РАСПЛАВЫ
6 · 2019
УДК 544.77
МАКРОСКОПИЧЕСКАЯ МОДЕЛЬ НУКЛЕАЦИИ ПРИ КОНДЕНСАЦИИ
ПАРОВ МЕДИ В СРЕДЕ ИНЕРТНОГО ГАЗА
© 2019 г. А. Е. Коренченкоa, b, *, А. Г. Воронцовc, А. А. Жуковаa
aПервый Московский государственный медицинский университет им. И.М. Сеченова,
Москва, Россия
bИнститут металлургии УрО РАН, Екатеринбург, Россия
cЮжно)Уральский государственный университет (НИУ), Челябинск, Россия
*e)mail: akorenchenko@gmail.com
Поступила в редакцию 18.05.2019 г.
После доработки 05.06.2019 г.
Принята к публикации 28.06.2019 г.
В рамках вычислительной схемы, подходящей для описания конденсации, про
веден статистический анализ результатов молекулярно динамических расчетов газо
фазной “самосборки” нанокластеров при конденсации паров металла. Выявлены за
кономерности столкновений и роста малых кластеров меди. Определены параметры
взаимодействия кластеров с атомами металла, позволяющие перенести информа
цию в макроскопическую модель нуклеации. Результаты могут быть использованы
при описании нуклеации для прогнозирования распределения наночастиц по разме
рам в промышленном производстве металлических нанопорошков.
Ключевые слова: нуклеация, конденсация, пары меди.
DOI: 10.1134/S0235010619060082
ВВЕДЕНИЕ
Использование термодинамических моделей нуклеации не позволяет добиться ко
личественного согласия с экспериментом [1, 2], так что данные могут различаться на
порядки [3]. Популярный в последние десятилетия метод Кинетического Монте Кар
ло (КМК) [4, 5] позволяет приблизиться к масштабам реального эксперимента, одна
ко скорости процессов адсорбции и десорбции атомов на зародыши, диффузии моно
меров к их поверхности определяются коэффициентами, значения которых подбира
ются так, чтобы наилучшим образом описать результаты эксперимента, или МД
симуляции [6, 7]. Подбор коэффициентов является слабым местом этих подходов, т.к.
не может быть строго обоснован. В [8] проведен анализ одиночных актов взаимодей
ствия атомов аргона и кластеров железа и статистическими методами получены значе
ния коэффициентов термической аккомодации и теплоотдачи для кластеров, содер
жащих различное количество атомов. Эти коэффициенты могут быть использованы
для определения теплоты конденсации в макроскопическом масштабе, однако по
строение кластеров в [8] производится искусственным путем - состояние кластера за
дается единожды и не меняется при моделировании. Стадия зародышеобразования в
работах [4-8] не рассматривается, поэтому нет возможности учесть, например, появ
ление устойчивых изомерных форм, “магических” размеров кластеров и т.д.
Цель настоящей работы - построение численной статистической модели, описыва
ющей столкновения кластеров с атомами меди, и пригодной для использования в
макроскопическом масштабе.
602
А. Е. Коренченко, А. Г. Воронцов, А. А. Жукова
СХЕМА МД ЭКСПЕРИМЕНТА
(ПОЛУЧЕНИЕ СТАТИСТИЧЕСКИХ ДАННЫХ)
Исследование формирования наночастиц проводилось методом молекулярной ди
намики в пакете LAMMPS [9]. Система состояла из атомов буферного газа (Аr) и ато
мов меди. Детали расчетов, связанные с особенностями потенциалов взаимодействия,
геометрией ячейки и выбором величины межатомного расстояния Δ, на котором ато
мы рассматриваются как принадлежащие одному кластеру, обсуждаются в [10-12].
В МД исследовании взаимодействий кластеров и атомов меди анализировались
энергетические соотношения между ними до и после взаимодействия, причем как в
лабораторной системе отсчета (ЛСО), так и в системе центра масс кластера.
Температура аргона выбиралась в интервале 300 K ≤ TAr ≤ 1500 K, размеры ячейки
подбирались так, чтобы плотность пара металла соответствовала экспериментальным
3
значениям и находилась в диапазоне
Термостатирование системы
≤ρ≤
0.1
0.3 кг м
происходило только для атомов инертного газа. В начальный момент времени темпе
ратура аргона и атомарного металлического пара были одинаковы, атомы имели рав
номерное распределение по пространству и максвелловское - по скоростям. Эволю
ция атомной системы рассматривалась в течение времен порядка 0.5 мс с шагом, рав
ным 3 фс.
СТАТИСТИЧЕСКАЯ МОДЕЛЬ КОНДЕНСАЦИИ
Описание состояний кластеров и атомов
Макроскопическая модель конденсации должна описывать процессы взаимодей
ствия атомов и кластеров в системах, содержащих число частиц, сравнимое с числом
Авогадро. В этих условиях возможно описание только самых общих закономерностей,
поэтому количество используемых параметров должно быть, по возможности, мини
мальным. Анализ эволюции системы проводился на основе законов сохранения энер
гии и импульса, предположения о беспорядочности теплового движения и допустимо
сти элементарных процессов вида:
M
k
+
M
1
→
M
k+1
,
(1a)
M
→
M'
+
M'.
