

Известия РАН. Серия биологическая, 2023, № 2, стр. 115-121
Параллелизмы в видообразовании и внутривидовой диверсификации микроба чумы Yersinia pestis
В. В. Сунцов *
Институт проблем экологии и эволюции им. А.Н. Северцова РАН
119071 Москва, Ленинский просп., 33, Россия
* E-mail: vvsuntsov@rambler.ru
Поступила в редакцию 09.03.2022
После доработки 05.04.2022
Принята к публикации 05.04.2022
- EDN: BPNSYT
- DOI: 10.31857/S1026347023010122
Аннотация
Молекулярные филогении микроба чумы Yersinia pestis противоречат экологическим фактам и не интерпретируются в понятиях концепции адаптациогенеза. Одной из причин противоречий видится недооценка молекулярным подходом параллелизмов в процессах видообразования и внутривидовой диверсификации микроба чумы. Экологические данные свидетельствуют о параллельном тритопном (почти) одновременном видообразовании трех исходных геновариантов (популяций, подвидов) Y. pestis 2.ANT3, 3.ANT2 и 4.ANT1 в трех географических популяциях монгольского сурка (Marmota sibirica), которое в МГ-подходе принимают за мягкую политомию (“Big Bang”). Параллелизмы также имели место при формировании внутривидового разнообразия Y. pestis. Самостоятельность трех филогенетических линий и связанные с этим эволюционные параллелизмы в формировании внутривидового разнообразия Y. pestis в молекулярном подходе не принимаются во внимание. Перспектива создания реального филогенетического дерева Y. pestis состоит в творческом синтезе методологий двух подходов – молекулярного и экологического.
Формо-видообразование патогенных микроорганизмов составляет актуальную проблему современного здравоохранения. Наглядным подтверждением этого тезиса может служить протекающая в современном мире пандемия ковид-19. Ее истоки остаются неизвестными, что в значительной мере сдерживает разработку средств и методов контроля пандемии, лечения и профилактики инфекции и выявление ее предикторов. Апокалиптической болезнью, хорошо известной с давних времен, является чума, оставившая неизгладимые следы в человеческой истории. Но, несмотря на более чем столетнюю историю изучения возбудителя этой болезни – микроба Yersinia pestis, открытого А. Йерсеном в Гонконге в 1894 г., вопрос о его происхождении и мировой экспансии остается не вполне решенным.
Возбудитель чумы – уникальный бактериальный патоген. Среда его обитания – паразитарная система грызун/пищуха–блоха. Теплокровные млекопитающие являются хозяевами инфекции, блохи переносят возбудителя между хозяевами. В то же время по биохимическим, генетическим и молекулярным признакам микроб чумы включен в семейство Enterobacteriaceae, объединяющее бактериальных обитателей пищеварительного тракта широкого круга животных, как позвоночных, так и беспозвоночных. У кишечных инфекций передача возбудителя от одного хозяина к другому осуществляется пищевым (алиментарным) способом. Микроб чумы – единственный вид в обширном семействе кишечных микробов, передающийся через укусы блох. То есть в этом семействе он уникален, что свидетельствует о каком-то особом эволюционном пути.
Yersinia pestis: ДВА ОТКРЫТИЯ
В последние два десятка лет в связи с бурным развитием молекулярно-генетических (МГ) технологий и их внедрением в эволюционную инфектологию реконструкция истории Yersinia pestis стала прерогативой МГ-подхода. Филогенетическую структуру выстраивают с помощью анализа нуклеотидных признаков-маркеров на основе, как правило, моделей нейтральной эволюции и с применением статистических методов и компьютерных технологий (Achtman et al., 1999, 2004). Преобразование популяции (клона) предкового кишечного микроба в популяцию микроба чумы связывают с сальтационными генетическими процессами: горизонтальным переносом плазмид вирулентности, генетических комплексов и отдельных генов от других микроорганизмов или из внешней среды, делециями и инактивациями генов, утративших функции в новой среде обитания, в меньшей мере с генетическими рекомбинациями (Achtman et al., 1999, 2004; Zhou et al., 2004).
