РАСПЛАВЫ
1 · 2019
УДК 669:541.1
КОМПЬЮТЕРНОЕ МОДЕЛИРОВАНИЕ МНОГОКОМПОНЕНТНЫХ
РАСПЛАВОВ МЕТАЛЛУРГИЧЕСКИХ ШЛАКОВ
© 2019 г. Э. В. Дюльдинаa, *, Б. Р. Гельчинскийb, В. Н. Селивановa
aМагнитогорский государственный технический университет им. Г.И. Носова,
455000 Россия, Магнитогорск, пр. Карла Маркса, 38
bИнститут металлургии УрО РАН, 620016 Россия, Екатеринбург, ул. Амундсена, 101
*e+mail: e.dyuldina@mail.ru
Поступила в редакцию 01.06.2018
Основываясь на экспериментальных данных о плотности, в приближении моде(
ли ионной связи в работе впервые осуществлено молекулярно(динамическое моде(
лирование многокомпонентных оксидно(фторидных металлургических шлакообра(
зующих систем: SiO2-CaO-Al2O3-MgO-CaF2-Na2O-K2O-FeO, проведено обсуж(
дение результатов и сопоставление с литературными экспериментальными и
расчетными данными. Полученная модель свидетельствует о слабой температурной
зависимости структуры исследуемого расплава. Выявлена повышенная диффузион(
ная подвижность ионов фтора, а также ионов щелочных металлов по сравнению с
другими элементами.
Ключевые слова: расплав, металлургический шлак, компьютерное моделирование,
структура, свойства.
DOI: 10.1134/S0235010618050055
ВВЕДЕНИЕ
Непрерывная разливка стали производится, как правило, с защитой поверхности
жидкого металла в промежуточном ковше и в кристаллизаторе шлаком, получаемым
из шлакообразующих смесей (ШОС). Все возрастающие требования к качеству слит(
ков вызывает необходимость совершенствования существующих и создания новых
ШОС. Для успешного решения этой задачи необходима информация о зависимости
физико(химических свойств и структуры получающегося шлака от химического со(
става ШОС. Эти данные могут быть получены из экспериментальных исследований,
однако для разработки более эффективных технологических процессов зачастую не(
обходимо знать фундаментальные закономерности, лежащие в их основе. Такую ин(
формацию может дать микроскопическая теория расплавов и современные методы
компьютерного моделирования. Большое число экспериментальных данных по свой(
ствам оксидных систем указывает на значительный вклад ионной связи. Примени(
мость ионной теории обуславливается значительной разницей электроотрицательно(
стей кислорода (3.5 по Полингу) и обычных металлов (0.9 у Na, 1.0 у Ca, 1.5 у Al и т.д.).
Выбор чисто ионного варианта имеет значительные преимущества. При этом удается
получить неплохое согласие с опытом для структуры и энергии моделей. В последнее
время ионная модель некристаллических оксидов применяется довольно широко, в
том числе для моделирования многих простых и сложных оксидов и окси(фторидов
[1]. В данной работе впервые проведено молекулярно(динамическое моделирование
расплава многокомпонентного окси(фторида, состоящего из 9 ионов (Si, Ca, Al, Mg,
K, Na, Fe, F, O), проведено обсуждение результатов и сопоставление с литературными
экспериментальными и расчетными данными.
Компьютерное моделирование многокомпонентных расплавов
95
МЕТОДИКА МОДЕЛИРОВАНИЯ
Построены молекулярно(динамические модели 8 компонентного окси(фторидного
расплава шлакообразующей системы (ШОС) размером от 2000 до 20000 атомов в ос(
новном кубе с периодическими граничными условиями для четырех температур (1257,
1473, 1573, 1673 К) при фиксированном объеме. Шаг по времени равнялся 0.05t0, где
t0 - внутренняя единица времени, равная 7.608 ⋅ 10-14 с. Для получения зависимостей
средних квадратов смещения частиц от времени просчитывали до 50000-60000 шагов.
Плотность расплавов брали из собственных экспериментальных данных [2]. Рассмат(
риваемые модели расплава ШОС соответствуют реальному составу в мас. % (35.35%-
SiO2; 30.79% - CaO; 8.58% - Al2O3; 1.26% - MgO; 13.73% - CaF2%; 7.57% - Na2O;
0.88% - K2O; 1.82% - FeO). С учетом этих концентраций был проведен пересчет со(
става на мольные доли и вычислено соответствующее число ионов каждого компо(
нента.