(1б)
k+1
k
1
Здесь
- кластер, содержащий k атомов металла. Штрихи в (1б) означают, что и
k
M
атом, и кластер могут изменить свое энергетическое состояние после взаимодействия.
В модели учитываются только столкновения кластер атом (1а) и распады на кластер и
атом (1б), т.е. коагуляцией, происходящей вследствие столкновений малых кластеров
между собой, и распадом на два кластера пренебрегается. Следует отметить, что
k атомный кластер в левой части (1а) может находиться в равновесии со средой, а мо
жет быть возбужденным в той или иной степени. Скорости атомов внутри кластеров
могут в 10 и более раз превышать тепловые, внутренние степени свободы в этом слу
чае являются “перегретыми” и нуждаются в отводе тепла. При определенных условиях
при столкновении кластер - атом металла может образоваться долгоживущий кластер
[11], который может быть “перегретым”, но таким, что времени его существования
будет достаточно для того, чтобы, сталкиваясь с атомами аргона, охладиться и затем
продолжить рост. В этом случае реакция (1б) не реализуется. В других обстоятельствах
при таких столкновениях может образоваться короткоживущий кластер, который
вскоре распадется по (1б). Часто при этом атом уносит часть избытка внутренней
энергии, т.е. происходит “охлаждение” кластера.
Макроскопическая модель нуклеации при конденсации паров меди
603
Состояние k атомного кластера описывалось двумя величинами: кинетической
энергией его центра масс в ЛСОT и внутренней энергией
, которая представляет
k
εk
собой сумму кинетической и потенциальной энергии каждого атома кластера, вычис
ленную в системе центра масс кластера и поделенную на количество атомов, так что
энергия кластера в ЛСО равна
Одиночные атомы металла и аргона характе
T kεk.
k
ризуются кинетической энергией, возможным изменением состояния атома - воз
буждением электронных уровней или ионизацией - пренебрегается.
Вероятности протекания реакций (1а) и (1б)
В работе [11] было показано, что вероятность того, что кластер просуществует до
статочное время, чтобы успеть “охладиться” столкновениями с атомами аргона и при
соединить атом металла, определяется только внутренней энергией этого кластера. В
[11] были построены зависимости этой вероятности отε,основанные на данных МД
k
моделирования и представляющие собой ступеньки, в которых вероятность плавно
изменяется от 1 в области низких энергий до 0 в области высоких энергий. На основа
д
нии анализа, приведенного в [11], был заполнен массив
в котором целочис
P
(k, ),
n
ленные индексы отвечают: k - за количество атомов в кластере, n - за значение внут
д
ренней энергии кластера
значение элемента массива
равно
ε
=nδ,
k
δ = 0.01эВ,
P
(k, )
n
вероятности того, что кластер, содержащий k атомов, с внутренней энергией
будет
nδ
долгоживущим. Таким образом, в работе [11] были получены статистические данные,
д
определяющие вероятность протекания отдельно реакции (1а) -
и вероят
P
(k, ),
n
д
ность последовательного протекания реакций (1а + 1б) - (1 -
).
P
(k, )
n
Определение количества долгоживущих кластеров
и их энергетических характеристик
Определим закономерности энергообмена при столкновении кластер атом метал
ла. Зная внутреннюю энергию кластера и кинетические энергии центра масс k атом
ного кластера и атома металла в ЛСО, определим внутреннюю энергию получившего
ся (k + 1) атомного кластера. Законы сохранения импульса и энергии в лабораторной
системе отсчета запишутся как
ЛСО
ЛСО
ЛСО
⎪mV
+
kmV
=
(k
+1)mV
,
(2a)
1
k
k+1
⎨
ЛСО
ЛСО
ЛСО
⎩T
1
+T
k
+ kε
k
=
T
k+1
+
(k
+1)ε
k+1
,
(2б)
ЛСО
ЛСО ЛСО
гдеT
, T
, T
- кинетические энергии центра масс атома и кластеров в ЛСО,
1
k
k
+1
ЛСО
ЛСО
m - масса атома металла,ε и
ε
- внутренние энергии кластеров,V
,V
- ско
k
k+1
1
k
ЛСО
рости центров масс атома и k атомного кластера до взаимодействия,V
k+1
- скорость
(k + 1) атомного кластера после взаимодействия.
Реализуется следующая последовательность вычислений:
• выражая из (2) энергию
ε
k+
1,
получаем
2
2
m k
ЛСО
ЛСО
ЛСО ЛСО
k
ε
=
(
V
)
+
(
V
)
−
2V
V
cosϕ
+
ε
,
(3)
k+1
2
1
k
1
k
)
k
2
(k
+1)
k
+1
ЛСО
• подстановкой (3) в (2б) получаем значение кинетической энергии
+1
k
T
604
А. Е. Коренченко, А. Г. Воронцов, А. А. Жукова
Как видно из (3), внутренняя энергия образованного кластера зависит, помимо все
ЛСО
го прочего, от ϕ - угла между векторами скоростей
и
Ввиду беспорядоч
V
ЛСО.
V
1
k
ности теплового движения можно считать, что относительные скорости распределены
равномерно по всем направлениям, поэтому вероятность того, что ϕ заключен в ин
тервале
(ϕ,ϕ + dϕ),
составит
sinϕ
P
(ϕ, ϕ+dϕ)
=
dϕ
(4)
2
Таким образом, внутренняя и кинетическая энергии частицы, полученной в реакции
(1а), определяются через энергетические характеристики частиц исходных частиц.