Молекулярные технологии позволили сделать два важных вклада в решение проблемы происхождения и мировой экспансии микроба чумы. Во-первых, вопреки положениям “классической” теории природной очаговости чумы о древности возбудителя (олигоцен–плиоцен), была доказана его эволюционная молодость. МГ методами показано, что дивергенция чумного микроба от предковой формы произошла в эволюционном масштабе времени недавно, не ранее 30 тыс. лет назад (Achtman et al., 1999, 2004; Morelli et al., 2010; Cui et al., 2013). Более того, МГ-методы позволили оценить скорость эволюции отдельных филогенетических ветвей и создать стройные филогенетические деревья, ставшие современной иллюстрацией истории происхождения и эволюции чумного микроба (Cui et al., 2013; Demeure et al., 2019; Pisarenko et al., 2021).
Во-вторых, выявлен прямой предок чумного микроба, им оказался убиквитарный психрофильный псевдотуберкулезный микроб Yersinia pseudotuberculosis 0:1b, а точнее возбудитель дальневосточной скарлатиноподобной лихорадки (ДСЛ), широко распространенный в холодных районах северной и центральной Азии и на Дальнем Востоке (Fukushima et al., 1998, 2001; Skurnik et al., 2000). Это открытие позволило охарактеризовать корень молодого филогенетического дерева Y. pestis: стало ясно, что некая популяция (клон) возбудителя ДСЛ, обитающая в холодных районах Азии, в недалеком историческом прошлом попала в какие-то уникальные условия, в которых прошло ее преобразование в популяцию микроба чумы. Возникла необходимость выявить эту стартовую анцестральную популяцию и условия, в которые она попала.
МГ-НОМЕНКЛАТУРА ВНУТРИВИДОВЫХ ФОРМ Yersinia pestis
Предложена специфическая номенклатура внутривидовых форм микроба чумы, удобная для конструирования МГ филогенетических схем, которая используется во многих лабораториях мира (Achtman et al., 2004; Morelli et al., 2010; Cui et al., 2013). Популяции/геноварианты/подвиды микроба чумы, циркулирующие в природных очагах имеют цифровые и буквенные обозначения, например 0.PE2 (как считают в МГ-подходе – это один из более древних возбудителей, имеющий пониженную вирулентность, циркулирующий в популяциях обыкновенной полевки на Кавказе). Первая цифра (0–4) обозначает филогенетическую ветвь, ветвь 0 наиболее древняя. Аббревиатура не строго обозначает биохимические биоварианты Pestoides (PE), Antiqua (ANT), Mediaevalis (MED), Intermedium (IN), Orientalis (ORI). Последняя цифра иногда с добавлением буквы обозначает конкретный природный очаг (1–10) (рис. 1).
Рис. 1.
Наиболее популярные филогенетические схемы Y. pestis, построенные анализом SNP маркеров по: Cui et al., 2013 (а); по: Demeure et al., 2019 (б). Значки на концах филогенетических ветвей означают геноварианты (популяции, подвиды) чумного микроба, циркулирующие в природных и антропогенных очагах мира. В корне филогенетического дерева размещена статистически охарактеризованная абстрактная предковая форма чумного микроба MRCA (most recent common ancestor). Деревья построены на основе модели дивергенции голофилетической (без параллелизмов) группы.