Потенциалы межчастичного взаимодействия были выбраны в форме Борна-Майера:
uij(r) = ZiZje2/r + Bijexp(-r/ρij) + Cij/r6,
где r - расстояние между центрами ионов, Zi - заряд i(го иона в единицах элементар(
ного заряда e, Bij и ρij - параметры отталкивания ионных оболочек частиц i и j, Cij - па(
раметры эффективного диполь(дипольного взаимодействия.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Основными структурными характеристиками некристаллических многокомпо(
нентных систем являются парциальные парные корреляционные функции (ППКФ)
gij(r), описывающие вероятность обнаружения пары частиц сортов i и j на расстоянии r.
Они нормированы так, что при r → ∞ все gij(r) → 1. Эти функции непосредственно
рассчитываются в методе молекулярной динамики (МД). Введем обозначения: R1(ij),
g1(ij) - координата и высота первого пика ППКФ для пар ij.
Для всех типов частиц в зависимости от состава получены парциальные парные
корреляционные функции (ППКФ)), а также интегральная ПКФ от всего расплава.
Прежде всего следует отметить, что для изученного диапазона 1257-1673 К темпера(
турная зависимость ПКФ крайне незначительна. Положения пиков при всех темпера(
турах практически совпадают, а различия по их высоте невелики. В связи с этим для
анализа особенностей атомной структуры в дальнейшем можно остановиться на какой(
либо одной температуре, например, на температуре ликвидуса (Т = 1257 К).
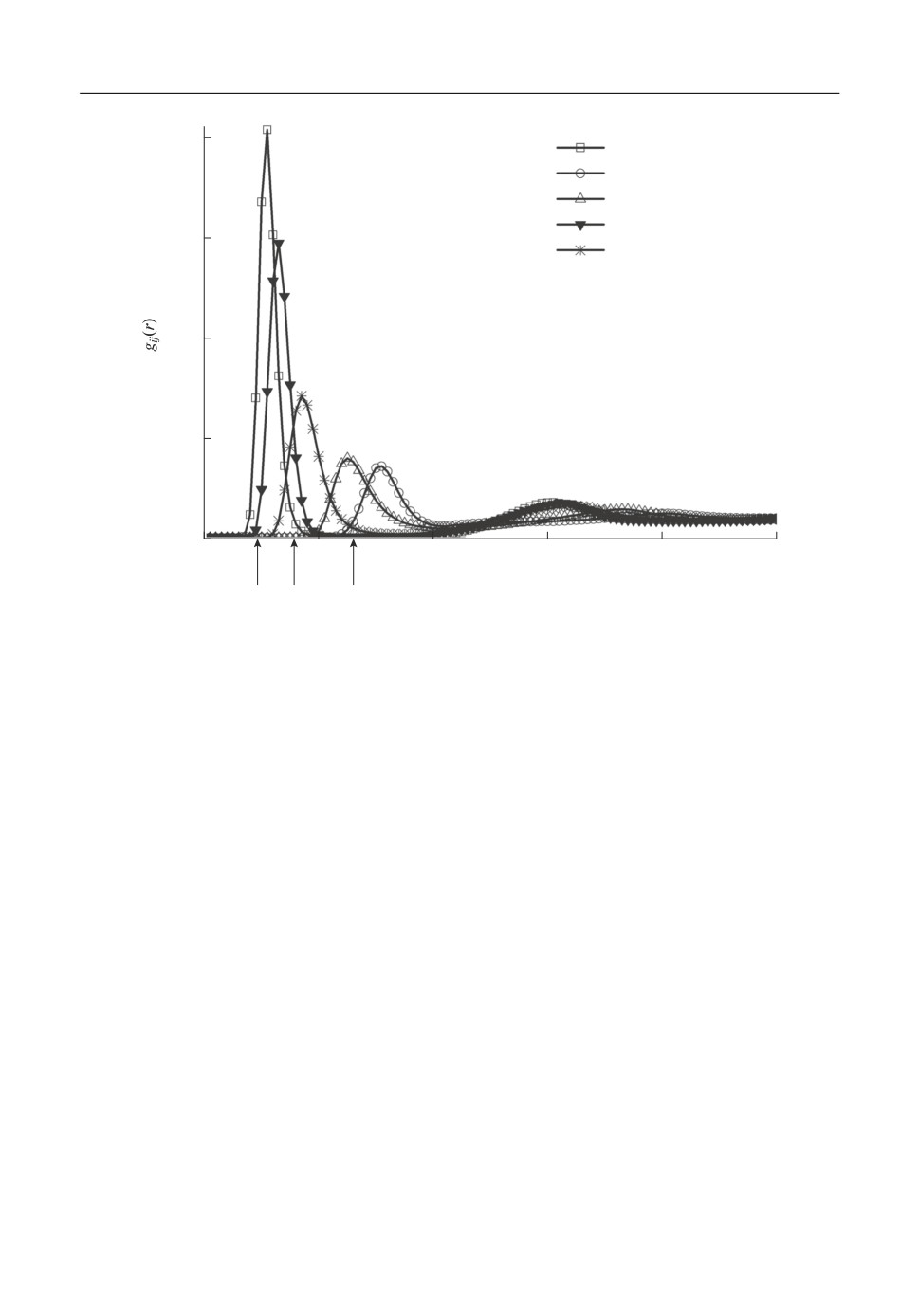
Рассмотрим корреляцию интегральной ПКФ и ППКФ оксидов, имеющих наи(
больший массовый процент в исследуемой системе, а именно Me-O, где Ме = Si, Ca,
Al, Mg (рис. 1). Хорошо видно, что парциальные ПКФ достаточно четко структуриро(
ваны по трем группам и положения пиков в этих группах коррелируют с указанными
на рисунке положениями пиков R1, R2 и R3, выявленных на интегральной ПКФ.
Все парциальные ПКФ имеют четко выраженные первые пики. Максимальная вы(