Обозначим
количество столкновений кластер - атом металла, в результате
k+1,n
Z
которых образуется (k + 1) атомный кластер с внутренней энергией
опреде
+
ε
=nδ
1
,
k
ленной из (3). Значение
может быть определено по формулам молекулярно ки
k+1,n
Z
нетической теории [16]. Тогда для определения числа образованных в реакции (1а)
д
долгоживущих кластеров нужно умножить
на вероятность
Таким об
k+1,n
Z
+
P
(k
1, ).
n
разом, модель позволяет можно оценить не только состояние, но и количество частиц,
получившихся в единицу времени после реакции (1а).
Определение количества распадов (1б)
и энергетических характеристик продуктов распада
Образованные в реакции
(1а) (k
+
1) атомные кластеры в количестве
д
Z
1
−
P
(k
+1,n)
не будут долгоживущими и вскоре распадутся на k атомный кла
k+
1,n
(
)
стер и атом (реакция (1б)), не успев столкнуться с другими частицами. Для описания
энергетического эффекта реакции (1б) следует определить внутреннюю энергию
εk
образованного k атомного кластера, кинетическую энергию движения его центра
масс, кинетическую энергию отделившегося атома и угол разлета, считая известными
кинетическую энергию движения центра масс (k + 1) атомного кластера в ЛСО
и
k+1
T
его внутреннюю энергию
ε
На две известных величины в реакции (1б) приходятся
k
+1
4 неизвестных, поэтому для их определения нужно привлечь 2 дополнительных условия.
Анализ результатов МД моделирования показывает, что взаимодействие между
кластером и атомом металла занимает интервал времени значительно больший, чем,
если бы, атом просто пролетал через кластер. За время, проведенное внутри кластера,
налетевший атом взаимодействует с другими атомами кластера и энергия перераспре
деляется между ними так, что их состояния уравниваются. За это предположение го