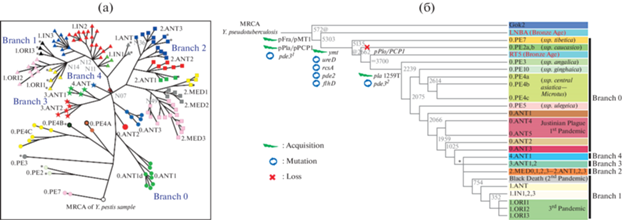
К сожалению, МГ-подход не дает возможности конкретизировать и подробно охарактеризовать исходную популяцию чумного микроба после ее дивергенции от возбудителя ДСЛ и генеалогические линии (интерноды) от анцестрального вида к локальным геновариантам/подвидам. В то же время понятно, что видообразование – популяционно-генетический процесс: популяция предкового вида порождает популяции нового производного вида и интерноды должны представлять определенный геновариант. Статистическая методология (SNP-анализ) позволяет обозначить только безликую экологически не характеризуемую абстрактную форму наиболее современного общего предка MRCA (most recent common ancestor) (Achtman et al., 1999, 2004). “Популяцию” этой абстрактной формы принимают в качестве единого предка всего внутривидового разнообразия чумного микроба, как голофилетическую группу, адекватную МГ методологическим манипуляциям. Но на краеугольные вопросы где, когда, каким образом и при каких обстоятельствах произошло преобразование популяции возбудителя ДСЛ в “статистическую” популяцию MRCA МГ-подход ответа не дает. И, по-видимому, дать не может, так как видообразование – это популяционно-генетический адаптивный процесс, прерогативой изучения которого обладает прежде всего экология (в широком понимании).
ИСТОРИЧЕСКАЯ ИНТЕРПРЕТАЦИЯ МГ ФИЛОГЕНЕТИЧЕСКИХ СХЕМ
Как следует из МГ-логики, MRCA предпринял двухэтапную мировую экспансию в популяциях норовых грызунов (Rodentia) и пищух (Lagomorpha, Ochotona) (Achtman et al., 2004; Cui et al., 2013; Demeure et al., 2019; Сунцов, 2021). Сначала от MRCA отделилась филогенетическая ветвь/ кластер 0.PE, представленная биоваром Pestoides, циркулирующим в популяциях сибирского тушканчика (Allactaga sibirica), нескольких видов полевок (Microtina) и монгольской пищухи (Ochotona pallasi pricei) (рис. 1). Геноварианты/подвиды этого биовара обладают избирательной вирулентностью и слабо или не патогенны для сурков, сусликов, песчанок и человека. Ареал представителей ветви/кластера 0.PE охватил, как полагают, обширные пространства Азии от Маньчжурии и восточного Тибета на востоке до Предкавказья и Ближнего Востока на западе и от Забайкалья и северного Прикаспия на севере до юга Индостана.
Через тысячи лет после формирования первичного ареала геновариантов 0.PE вторая волна экспансии природных очагов чумы уже с высоковирулентным возбудителем началась из тянь-шаньских популяций алтайского сурка (Marmota baibacina) накануне первой пандемии (чума “Юстиниана” в Европе и Средиземноморье, 6–8 века) и распространилась в пределах ареала “древней” “полевково/пищуховой” чумы. Основателем высоковирулентных геновариантов называют 0.PE5 – самого молодого геноварианта среди представителей биовара Pestoides, циркулирующего в географической популяции монгольской пищухи на Монгольском и Гобийском Алтае. По молекулярной структуре (CRISPR) он наиболее близок к высоковирулентным “сурочьим” геновариантам ветви 0.ANT (рис. 1б) (Riehm et al., 2012; Demeure et al., 2019). Природные события, которые обеспечили дальний “транзит” микроба чумы с Монголо-Гобийского Алтая из популяций монгольской пищухи в популяции алтайского сурка на Тянь-Шане и экологические процессы, которые привели к преобразованию геноварианта 0.PE5 в геноварианты линии 0.ANT в популяциях алтайского сурка накануне 1-й пандемии, МГ-подход оставляет без внимания. Двухэтапная экспансия и гостальная адаптация слабовирулентных и высоковирулентных возбудителей, согласно МГ-подходу, привела к внутривидовой диверсификации – возникновению многочисленных геновариантов (популяций, подвидов), сформировавших в Евразии в последние 1500–20 000 лет природные очаги различного ранга, от крупных полигостальных, занимающих обширные пространства мегаочагов до местных (элементарных) очагов, занимающих локальные урочища и биотопы.
