 моль/л H2O2+
85
180
24.8
1.0 моль/л (NH4)2S2O8
1.0 моль/л NH4HCO3
1.0 моль/л (NH4)2S2O8
85
120
2.1
Разовая подача кристаллического
(NH4)2S2O8
пероксидов Pu(IV) в содовых растворах образуется
В отсутствие добавок окислителя при длитель-
малорастворимое пероксо-карбонатное соединение
ной (в течение 24 ч) выдержке порошка NpO2 в рас-
Pu(IV) - Na3Pu2(O2)2(CO3)6∙12H2O [13].
творе 0.25 моль/л Na2CO3 с периодическим переме-
шиванием при температуре 20 ± 2°С, Т : Ж = 1 : 430
Окислительное растворение диоксида нептуния
в раствор переходит не более 1.4% от исходного ко-
в карбонатных средах
личества NpO2. После добавления в карбонатный
В окислительных условиях растворимость NpO2
раствор 0.5 моль/л H2O2 концентрация нептуния в
в нейтральных и основных растворах возрастает за
карбонатном растворе в течение 60 мин перемеши-
счет образования более растворимых окисленных
вания уменьшалась с 29 до 6.9 мг/л, что соответ-
форм. Для окисления Np(IV) до Np(V) и Np(VI) и
ствовало извлечению 0.3% от исходной массы NpO2.
перевода его в более растворимые в щелочных и
При увеличении времени перешивания до 120 мин
карбонатных средах формы могут быть использо-
и добавлении дополнительных порции 30%-ного
ваны кислород воздуха, озон, перманганат-, пер-
H2O2 в карбонатный раствор не наблюдали повы-
сульфат-, перхлорат-, пербромат-, ферроцианид-,
шения растворения порошка NpO2. Через 4 сут кар-
феррат- и хромат-ионы [45]. В карбонатно-перок-
бонатный раствор был окрашен в бледно-желтый
сидных растворах нептуний в различных степенях
цвет, концентрация нептуния в растворе составила
окисления участвует в окислительно-восстано-
9.1 мг/л, что соответствовало α(NpO2) = 0.4%.
вительных реакциях с образованием соединений
После выдержки порошка NpO2 без перемеши-
Np(V). Нептуний(VI) в составе карбонатного ком-
вания в течение 1 ч в 30%-ном растворе H2O2 при
плекса [NpVIO2(CO3)3]4- в присутствии стехиоме-
Т : Ж = 1 : 400 и комнатной температуре в раствор
трического количества H2O2 восстанавливается
переходит не более 1.0% NpO2. После выдержки в
до Np(V), а Np(IV) окисляется до Np(V). В обоих
течение 4 сут в раствор перешло около 1.6% NpO2.
случаях происходит образование карбонатного ком-
Незначительный переход нептуния в раствор мо-
плекса [NpVO2(CO3)3]5-. При добавлении избытка
жет быть обусловлен радиолитическим растворе-
H2O2 происходит образование аморфного осадка
нием NpO2 под действием продуктов радиолиза в
коричневого цвета [46], что приводит к снижению
отсутствие CO2-. При добавлении к выдержанно-
содержания нептуния в карбонатном растворе.
му в течение 4 сут порошку NpO2 водного раство-
РАДИОХИМИЯ том 65 № 3 2023
222
ЧЕРВЯКОВ и др.
Таблица 4. Растворение порошка NpO2 в смеси
урана и ТУЭ, а частности NpO2 и PuO2, в водных
Na2CO3-H2O2. [Na2CO3]исх
= 0.5 моль/л, t = 20 ± 2°С,
щелочных и карбонатных средах [48, 49]. В усло-
Т : Ж = 1 : 500
виях УЗО растворов Na2CO3-H2O2 может происхо-
[Na2CO3]равн
,
[H2O2]исх,
дить образование карбонатных радикалов, позволя-
τ, мин
[Np], мг/л
α, %
моль/л
моль/л
ющих ускорять химические процессы с участием
Без УЗО
нептуния.