ворит, например, факт, что, согласно результатам МД расчетов, вылетающий из кла
стера атом металла не обязательно будет тем же, который налетел. Поэтому, в основу
анализа энергообмена в реакции (1б) положим два предположения:
1) Будем считать, что средняя кинетическая энергия вылетающего атома в системе
центра масс (k + 1) атомного кластера (далее, Ц система) определяется только внут
ренней энергией этого кластера. Это предположение подтверждается результатами
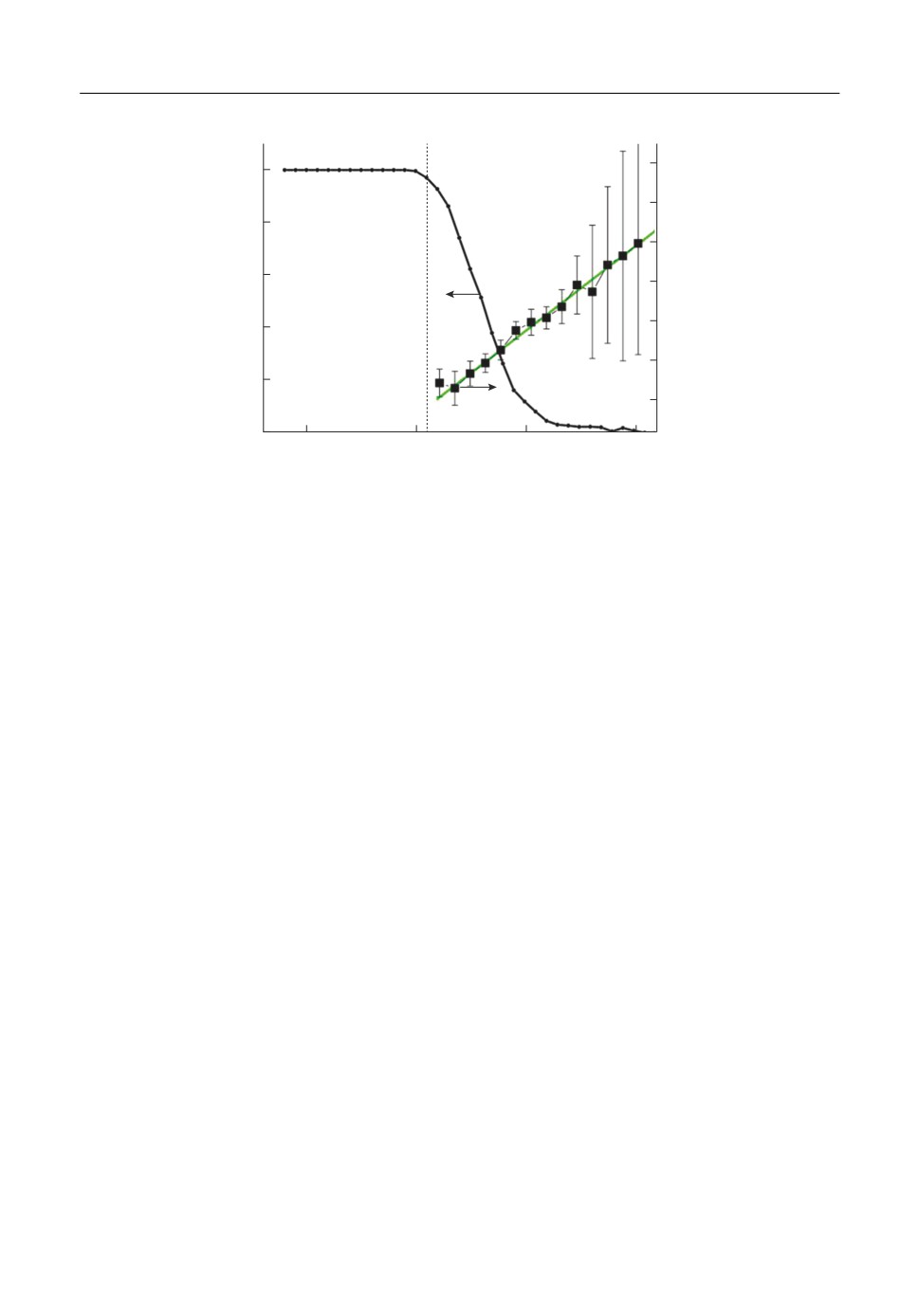
МД моделирования. На рис. 1 показаны зависимости вероятности того, что кластер
является долгоживущим (левая шкала) и средней кинетической энергии атома меди
(правая шкала) от внутренней энергии 6 ти атомного кластера. Как видно из рисунка,
график для
слева ограничен энергией, при которой вероятность образования
T1
(ε6)
долгоживущего кластера становится равной 1, так как для долгоживущих кластеров
реакция (1б) не реализуется. Интервалы, показанные на графике, соответствуют раз
бросу данных, они малы для низких значений внутренней энергии и возрастают почти
Макроскопическая модель нуклеации при конденсации паров меди
605
T1, эВ
P
0.8
1.0
0.7
0.8
0.6
0.6
0.5
0.4
0.4
0.3
0.2
0.2
0
-2.5
-2.0
-1.5
-1.0
ε6, эВ/атом
Рис. 1. Зависимость вероятности того, что 6 ти атомный кластер является долгоживущим (левая шкала) и
средняя кинетическая энергия вылетающего атома меди (правая шкала) от внутренней энергии кластера.
до 100% вблизи верхней границы существования кластера. График хорошо аппрокси
мируется линейной зависимостью, показанной сплошной линией.
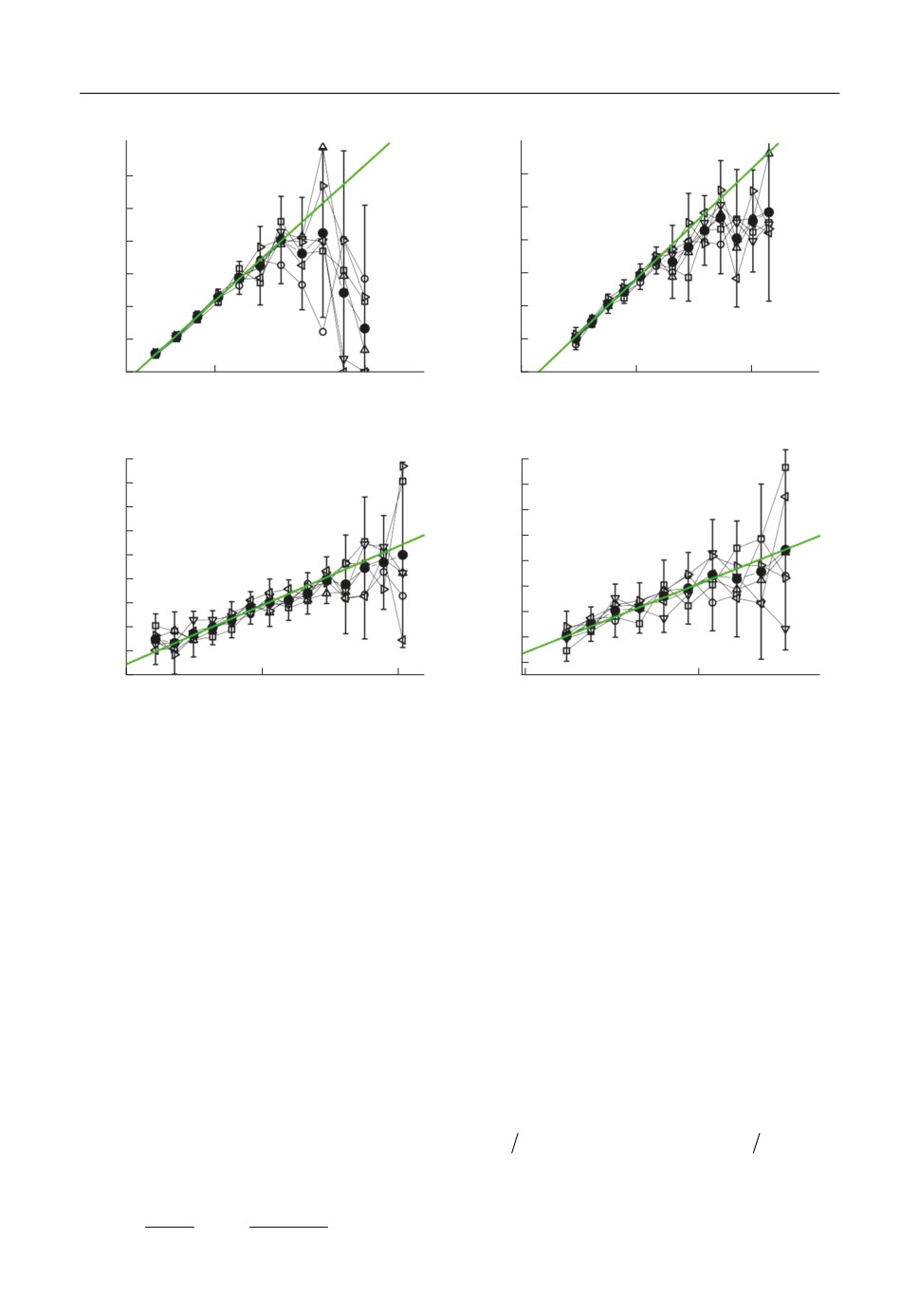
На рис. 2 изображены графики зависимости средней кинетической энергии выле
тевшего атома меди, вычисленной в Ц системе, от внутренней энергии для кластеров,