Приведенный сценарий исторических событий, постулируемых МГ-подходом, не находит поддержки фактами и закономерностями, накопленными и установленными широким кругом релевантных естественных наук. Двухэтапная недавняя в историческом времени естественная экспансия чумного микроба по принципу “дальних транзитов” в границах одного и того же Евразийского ареала с точки зрения биологической (эпизоотологической) логики представляется совершенно невозможной: бесспорно, что эпизоотии в естественных условиях распространяются по принципу “масляного пятна” за счет внутрипопуляционных, внутривидовых и межвидовых паразитарных контактов животных через укусы блох.
ЭКОЛОГИЧЕСКАЯ ИНТЕРПРЕТАЦИЯ ИСТОРИИ Yersinia pestis
Два выше указанных открытия МГ (эволюционная молодость вида Y. pestis и его происхождение от возбудителя ДСЛ) позволили пересмотреть положения классической теории природной очаговости чумы и создать новый более доверительный непротиворечивый экологический (ЭКО) сценарий видообразования и мировой экспансии возбудителя (Сунцов, Сунцова, 2000, 2006; Сунцов 2020). Согласно ЭКО-сценарию, тритопное видообразование вида Y. pestis произошло (почти) одновременно в трех географических популяциях монгольского сурка (Marmota sibirica) под влиянием тривиальных физико-климатических факторов – нарастания сухости и суровости климата в Центральной Азии во второй половине кайнозоя. Аридность климата вызвала формирование защитного поведения монгольского сурка, приведшего к накоплению псевдотуберкулезного микроба в его организме во время зимней спячки без проникновения в лимфо-миелоидный комплекс. Суровость климата в сартанское время, 22 000–15 000 лет назад, вызвала переход личинок сурочьей блохи Oropsylla silantiewi от детритофагии в подстиле гнезда к факультативной гематофагии на теле спящих сурков в холодное время года и, как следствие, заражение популяции монгольского сурка кишечной псевдотуберкулезной инфекцией неадаптивным механическим, травматическим (но не традиционным алиментарным!) способом. Таким образом, уникальный способ заражения монгольского сурка возбудителем ДСЛ привел к появлению уникального патогена.
ПАРАЛЛЕЛИЗМ ВИДООБРАЗОВАНИЯ Yersinia pestis
Параллелизмы в природе распространены очень широко. Факт параллельного формирования сходных признаков у близкородственных форм известен еще со времен Дарвина (Медников, 1980). Толчок в развитии этого научного направления был сделан Н. Вавиловым (1935). Он установил закономерность, согласно которой многие виды характеризуются сходными и параллельными рядами (сериями) наследственной изменчивости. В проблеме видообразования и внутривидовой диверсификации микроба чумы понятие о параллелизме формирования сходных признаков у родственных форм/геновариантов специально не разрабатывалось, хотя некоторые авторы указывали на параллельное или конвергентное формирование некоторых внутривидовых форм (геновариантов, биоваров) (Achtman, 2004; Zhou et al., 2004). Исследователи, занимающиеся МГ-реконструкциями филогенеза микроба чумы, основываются на эволюционной модели, предписывающей наличие единого корня и строгую дихотомию голофилетических групп. При этом группы, предположительно возникающие параллельно на основе гомоплазии, из филогенетических построений исключают как не соответствующие требованиям узкой монофилии – базовому требованию в МГ филогенетической методологии (Achtman et al., 2004). Только монофилетические в узком смысле, т.е. голофилетические группы подлежат МГ-анализу для реконструкции филогенетического процесса Y. pestis.