60
0.38
2.3
2.6
0.14
Обработка в ультразвуковой ванне суспензии по-
150
0.34
1.0
15.0
0.87
рошка NpO2 в растворе 0.5 моль/л Na2CO3 в присут-
С УЗО
ствии H2O2 при прочих равных условиях позволяет
210
0.34
1.0
438.0
26.3
повысить растворимость NpO2 с 0.9 до ~ 26.3% за
210 мин (табл. 4). Однако увеличение времени УЗО
270
0.31
378.1
25.1
с 210 до 450 мин приводило к постепенному сниже-
330
0.28
338.0
24.6
нию α(NpO2) с 26.3 до 24.1%. Карбонатный раствор
390
0.28
334.7
24.4
после 450 мин УЗО был окрашен в желтый цвет и
450
0.28
330.1
24.1
содержал 330.1 мг/л Np. Через 24 ч концентрация
510
0.26
160.2
12.7
Np(VI) в карбонатном растворе снизилась с 330.1
) = 1.5%,
до 18.4 мг/л, что соответствовало α(NpO2
ра 2.0 моль/л Na2CO3 и последующей выдержке в
при этом наблюдали образование осадка желтого
ультразвуковой ванне в течение 30 мин извлечение
цвета. Снижение концентрации нептуния может
NpO2 в карбонатный раствор составило 7.2%. С по-
быть связано с гидролизом его карбонатных соеди-
вышением времени выдержки в ультразвуковой
нений в относительно разбавленных карбонатных
ванне извлечение NpO2 в карбонатный раствор сни-
растворах после сонохимического растворения и
жалось до 4.0% за 60 мин и до 3.7% за 120 мин, но
образованием малорастворимых продуктов гидро-
далее не изменялось. Повышение извлечения NpO2
лиза, выделяющихся из растворов в виде вторично-
в данных условиях может быть обусловлено воздей-
го осадка.
ствием ультразвуковых волн. Как отмечалось выше,
Рассмотренные варианты окислительного рас-
УЗО позволяет существенно ускорять окислитель-
творения порошка NpO2 в растворах Na2CO3 по-
ное растворение U3O8 в водных растворах Na2CO3
зволяют говорить о возможности перевода NpO2
и Na2CO3-H2O2, а также способствует получению
в карбонатные растворы в присутствии H2O2. Рас-
более концентрированных карбонатных растворов
творимость NpO2 в изученной карбонатной системе
U(VI) [4, 11]. Возникающие при прохождении уль-
существенно возрастает при УЗО, которая, по-ви-
тразвука через жидкие и жидко-дисперсные среды
димому, приводит к ускорению окисления NpO2 и
акустические течения улучшают перемешивание
последующего растворения окисленных форм неп-
и гомогенизацию системы, приводят к снижению
туния в карбонатном растворе. В то же время необ-
толщины диффузионного слоя на границе твердое-
ходимо проведение более детального исследования
жидкое, таким образом ускоряя массообменные
для уточнения протекающих в данной системе про-
процессы. Ультразвуковая кавитация, воздействуя
цессов и установления механизма сонохимического
на твердую фазу, измельчает ее, вызывая разруше-
растворения NpO2 и оксидов других ТУЭ в карбо-
ние, обновление и увеличение площади реакцион-
натных средах.
ной поверхности [47]. При сонолизе воды, как и в
случае радиолиза, происходит образование H2O2,
ЗАКЛЮЧЕНИЕ
O2, O3, а также радикалов O• и OH•, которые, несмо-
тря на низкий выход, обладают чрезвычайно силь-
Проведены модельные эксперименты по пря-
ными окислительными свойствами в щелочных
мому окислительному растворению порошков ок-
средах и позволяют существенно ускорять окис-
сидов UO2, U3O8, PuO2 и NpO2 в водных растворах
лительно-восстановительные реакции с участием
NaHCO3/Na2CO3-H2O2/2Na2CO3·3H2O2/М2S2O8,
ТУЭ [48]. Таким образом, создаются предпосылки
где M = Na+, K+ или NH+. Экспериментально под-
к варианту сонохимического растворения оксидов
тверждена принципиальная возможность раство-
РАДИОХИМИЯ том 65 № 3 2023
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
223
рения порошков PuO2 и NpO2 в растворах Na2CO3
которых накапливаются вторичные Np/Pu/U-содер-
в окислительных условиях. Установлено, что при
жащие осадки. Стабилизация карбонатных раство-
подборе оптимальных условий и режимов процесса
ров актиноидов и предотвращение накопления вто-
окислительного растворения возможно достижение
ричных осадков могут быть организованы за счет
полноты растворения оксидов ТУЭ в карбонатных
введения дополнительных комплексообразующих
средах. К факторам, способствующим повышению
агентов и корректировки рН карбонатных растворов
растворимости оксидов ТУЭ в карбонатных средах,
газацией углекислым газом. Таким образом, факто-
относятся повышение температуры среды, концен-
рами повышения скорости окислительного раство-
трации карбонатного реагента и окислителя, орга-
рения оксидов актиноидов в карбонатных средах и
низация режима дробной подачи окислителя для
повышения устойчивости образующихся раство-
поддержания постоянного значения ОВП, а также
ров могут быть: 1) повышение окислительно-вос-
использования способов дополнительной химиче-
становительного потенциала среды растворения;
ской (корректировка рН карбонатного раствора или
2) регулировка рН среды или добавка комплексо-
добавка сильного комплексообразователя) и меха-
образователей, эффективно удерживающих ТУЭ в
нической (ультразвуковой) интенсификации.