содержащих различное количество атомов. Данные, отмеченные пустыми символами,
получены при различных значениях температуры смеси паров меди и аргона, закра
шенными кругами показано среднее значение. Как видно из рисунка, зависимости,
полученные для различных начальных температур газовой смеси, практически совпа
дают, разброс данных мал для кластеров с низкой энергией и возрастает при увеличе
нии энергии. На основании приведенного анализа был сформирован массив T1(k, n), в
котором k есть количество атомов в кластере, n характеризует внутреннюю энергию
кластера, значение элемента массива равно среднему значению кинетической энер
гии вылетающего атома в Ц системе.
2) Предположим, что направление, в котором вылетает атом в Ц системе, является
произвольным, не связанным с направлением движения центра масс этого кластера.
Это означает, что после образования (k + 1) атомного кластера в реакции (1а) в нем
происходит уравнивание не только энергий атомов в Ц системе, но и направлений, в
которых движутся атомы. Поэтому прямая, на которой лежат скорости разлетающих
ся частиц из правой части (1б) в Ц системе, может быть произвольно ориентирована
относительно направления скорости центра масс (k + 1) атомного кластера из левой
части (1б). Тогда угол α между ними будет распределен как
sinα
P(α, α+dα)
=
dα
(5)
2
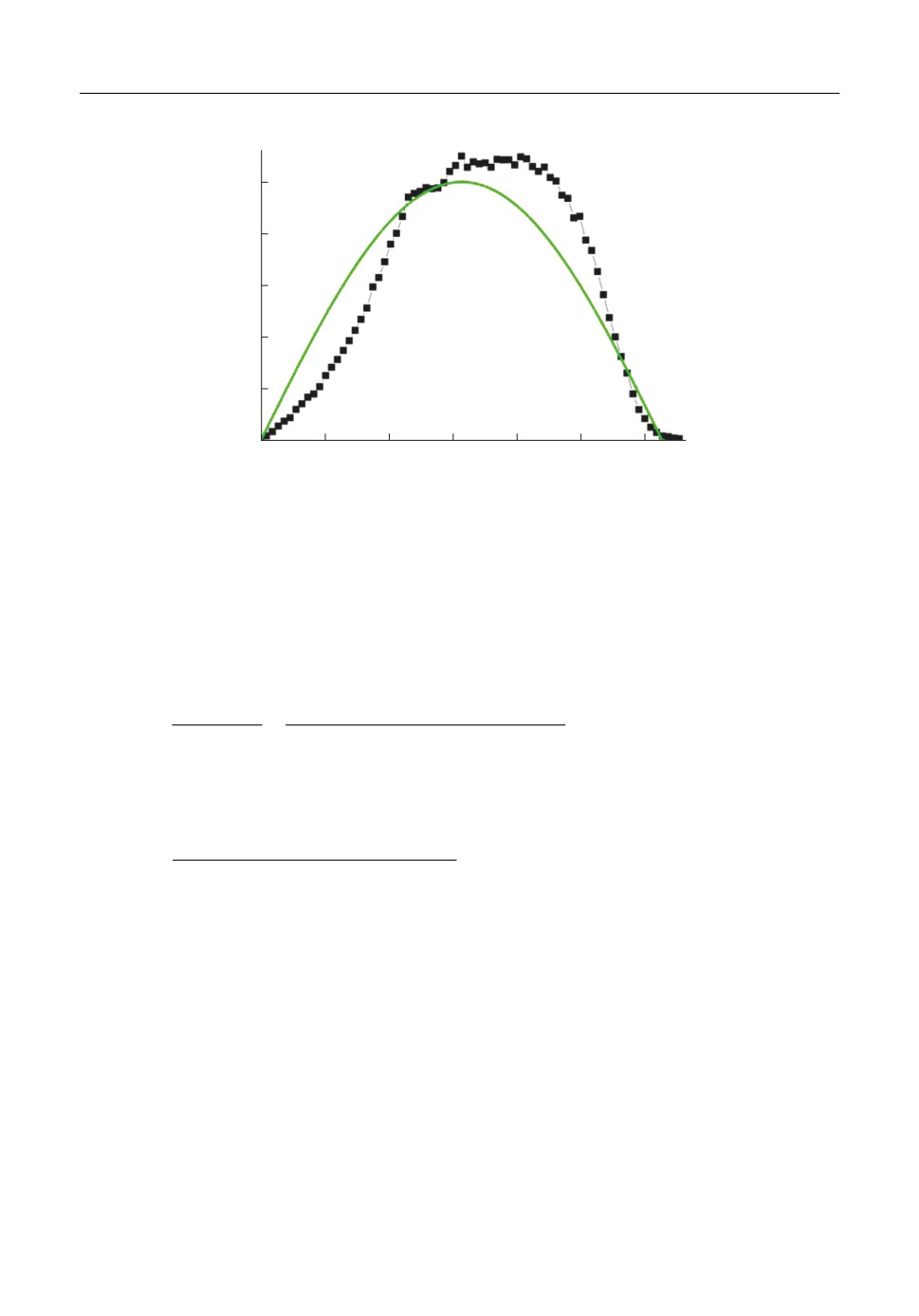
Результаты МД моделирования, показанные на рис. 3 подтверждают это предположе
ние. На рис. 3 закрашенными квадратами показана функция распределения угла
α
между скоростью димера и атома меди, образовавшихся при распаде 3 х атомного кла
стера. Как видно из рисунка, формы графиков функций похожи между собой, отличие
молекулярно динамической функции от теоретической (5) составляет около 10%.