Как мы показали (Сунцов, 2020, 2021), триггером видообразования Y. pestis послужило наступление максимального сартанского похолодания в Центральной Азии, которое одновременно охватило ареал монгольского сурка, включающего три географические популяции с обитающими в них подвидами M. sibirica sibirica, M. sibirica caliginosus и M. sibirica ssp. В трех географических популяциях сложились равные условия для травматического внедрения возбудителя ДСЛ в кровь спящих сурков и запуска процесса адаптации возбудителя к организму нового хозяина по принципу “квантового” видообразования (Сунцов, 2018). Территориальная гетерогенность возбудителя ДСЛ, циркулирующего в географических популяциях монгольского сурка, и в некоторой степени различные биотические условия среды его обитания привели, по нашему мнению, к (почти) одновременному формированию трех различных геновариантов чумного микроба: 2.ANT3, 3.ANT2 и 4.ANT1 (рис. 2а) (Сунцов, 2020). В МГ-подходе одновременное образование этих геновариантов рассматривают как возникновение политомии (N07, “Big Bang”) (рис. 1б), вызванной неизвестными природными событиями накануне 2-й пандемии (“Черной смерти”, XIV–XVIII вв.).
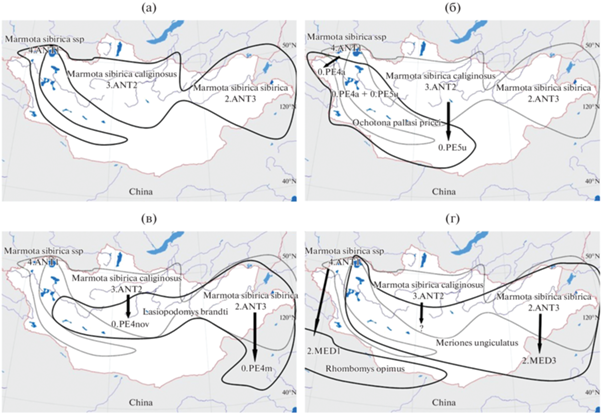
Рис. 2.
Параллелизмы в видообразовании и внутривидовой диверсификации микроба Yersinia pestis. (а) – одновременное формирование трех геновариантов/популяций 2.ANT3, 3.ANT2 и 4.ANT1 чумного микроба в трех географических популяциях монгольского сурка (Marmota sibirica sibirica, M. sibirica caliginosus и M. sibirica ssp.) из различных популяций возбудителя ДСЛ, циркулирующих в популяцих сурка на Хэнтэе, Хангае и Хархира-Монгун-Тайгинском горном поднятии (по: Сунцов, 2020, 2021); (б) – проникновение “сурочьих” геновариантов микроба чумы 3.ANT2 и 4.ANT1 (здесь и далее показано стрелками) в географическую популяцию монгольской пищухи (Ochotona pallasi pricei) и параллельное формирование очагов чумы с геновариантами 0.PE5u (улегейский подвид) и 0.PE4a (алтайский подвид); в промежуточной зоне в Баян-Ульгийском аймаке Монголии подвиды циркулируют в условиях симпатрии; (в) – параллельное проникновение сурочьих геновариантов микроба чумы 3.ANT2 и 2.ANT3 в различные части ареала полевки Брандта (Lasiopodomys brandti) и формирование очагов чумы с геновариантами 0.PE4m (подвид microtus) во Внутренней Монголии (Китай) и 0.PE4nov (неописанный подвид) на Хангае (Монголия); (г) – параллельное проникновение “сурочьих” геновариантов микроба чумы 2.ANT3 и 4.ANT1 в популяции монгольской (Meriones unguiculatus) и большой (Rhombomys opimus) песчанок с последующим формированием геновариантов 2.MED3 и 2.MED1 во Внутренней Монголии и в Джунгарии, соответственно.