карбонатных растворах и позволяющих переводить
вторичные малорастворимые соединения нептуния,
Низкая скорость окислительного растворения
плутония и урана в растворимые формы; 3) прове-
порошков NpO2 и PuO2 обусловлена низким значе-
дение окислительного растворения оксидов акти-
нием ОВП карбонатно-пероксидных систем, а так-
ноидов в карбонатных растворах в условиях меха-
же неустойчивостью карбонатных растворов вслед-
нического обновления поверхности растворяемой
ствие гидролиза карбонатных комплексов ТУЭ.
твердой фазы, например, ультразвуковая обработка,
Образование вторичных (гидролитические продук-
и снижения диффузионного сопротивления при до-
ты и продукты окисления) малорастворимых форм
ставке реагирующих компонентов к реакционным
Np(IV), Np(V), и Pu(IV) приводит к увеличению
центрам на поверхности оксидной фазы.
диффузионного сопротивления в гетерогенной
системе и замедляет скорость процесса окисле-
Полученные результаты являются научной базой
ния/растворения порошков NpO2, PuO2, а также
для обоснования и разработки вариантов как со-
UO2 и U3O8.
вместного, так и раздельного растворения урана и
ТУЭ за счет комбинации окислителей, изменением
Воздействие ультразвуковой кавитации на су-
условий и режимов проведения окислительного рас-
спензию оксидов NpO2 или PuO2 в карбонатном рас-
творения уранового и смешанного уран-плутоние-
творе позволяет снизить диффузионное сопротив-
вого ОЯТ в карбонатных средах в КАРБЭКС-про-
ление в гетерогенной системе за счет механическо-
цессе. На основании полученных результатов могут
го обновления поверхности растворяемой твердой
быть разработаны физико-химические основы аль-
фазы оксида актиноида и интенсификации массооб-
тернативного метода выщелачивания урана, плуто-
мена. Ультразвуковое воздействие, по-видимому, не
ния и нептуния из ОЯТ в карбонатные растворы.
ограничивается только механическим действием на
твердую фазу оксида актиноида. Химическое воз-
действие ультразвука на жидкую фазу связано с об-
БЛАГОДАРНОСТИ
разованием активных частиц, интенсифицирующих
химические процессы окисления и растворения и
Авторы выражают благодарность Перевалову С.А.
влияющих на химические равновесия в карбонат-
за помощь в проведении исследований окислитель-
ных окислительных системах, содержащих акти-
ного растворения диоксида нептуния в карбонатных
ноиды. Полученные результаты подтверждают воз-
средах.
можность организации варианта сонохимического
растворения оксидов актиноидов в карбонатных
ФОНДОВАЯ ПОДДЕРЖКА
средах, однако в этой области требуется проведение
дальнейших детальных исследований. В то же вре-
Данная работа выполнена при финансовой под-
мя УЗО ускоряет вторичные процессы гидролиза
держке Российского научного фонда, соглашение
карбонатных комплексов актиноидов, в результате
№ 20-63-46006.
РАДИОХИМИЯ том 65 № 3 2023
224
ЧЕРВЯКОВ и др.
КОНФЛИКТ ИНТЕРЕСОВ
16. Kim K.W., Lee J.W., Chung D.Y., Lee E.H., Kang K.H.,
Lee K.W., Song K.C., Yoo M.J., Park G.I., Moon J.K. //
J. Radioanal. Nucl. Chem. 2012. Vol. 292. P. 909-916.
Авторы заявляют об отсутствии конфликта ин-
тересов.
17. Kim K.W., Hyun J.T., Lee E.H., Park G.I., Lee K.W.,
Yoo M.J., Song K.C., Moon J.K. // J. Radioanal. Nucl.
Chem. 2011. Vol. 418. P. 93-97.
СПИСОК ЛИТЕРАТУРЫ
18. Kweto B., Groot D.R., Stassen E., Suthiram J.,
Zeevaart J.R.
// J. Radioanal. Nucl. Chem.
2014.
1.