сота пиков парциальных ПКФ наблюдается для пар Si-O (~20), а также Al-O и Mg-O;
положение этих пиков соответствует расстоянию R1.
С расстоянием R2 коррелирует пик, характерный для связей Mg-O, имеющие зна(
чительно меньшую высоту - порядка 6-7.
Для остальных пар Ca-O и O-O первые пики также явно выражены, но имеют
большой разброс по высоте (от 3 до 4). Они формируют атомную структуру, дающую
вклад в третий пик интегральной ПКФ, характеризуемый расстоянием R3. Пики этих
парциальных ПКФ более широкие, чем первые, их полуширина составляет 0.4-0.6 Å.
Все это свидетельствует о слабой корреляции расположения ионов этой группы.
96
Э. В. Дюльдина, Б. Р. Гельчинский, В. Н. Селиванов
20
Si-O
O-O
Ca-O
Al-O
15
Mg-O
10
5
0
1
2
3
4
5
6
r, Å
R1
R2
R3
Рис. 1. Парциальные ПКФ, для O-O и оксидов Ме-О, где Ме = Si, Ca, Al, Mg.
Можно полагать, что ионы металла хаотически движутся внутри расплава, находясь
преимущественно в кислородном окружении.
Следует отметить, что положение и высота пиков ППКФ для ряда оксидов, дающих
основной вклад в интегральную ПКФ, например, SiO2, FeO, MgO, CaO, Al2O3, Na2O
хорошо согласуются с данными дифракционного эксперимента для соответствующих
расплавов [3].
Существует ряд других важных свойств, которые часто исследуются на моделях.
Сюда относится, например, расчет коэффициентов самодиффузии частиц, вязкости,
автокорреляционных функций скоростей, потоков и т.д., которые позволяют опреде(
лить колебательные спектры системы и провести сопоставление с экспериментальны(
ми данными по упругому и неупругому рассеянию излучения. Нами определена диффу(
зионная подвижность частиц в модели, которая оказалась очень малой, что затрудняет
их количественную оценку. Более или менее надежные данные для коэффициента са(
модиффузии можно получить только для ионов натрия (1.1 ⋅ 10-5 см2/с) и фтора
(2.3 ⋅ 10-5 см2/с), в меньшей степени, для ионов калия, для которых наблюдаются за(
метные перемещения частиц в МД(прогоне.