ПАРАЛЛЕЛИЗМЫ ВНУТРИВИДОВОЙ ДИВЕРСИФИКАЦИИ Yersinia pestis
МГ филогенетический подход рассматривает чумоподобных возбудителей биовара Pestoides как наиболее древнюю группу, геноварианты которой возникли параллельно из абстрактной популяции MRCA (рис. 1) (Cui et al., 2013; Кисличкина и др., 2019). MRCA после отделения от возбудителя ДСЛ или на Кавказе (Pisarenko et al., 2021), или на Цинхайском плато Тибета (Cui et al., 2013) широко распространился в Евразии. Затем по неизвестным причинам произошла инсуляризация древнего обширного ареала и образовалось локальные природные очаги в популяциях сибирского тушканчика, полевок и монгольской пищухи. Экологическую характеристику этой ветви МГ-подход не дает. Согласно ЭКО-сценарию, территориальная экспансия трех исходных геновариантов 2.ANT3, 3.ANT2 и 4.ANT1 осуществлялась параллельными самостоятельными маршрутами (Сунцов, 2020), на которых происходила адаптация микроба к новым хозяевам.
Обширный вытянутый с юго-востока на северо-запад ареал монгольской пищухи совмещается с хангайской и хархира-монгун-тайгинской географическими популяциями монгольского сурка, в которых циркулируют два разных геноварианта микроба чумы: 3.ANT2 и 4.ANT1. В районах совместного обитания сурка и пищухи “сурочьи” геноварианты перешли в популяцию монгольской пищухи и на дистальных концах ее ареала параллельно возникли очаги чумы с разными геновариантами: 0.PE5u (Гобийский Алтай) и 0.PE4а (Горный Алтай). В промежуточной зоне в Баян-Ульгейском аймаке Монголии на Монгольском Алтае от монгольской пищухи в местных очагах чумы выделяют оба геноварианта/подвида микроба (рис. 2б).
То же самое можно предположить относительно геновариантов 0.PE4m, циркулирующего в популяциях полевки Брандта на Хэнтэе и во Внутренней Монголии (Китай), и нового, пока не описанного геноварианта 0.PE4nov, обнаруженного в популяциях полевки Брандта на Хангайском нагорье (Монголия) (Куклева и др., 2015; Платонов и др., 2015). Согласно экологической логике, анцестральной формой 0.PE4m был геновариант 2.ANT3, а геноварианта 0.PEnov – 3.ANT2 (рис. 2в). Реальность существования геноварианта 0.PE4nov следует подтвердить МГ-методами.
Филогенетическая ветвь (кластер) 2.MED. В МГ-подходе принято считать, что возбудитель чумы биовара Mediaevalis формирует единую филогенетическую ветвь 2.MED, корневым более древним геновариантом которой является 2.MED0, циркулирующий в популяциях горного суслика (Spermophilus musicus) на Кавказе (Носов и др., 2016; Pisarenko et al., 2021). В процессе экспансии далеко на восток геновариант 2.MED0 дивергировал с образованием геновариантов 2.MED1, 2.MED2, 2.MED3 и 2.MED4, образовав, как полагают, голофилетическую группу 2.MED биовара Mediaevalis.
Согласно ЭКО-сценарию, биовар Mediaеvalis и геноварианты группы 2.MED не составляют голофилетическую группу, но возникли параллельными путями из разных исходных “сурочьих” геновариантов 2.ANT3 и 4.ANT1 (рис. 2г). На западном маршруте экспансии геноварианта 4.ANT1 в популяциях большой песчанки (Rhombomys opimus) в Джунгарии сформировался геновариант 2.MED1, а на восточном маршруте на Хэнтэе и во Внутренней Монголии (Китай) в популяциях монгольской песчанки (Meriones unguiculatus) сформировался геновариант 2.MED3 (Zhou et al., 2004; Павлова и др., 2012; Zhang et al., 2018; Сунцов, 2020).