606
А. Е. Коренченко, А. Г. Воронцов, А. А. Жукова
T1, эВ
T1, эВ
0.6
0.6
0.5
0.5
0.4
0.4
0.3
0.3
0.2
0.2
0.1
0.1
0
0
-0.5
0
-1.5
-1.0
-0.5
ε3, эВ/атом
ε4, эВ/атом
1.0
1.0
0.9
0.9
0.8
0.8
0.7
0.7
0.6
0.6
0.5
0.5
0.4
0.4
0.3
0.3
0.2
0.2
0.1
-2.0
-1.5
-1.0
-2.5
-2.0
ε6, эВ/атом
ε12, эВ/атом
Рис. 2. Зависимости энергии атома меди (реакция (1б)) от внутренней энергии кластера, полученные при
различных температурах газовой смеси и для кластеров, содержащих различное количество атомов.
- 300;
- 500;
- 750; ∇ - 1000;
⊲
- 1250;
⊳
- 1500 К.
В принятых предположениях результаты распада возбужденного кластера (1б) опи
сываются следующим образом. Законы сохранения импульса и энергии, записанные в
Ц системе, выглядят как
⎧0
=
m
1
V
1
+
m
Vk k
,
(6а)
⎨
⎩(k
+1
)ε
k+1
=
T
1
+
k
T kε
k
(6б)
Здесь и далее переменные без верхнего индекса определяются в Ц системе. В (6) пред
полагается, что атом удалился на достаточное расстояние от кластера, так, что энерги
ей их взаимодействия можно пренебречь. Реализуется следующая последовательность
вычислений:
• определяем T из массива T1 (k + 1, n),
n
=
[ε
δ]
Из (6а) получимT
=
T
k
Из за
1
k+1
k
1
кона сохранения энергии (6б) определим внутреннюю энергию k атомного кластера
(k
+1
)
T
1
(k
+1,n)
ε
k
=
ε
k+1
−
,
2
k
k
Макроскопическая модель нуклеации при конденсации паров меди
607
f(
, рад-1
0.5
0.4
0.3
0.2
0.1
0
0.5
1.0
1.5
2.0
2.5
3.0
, рад
Рис. 3. Функции распределения угла между скоростями разлета частиц в Ц системе 3 х атомного кластера и
скоростью центра масс распадающегося кластера;
- полученная из молекулярно динамических расчетов;
— - теоретическая (5).
ЛСО
ЛСО
• скорость атома в ЛСО выражается формулой
V
1
=V
1
+V
k+1
Тогда кинетическая
энергия атома в ЛСО будет равна
2
2
2
ЛСО
ЛСО
ЛСО
m
V
+
V
+
2V
V
cosα
m
V
1
(
1
(
k+1
)
1
k+1
)
ЛСО
1
(
1
)
T
1
=
=
,
(7а)
2
2
где угол α распределен как (5). Кинетическая энергия центра масс k атомного класте
ра в ЛСО
2
2
ЛСО
ЛСО
m
V
+
V
−2V
V
cosα
1
(
k
(
k+1
)
k k+1
)
ЛСО
T
k
=
(7б)
2
Таким образом можно проследить перемещение кластеров по осям энергии - внут
ренней и кинетической энергии центра масс, а также изменение числа атомов, проис
ходящее в результате столкновений кластер-атом металла.
ЗАКЛЮЧЕНИЕ
На основе данных МД моделирования проведен анализ зарождения и роста класте
ров меди, исследованы взаимодействия кластеров с атомами меди и получены следую
щие результаты.
1. Предложена математическая модель взаимодействия между атомом меди и мед
ным кластером, пригодная для описания макроскопических систем. В качестве пара
метров взаимодействия выбраны внутренняя энергия кластера и кинетические энер
гии центра масс кластера и атома.