Asanuma N., Asano Yu., Tomiyasu H.
// Proc.
5th
Vol. 302. P. 131-137.
Int. Conf. Recycling, Conditioning and Disposal
(RECOD 98). Bern: ENS, 1998. P. 709-716.
19. Stassen L., Suthiram J. // J. Radioanal. Nucl. Chem.
2015. Vol. 305. P. 41-50.
2.
Goff G.S., Brodnax L.F., Cisneros M.R., Williamson K.S.,
Taw F.L., May I., Runde W. // AIChE Annual Meet.
20. Stassen L., Suthiram J., Topkin J.
// Annual Waste
Conf. Proc. Salt Lake City: AIChE, 2007. 393c.
Management Symp. (WM2014). Arizona: Curran
Associates, 2014. Paper № 14399.
3.
Kim K.W., Chung D.Y., Yang H.B., Yang J.K., Lim E.H.,
Lee K.C., Song K.S. // Nucl. Technol. 2009. Vol. 166,
21. Lee E.H., Yang H.B., Lee K.Y., Kim K.W., Chung D.Y.,
N 2. P. 170-179.
Moon J.K. // J. Korean Radioact. Waste Soc. 2013.
Vol. 11, N 2. P. 85-93.
4.
Stepanov S.I., Chekmarev A.M. // Dokl. Chem. 2008.
Vol. 423, N 1. P. 276-278.
22. Lee E.H., Lee K.Y., Chung D.Y., Kim K.W., Lee K.W.,
Moon J.K. // J. Korean Radioact. Waste Soc. 2012.
5.
Stepanov S.I., Boyarintsev A.V., Vazhenkov M.V., Myaso-
Vol. 10, N 2. P. 77-85.
edov B.F., Nazarov E.O., Safiulina A.M., Tananaev I.G.,
Hen Vin So, Chekmarev A.M., Civadze A Yu. // Russ. J.
23. Soderquist C.Z., McNamara B.K., Oliver B.М. // J. Nucl.
Gen. Chem. 2011. Vol. 81, N 9. P. 1949-1959.
Mater. 2008. Vol. 378, N 3. P. 299-304.
6.
Stepanov S.I., Boyarintsev A.V. // Nucl. Eng. Technol.
24. Steward S.A., Gray W.J. // Proc. 5th Annual Int. Conf.
2022. Vol. 54, N. 7. P. 2339-2358.
High Level Radioactive Waste Management. New York:
ASCE, 1994.
7.
Peper S.M., Brodnax L.F., Field S.E., Zehnder R.A.,
Valdez S.N., Runde W.H. // J. Ind. Eng. Chem. Res. 2004.
25. Steward S.A., Mones E.T. // MRS Online Proc. Library.
Vol. 43. P. 8188-8193.
1996. Vol. 465. P. 557-564.
8.
Smith S.C., Peper S.M., Douglas M., Ziegelgruber K.L.,
26. Allen G.C., Tucker P.M., Tyler J.W. // J. Phys. Chem.
Finn E.C. // J. Radioanal. Nucl. Chem. 2009. Vol. 282.
1982. Vol. 86. P. 224-230.
P. 617-621.
27. Chervyakov N.M., Boyarintsev A.V., Andreev A.V.,
9.
Chung D.Y., Seo H.S., Lee J.W., Yang H.B., Lee E.H.,
Stepanov S.I. // RAD Conf. Proc. 2021. Vol. 5. P. 68-74.
Kim K.W. // J. Radioanal. Nucl. Chem. 2010. Vol. 284.
28. Gotcu-Freis P. High Temperature Thermodynamic
P. 123-129.
Studies on the Transuranium Oxides and Their Solid
10. Goff G.S., Long K.M., Reilly S.D., Jarvinen G.D.,
Solutions. Amsterdam: IOS, 2011. P. 180.
Runde W.H.
//
36th Actinide Separations Conf.
29. Asanuma N., Harada M., Ikeda Y., Tomiyasu H. // J.
Chattanooga: PNNL, 2012. LA-UR-12-21528.
Nucl. Sci. Technol. 2001. Vol. 38, N 10. P. 866-871.
11. Stepanov S.I., Boyarintsev A.V., Chekmarev A.M.
//
30. Pehrman R.,
Ammea M., Roth O., Ekeroth E.,
Dokl. Chem. 2009. Vol. 427, N 2. P. 202-206.
Jonsson M. // J. Nucl. Mater. 2010. Vol. 397, N 1-3.
12. Clark D., Hobart D., Neu M. // Chem. Rev.
1995.