ЗАКЛЮЧЕНИЕ
МГ-филогении Y. pestis выстраиваются на основе моделей нейтральной эволюции и статистических методов анализа и не интерпретируются в экологических терминах, тем самым выхолащивается их эволюционный смысл. Статистическое толкование молекулярной эволюции чумного микроба представляется крайне редукционистским, не отражающим естественные эволюционные события, МГ-реконструкции филогенеза чумного микроба противоречат очевидным экологическим фактам. Создание полноценных нарративов исторического развития возбудителя чумы в МГ-подходе невозможно. Но хотя МГ филогенетическое дерево отражает не реальную историю микроба, а является упрощенным “маркерным” филогенетическим конструктом, в нем все-таки можно усмотреть некоторые важные эволюционные события. Например, МГ дендрограмма хотя и не объясняет, но с очевидностью констатирует наличие политомии “Big Bang” и парафилетические отношения в ветви/кластере 0.PE. Достижения МГ-подхода, а именно два открытия – эволюционной молодости возбудителя чумы и его происхождение от возбудителя ДСЛ – позволили пересмотреть положения классической теории природной очаговости чумы и по-новому взглянуть на историю этого патогена. Понимание эволюционной молодости микроба чумы нацелило на поиск экологических факторов, которые ожидаемо сохранились и могут свидетельствовать о механизмах и принципах видообразовательного процесса. Такие факторы и принципы были обнаружены и установлены экологическими исследованиями. Экологические признаки оказались высокоинформативными для реконструкции истории чумы. Как следствие, помимо доминирующего МГ-подхода в филогенетике Y. pestis сформировался альтернативный ЭКО-подход, более наглядный и более доверительный. Сопоставление экологических и молекулярных реконструкций позволяет создать более совершенную эволюционную модель филогенеза микроба чумы, интегрирующую МГ и ЭКО методологии филогенетических построений. Можно надеяться, что создание и принятие на вооружение новой интеграционной эволюционной модели филогении чумного микроба, учитывающей наличие многочисленных параллелизмов, станет основой дальнейшего развития теории происхождения и мировой экспансии не только возбудителя чумы, но и многих других патогенов.
Список литературы
Вавилов Н.И. Закон гомологических рядов в наследственной изменчивости. М.–Л.: ОГИЗ-Сельхозгиз, 1935. 56 с.
Кисличкина А.А., Платонов М.Е., Вагайская А.С., Богун А.Г. и др. Рациональная таксономия Yersinia pestis // Мол. генет., микробиол. вирусол. 2019. Т. 37. № 2. С. 76–82.
Куклева Л.М., Шавина Н.Ю., Одиноков Г.Н., Оглодин Е.Г., Носов Н.Ю. Анализ разнообразия и определение геновариантов штаммов возбудителя чумы из очагов Монголии // Генетика. 2015. Т. 51. № 3. С. 298–305.
Медников Б.М. Закон гомологической изменчивости (К 60-летию со дня открытия Н.И. Вавиловым закона) М.: Знание, 1980. 64 с.
Носов Н.Ю., Оглодин Е.Г., Краснов Я.М., Куклева Л.М., Шавина Н.Ю. и др. Филогенетический анализ штаммов Yersinia pestis средневекового биовара из природных очагов чумы Российской Федерации и сопредельных стран // Пробл. особо опасн. инфекций. 2016. № 2. С. 75–78.
Павлова А.И., Ерошенко Г.А., Одиноков Г.Н., Куклева Л.М., Шавина Н.Ю. и др. Анализ генетической изменчивости штаммов Yersinia pestis средневекового биовара из природных очагов чумы России и Монголии // Проблемы особо опасн. инф. 2012. № 114. С. 49–53.
Платонов М.Е., Евсеева В.В., Ефременко Д.В., Афанасьев М.В., Вержуцкий Д.Б. и др. Внутривидовая принадлежность рамнозопозитивных штаммов Yersinia pestis из природных очагов чумы Монголии // Мол. генетика, микробиол. вирусол. 2015. № 1. С. 23–28.
Сунцов В.В. “Квантовое” видообразование микроба чумы Yersinia pestis в гетероиммунной среде – популяциях гибернирующих сурков-тарбаганов (Marmota sibirica) // Сиб. экол. журн. 2018. № 4. С. 379–394.