2. Модель основана на предположении, что кластер, образованнный при взаимо
действии атома и кластера металла, с вероятностью, зависящей от внутренней энер
гии, может существовать достаточное время, чтобы продолжить рост (долгоживущий).
Указанные вероятности определены из результатов МД моделирования и хранятся в
608
А. Е. Коренченко, А. Г. Воронцов, А. А. Жукова
д
двумерном массиве
где k - количество атомов в кластере,
- пара
P
(k, ),
n
n
= [εk δ]
метр, зависящий от внутренней энергии, δ - шаг по энергии, в настоящей модели вы
брано
δ = 0.01 эВ.
3. Часть кластеров, образованных при взаимодействии атома и кластера металла, с
вероятностью
распадается, не успев столкнуться с другими частицами. Мо
-Pд
1
(k, )
n
дель позволяет оценить энергии продуктов распада с помощью предположения о том,
что за время, проведенное внутри кластера, налетевший атом взаимодействует с дру
гими атомами кластера и энергия перераспределяется между ними так, что их состоя
ния уравниваются. Тогда средняя кинетическая энергия атома меди, вылетевшего при
распаде, в системе отсчета, связанной с центром масс распадающегося кластера, будет
определяться его внутренней энергией. На основании результатов МД моделирова
ния был сформирован массив T1(k, n), в котором k есть количество атомов в кластере,
n характеризует внутреннюю энергию кластера, значение элемента массива равно
среднему значению кинетической энергии вылетающего атома.
Описание “в среднем” не дает возможности предсказать энергетические соотноше
ния в отдельно взятом акте взаимодействия, но, ожидается, что такой подход правдопо
добно опишет начальный этап газофазной нуклеации в макроскопических системах.
СПИСОК ЛИТЕРАТУРЫ
1. K o r e n c h e n k o A . E . , G e l ’ c h u n s k i i B . R . Mathematical simulation of the forma
tion of metallic nanoparticles during the condensation of molten metal vapors // Russian metallurgy.
2011. № 8. Р. 723-728.
2. В а р а к с и н А . Ю . Кластеризация частиц в турбулентных и вихревых двухфазных по
токах // ТВТ. 2014. 52. № 5. С. 777-789.
3. Ф и с е н к о С . П . Микроструктура поля пересыщения при гомогенной нуклеации в па
рогазовой смеси // Журн. технической физики. 2013. 83. № 5. С. 35-40.
4. G o u d e l i E . , P r a t s i n i s S . E . Gas Phase Manufacturing of Nanoparticles: Molecular
Dynamics and Mesoscale Simulations // Particul Sci Techno. 2016. 34. № 4. P. 483-493.
5. G o r s h k o v V. , K u z m e n k o V. , P r i v m a n V. Modeling of Growth Morphology of
Core Shell Nanoparticles // J. Phys. Chem. C. 2014. 118. P. 24959.
6. H e a t h Tu r n e r C . , L e i Yu , Yu p i n g B a o . Modeling the Atomistic Growth Behavior
of Gold Nanoparticles in Solution // Nanoscale. 2016. 8. № 17. P. 9354-9365.
7. Pa r a m i t a H a l d a r, A b h i j i t C h a t t e r j e e . Seeking kinetic pathways relevant to the
structural evolution of metal nanoparticles // Modell. Simul. Mater. Sci. Eng. 2015. 23. P. 025002.
8. Л е н ё в Д . Ю . , Н о р м а н Г. Э . Молекулярное моделирование термической аккомо
дации атомов аргона на кластерах атомов железа // ТВТ. 2019. 57. № 4. С. 534-542.
9. P l i m p t o n S . Fast Parallel Algorithms for Short-Range Molecular Dynamics // J. Compu
tational Physics. 1995. 117. P. 1-19.
10. К о р е н ч е н к о А . Е . , В о р о н ц о в А . Г. , Ге л ь ч и н с к и й Б . Р. , Жу к о в а А . А .
Определение радиусов малых кластеров меди на основе моделирования процесса газофазной
конденсации // ТВТ. 2019. 57. № 2. С. 304-307.
11. В о р о н ц о в А . Г. , К о р е н ч е н к о А . Е . , Ге л ь ч и н с к и й Б . Р. Анализ стабиль
ности малых металлических кластеров при конденсации паров металла // ТВТ. 2019. 57. № 3.
С. 404-407.
12. K orenchenko A .E ., Vo rontsov A .G ., G el’chinskii B .R ., S annikov G .P.
Statistical analysis of dimer formation in supersaturated metal vapor based on molecular dynamics
simulation // Physica A. 2018. 496. P. 147-155.
13. F o i l e s S . M . , B a s k e s M . I . , D a w M . S . Embedded atom method functions for the
fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Phys. Rev. B. 1986. 33. Р. 7983-7991.
14. M o r s e M . D . Clusters o f transition metal atoms // Chem. Revs. 1986. 86. P. 1049-1109.
Макроскопическая модель нуклеации при конденсации паров меди
609
15. К о р е н ч е н к о А . Е . , В о р о н ц о в А . Г. , Ге л ь ч и н с к и й Б . Р. Статистический
анализ образования и релаксации атомных кластеров по данным молекулярно динамическо
го моделирования газофазной нуклеации металлических наночастиц // ТВТ. 2016. 54. № 2.
С. 243-248.