На рис. 2 представлены парциальные ПКФ для оксидов, массовый процент кото(
рых в расплаве невелик. По виду ППКФ для системы Сa-F и оксидов щелочных ме(
таллов можно полагать, что в этих системах довольно слабая корреляция в расположе(
нии ионов, как следствие более слабого межчастичного взаимодействия по сравнению
с оксидами, дающими основной вклад в интегральную ПКФ. Это вполне может быть
причиной повышенной диффузионной подвижности ионов фтора, натрия и калия.
Компьютерное моделирование многокомпонентных расплавов
97
6
K-O
5
Na-O
Fe-O
4
Ca-F
3
2
1
0
1
2
3
4
5
6
7
8
r, Å
Рис. 2. Парциальные ПКФ для Ca-F и оксидов Me-О, где Me = Fe, Na, K.
ЗАКЛЮЧЕНИЕ
Основываясь на экспериментальных данных о плотности, в приближении модели
ионной связи проведено молекулярно(динамическое моделирование расплава много(
компонентной окси(фторидной шлакообразующей смеси при нескольких температу(
рах. Полученная модель свидетельствует о слабой температурной зависимости струк(
туры этого расплава. Показано, что компьютерная модель позволяет получить доста(
точно реалистичную картину атомной структуры шлакового расплава, которая по
основным параметрам (положение и высота пиков парциальных ПКФ) хорошо согла(
суется с данными дифракционного эксперимента. Обнаружена повышенная диффу(
зионная подвижность ионов фтора и натрия (несколько меньше ионов калия) по
сравнению с другими элементами, которая коррелирует с видом ППКФ соответству(
ющих окси(фторидов.
СПИСОК ЛИТЕРАТУРЫ
1. Б е л а щ е н к о Д . К . Компьютерное моделирование жидких и аморфных веществ //
М.: МИСИС, 2005. 408 с.
2. Д ю л ь д и н а Э . В . , С е л и в а н о в В . Н . , Л о з о в с к и й Е . П . , И с т о м и н С . А . ,
Рябов В.В., Ченцов В.П. Физико(химические свойства расплавов шлакообразующих
смесей, используемых при непрерывной разливке стали // Расплавы. 2009. № 6. С. 3-10.
3. П а с т у х о в Э . А . , В а т о л и н Н . А . , Л и с и н В . Л . и д р . Дифракционные ис(
следования строения высокотемпературных расплавов. Екатеринбург: УрО РАН. 2003. 355 с.
98
Э. В. Дюльдина, Б. Р. Гельчинский, В. Н. Селиванов
Computer Simulation of Multicomponent Melts of Metallurgical Slags
E. V. Dyul’dina1, B. R. Gelchinski2, V. N. Selivanov1
1Magnitogorsk State Technical University G.I. Nosova, 455000 Russia, Magnitogorsk, Karl Marx av., 38
2Institute of Metallurgy, Ural Branch of the Russian Academy of Sciences,
620016 Russia, Yekaterinburg, Amundsena st., 101
Based on the experimental data on the density, the molecular dynamics simulation of
multicomponent oxide(fluoride metallurgical slag(forming systems: SiO2-CaO-Al2O3-
MgO-CaF2-Na2O-K2O-FeO was carried out for the first time in the approximation of the
ionic bond model. The results were discussed and compared with literary experimental and
calculated data. The obtained model indicates a weak temperature dependence of the struc(
ture of the melt. An increased diffusion mobility of fluorine ions as well as ions of alkali met(
als in comparison with other elements has been revealed.
Keywords: melt, metallurgical slag, computer simulation, structure, properties
REFERENCES
1. Belashchenko D.K. Computer modeling of liquid and amorphous substances. [Komp’yuternoye
modelirovaniye zhidkikh i amorfnykh veshchestv] // Moscow: MISIS. 2005. 408 p. [In Rus.].
2. Dul’dina E.V., Selivanov V.N., Lozovskii E.P. et al. Physicochemical properties of the melts of
slag(forming mixtures used for continuous casting of steel. [Fiziko+khimicheskiye svoystva rasplavov
shlakoobrazuyushchikh smesey, ispol’zuyemykh pri nepreryvnoy razlivke stali] // Rasplavy. 2009. № 6.
Р. 3-10. [In Rus.]
3. Pastukhov E.A., Vatolin N.A., Lisin V.L. et al. Diffraction investigations of the structures of
high(temperature melts. [Difraktsionnyye issledovaniya stroyeniya vysokotemperaturnykh rasplavov].
UrO RAN, Yekaterinburg, 2003. [in Rus.].