Сунцов В.В. Гостальный аспект территориальной экспансии микроба чумы Yersinia pestis из популяций монгольского сурка-тарбагана (Marmota sibirica) // Зоол. журн. 2020. Т. 99. № 11. С. 1307–1320.
Сунцов В.В. Политопное видообразование микроба чумы Yersinia pestis как причина филогенетической трихотомии в географических популяциях монгольского сурка-тарбагана (Marmota sibirica) // Журн. общей биол. 2021. Т. 82. № 6. С. 431–444.
Сунцов В.В., Сунцова Н.И. Экологические аспекты эволюции микроба чумы Yersinia pestis и генезис природных очагов // Изв. РАН. Сер. биол. 2000. № 6. С. 645–657.
Сунцов В.В., Сунцова Н.И. Чума. Происхождение и эволюция эпизоотической системы (экологические, географические и социальные аспекты). М.: КМК, 2006. 247 с.
Achtman M., Zurth K., Morelli G., Torrea G., Guiyoule A., Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis // PNAS. 1999. V. 96. № 24. P. 14043–14048.
Achtman M., Morelli G., Zhu P., Wirth T., Diehl I. et al. Microevolution and history of the plague bacillus, Yersinia pestis // PNAS. 2004. V. 101. № 51. P. 17837–17842.
Cui Y., Yu C., Yan Y., Li D., Li Y., Jombart T. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis // PNAS. 2013. V. 110. № 2. P. 577–582.
Demeure C.E., Dussurget O., Fiol G.M., Le Guern A.-S., Savin C., Pizarro-Cerda J. Yersinia pestis and plague: an updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics // Genes Immunity. 2019. V. 20. № 12. P. 1–14.
Fukushima H., Gomyoda M., Hashimoto N., Takashima I., Shubin F.N. et al. Putative origin of Yersinia pseudotuberculosis in western and eastern countries. A comparison of restriction endonuclease analysis of virulence plasmids // Intern. J. Medical Microbiol. 1998. № 288. P. 93–102.
Fukushima H., Matsuda Y., Seki R., Tsubokura M., Takeda N., et al. Geographical heterogeneity between Far Eastern and Western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains // J. Clinical Microbiol. 2001. № 10. P. 3541–3547.
Morelli G., Song Y., Mazzoni C.J., Eppinger M., Roumagnac P., et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity // Nature Genetics. 2010. V. 42. № 12. P. 1140–1145.
Pisarenko S.V., Evchenko A.Yu., Kovalev D.A., Evchenko Y.M., Bobrysheva O.V. et al. Yersinia pestis strains isolated in natural plague foci of Caucasus and Transcaucasia in the context of the global evolution of species // Genomics. 2021. V. 113 P. 1952–1961.
Riehm J.M., Vergnaud G., Kiefer D., Damdindorj T., Dashdavaa O. et al. Yersinia pestis Lineages in Mongolia // PLoS ONE. 2012. V. 7. № 2. E30624.
Skurnik M., Peippo A., Ervela E. Characterization of the O-antigen gene cluster of Yersinia pseudotuberculosis and the cryptic O-antigen gene cluster of Yersinia pestis shows that the plague bacillus is most closely related to and has evolved from Y.pseudotuberculosis serotype O:1b // Mol. Microbiol. 2000. V. 37. № 2. P. 316–330.
Zhang Y., Luo T., Yang C., Yue X., Guo R. et al. Phenotypic and molecular genetic characteristics of Yersinia pestis at an emerging natural plague focus, Junggar Basin, China // Am. J. Trop. Med. Hyg. 2018. V. 98. № 1. P. 231–237.
Zhou D., Han Y., Song Y., Huang P., Yang R. Comparative and evolutionary genomics of Yersinia pestis // Microbes and Inf. 2004. № 6. P. 1226–1234.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия биологическая