16. IUPAC, Compendium of Chemical Terminology; 2nd ed. the “Gold Book”, 1997.
Macroscopic Model of Nucleation in Condensation of Copper Vapors in an Inert Gas
A. E. Korenchenko1, 2, A. G. Vorontsov3, A. A. Zhukova1
1First Moscow State Medical University named after I.M. Sechenov, Moscow, Russia
2Institute of Metallurgy, Ural Branch of RAS, Yekaterinburg, Russia
3South Ural State University (NRU), Chelyabinsk, Russia
In the framework of a computational scheme suitable for describing condensation, a sta
tistical analysis of the results of molecular dynamics calculations of gas phase “self assem
bly” of nanoclusters during condensation of metal vapor was carried out. The patterns of
collisions and growth of small copper clusters are revealed. The interaction parameters of
clusters with metal atoms are determined, which allow transferring information to a macro
scopic model of nucleation. The results can be used in the description of nucleation to pre
dict the size distribution of nanoparticles in the industrial production of metal nanopowders.
Keywords: nucleation, condensation, copper vapor
REFERENCES
1. Korenchenko A.E., Gel’chunskii B.R. Mathematical simulation of the formation of metallic
nanoparticles during the condensation of molten metal vapors // Russian metallurgy. 2011. № 8.
Р. 723-728.
2. Varaksin A.YU. Klasterizatsiya chastits v turbulentnykh i vikhrevykh dvukhfaznykh potokakh
[Particle clustering in turbulent and vortex two phase flows] // TVT. 2014. 52. № 5. P. 777-789.
(in Russian).
3. Fisenko S.P. Mikrostruktura polya peresyshcheniya pri gomogennoy nukleatsii v parogazovoy
smesi [The microstructure of the supersaturation field during homogeneous nucleation in a vapor gas
mixture] // Zhurnal tekhnicheskoy fiziki. 2013. 83. № 5. P. 35-40. (in Russian).
4. Goudeli E., Pratsinis S.E. Gas Phase Manufacturing of Nanoparticles: Molecular Dynamics
and Mesoscale Simulations // Particul Sci Techno. 2016. 34. № 4. P. 483-493.
5. Gorshkov V., Kuzmenko V., Privman V. Modeling of Growth Morphology of Core Shell Nano
particles // J. Phys. Chem. C. 2014. 118. P. 24959.
6. Heath Turner C., Lei Yu, Yuping Bao. Modeling the Atomistic Growth Behavior of Gold Nano
particles in Solution // Nanoscale. 2016. 8. № 17. P. 9354-9365.
7. Paramita Haldar, Abhijit Chatterjee Seeking kinetic pathways relevant to the structural evolution
of metal nanoparticles // Modell. Simul. Mater. Sci. Eng. 2015. 23. P. 025002.
8. Lenov D.Yu., Norman G.E. Molekulyarnoye modelirovaniye termicheskoy akkomodatsii atom
ov argona na klasterakh atomov zheleza [Norman Molecular modeling of thermal accommodation of
argon atoms on clusters of iron atoms] // TVT. 2019. 57. № 4. P. 534-542. (in Russian).
9. Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics // J. Computation
al Physics. 1995. 117. P. 1-19.
10. Korenchenko A.Ye., Vorontsov A.G., Gel’chinskiy B.R., Zhukova A.A. Opredeleniye radiusov
malykh klasterov medi na osnove modelirovaniya protsessa gazofaznoy kondensatsii [Determination
of the radii of small copper clusters based on modeling of the gas phase condensation process] // TVT.
2019. 57. № 2. P. 304-307. (in Russian).
11. Vorontsov A.G., Korenchenko A.Ye., Gel’chinskiy B.R. Analiz stabil’nosti malykh metalli
cheskikh klasterov pri kondensatsii parov metalla [Analysis of the stability of small metal clusters dur
ing condensation of metal vapor] // TVT. 2019. 57. № 3. P. 404-407. (in Russian).
610
А. Е. Коренченко, А. Г. Воронцов, А. А. Жукова
12. Korenchenko A.E., Vorontsov A.G., Gel’chinskii B.R., Sannikov G.P. Statistical analysis of
dimer formation in supersaturated metal vapor based on molecular dynamics simulation // Physica A.
2018. 496. P. 147-155.
13. Foiles S.M., Baskes M.I., Daw M.S. Embedded atom method functions for the fcc metals Cu,
Ag, Au, Ni, Pd, Pt, and their alloys. Phys. Rev. B. 1986. 33. Р. 7983-7991.
14. Morse M.D. Clusters o f transition metal atoms // Chem. Revs. 1986. 86. P. 1049-1109.
15. Korenchenko A.Ye., Vorontsov A.G., Gel’chinskiy B.R. Statisticheskiy analiz obrazovaniya i
relaksatsii atomnykh klasterov po dannym molekulyarno dinamicheskogo modelirovaniya gazofaznoy
nukleatsii metallicheskikh nanochastits [Statistical analysis of the formation and relaxation of atomic
clusters according to molecular dynamics modeling of gas phase nucleation of metal nanoparticles] //
TVT. 2016. 54. № 2. P. 243-248. (in Russian).
16. IUPAC, Compendium of Chemical Terminology; 2nd ed. the “Gold Book”, 1997.