P. 128-131.
Vol. 95. P. 25-48.
31. Roth O. Redox chemistry in radiation induced
13. Goff G.S., Brodnax L.F., Cisneros M.R., Runde W.H. //
dissolution of spent nuclear fuel: from elementary
AIChE Annual Meet. Conf. Proc. Salt Lake City:
reactions to predictive modeling: Doctoral Thesis in
AIChE, 2007. 271e.
Chemistry. Stockholm: KTH, 2008. P. 73.
14. Shilov V.P., Yusov A.B., Gogolev A.V., Fedoseev A.M. //
32. Buck E.C., Hanson B.D. McNamara B.K. // Geological
Radiochemistry. 2005. Vol. 47, N 6. P. 558-562.
Society, London, Special Publications. 2007. Vol. 236,
N 1. P. 65-88.
15. Soderquist C.Z.,
Johsen A.M.,
McNamara B.K.,
Hanson B.D., Chenault J.W., Carson K.J., Peper S.M. //
33. Kleykamp H. // J. Nucl. Mater. 1985. Vol. 131, N 2-3.
J. Ind. Eng. Chem. Res. 2011. Vol. 50. P. 1813-1818.
P. 221-246.
РАДИОХИМИЯ том 65 № 3 2023
РАСТВОРЕНИЕ ОКСИДОВ АКТИНОИДОВ В КАРБОНАТНЫХ РАСТВОРАХ
225
34. Thomas L.E., Beyer C.E., Chariot L.A. // J. Nucl. Mater.
43. Nikonov M.V., Tananayev I.G., Myasoyedov B.F.
//
1992. Vol. 188. P. 80-89.
Radiochemistry. 2010. Vol. 52, N 1. P. 27-30.
35. Марков В.К., Виноградов А.В., Елинсон С.В. Уран,
44. Поляков С.А., Вольф А.С., Костикова Г.В., Боярин-
методы его определения. М.: Атомиздат, 1960. 265 с.
цев А.В., Степанов С.И., Чекмарев А.М. // Успехи в
36. Marczenko Z., Balcerzak M. Analytical Spectroscopy
химии и химической технологии. 2017. Т. 31, № 10.
Library. Vol. 10: Separation, Preconcentration, and
С. 73-75.
Spectrophotometry in Inorganic Analysis. New York:
Elsevier, 2000. P. 521.
45. Тананаев И.Г. Химическое поведение нептуния, плу-
37. Бояринцев А.В. Окислительное растворение U3O8
тония, америция в щелочных средах: Дис
д.х.н.
в карбонатных растворах при переработке ОЯТ
М.: ИФХЭ им. А.Н. Фрумкина РАН, 1998. С. 356.
в КАРБЭКС-процессе: Дис
к.х.н. М.: РХТУ
им. Д.И. Менделеева, 2009. С. 169.
46. Jarvinen G.D., Runde W.H., Goff G.S. // Proc. Symp.
38. Boyarintsev A.V., Stepanov S.I., Kostikova G.V., Zhi-
Emerging Trends in Separation Science and Technology
lov V.I., Chekmarev A.M., Tsivadze A.Y. // Nucl. Eng.
(SESTEC-2010). Mumbai: Bhabha Atomic Research
Technol. 2019. Vol. 51, N 7. P. 1799-1804.
Centre, 2010.
39. Hou C., He M., Fang H., Zhang M., Gao Y., Jiao C.,
47. Агранат Б.А., Дубровин М.Н., Хавский Н.Н., Эс-
He H. // 2023. Vol. 55, N 1. P. 63-70.
кин Г.И. Основы физики и техники ультразвука. М.:
40. Clifford W.E.,
Bullwinkel E.P.,
McClaine L.A.,
ВШ, 1987. 352 c.
Noble Jr.P. // J. Am. Chem. Soc. 1958. Vol. 80, N 12.
48. Nikonov M.V., Shilov V.P., Krot N.N. // Abstracts of Int.
P. 2959-2961.
Conf. Actinides-89. Moscow: Nauka, 1989. P. 359.
41. Nitsche H. // Inorg. Chim. Acta. 1987. Vol. 127. P. 121-
49. Никонов M.B., Шилов В.П. // Тез. докл. III Всесо-
128.
юзн. конф. «Химия нептуния и плутония». Л.: Наука,
42. Clark D.L., Hobart D.E., Neu M.P. // Chem. Rev. 1995.
Vol. 95. P. 25-48.
1987. С. 67.
РАДИОХИМИЯ том 65 № 3 2023