

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 1, стр. 3-9
Применение теории поля лигандов для моделирования предкраевой структуры спектров рентгеновского поглощения аморфных систем
Д. М. Пашков a, b, *, Д. С. Рубаник a, М. В. Киричков a, А. А. Гуда a, **, С. А. Гуда a, b, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b Институт математики, механики и компьютерных наук им. И.И. Воровича,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: pashkov@sfedu.ru
** E-mail: guda@sfedu.ru
Поступила в редакцию 28.02.2020
После доработки 25.03.2020
Принята к публикации 30.03.2020
Аннотация
В ходе данной работы построена теоретическая база спектров рентгеновского поглощения за K-краем железа в предкраевой области и описаны алгоритмы ее применения для количественного анализа экспериментальных спектров XANES. В рамках ab initio-подхода теории поля лигандов были рассчитаны более 60 теоретических спектров для железа с различным координационным числом, степенью окисления и межатомным расстоянием между центральным атомом и ближайшими атомами кислорода. Были применены различные методы машинного обучения для решения задач определения степени окисления атома железа и координационного числа по полученным спектрам. Оценка качества моделей проводилась методом Leave-One-Out, точность определения степени окисления составила 100%; точность определения координационного числа составила 75% для предсказаний по всей выборке, 96% и 68% по подвыборкам с фиксированными степенями окисления атома железа 2+ и 3+ соответственно.
ВВЕДЕНИЕ
Исследование локальной атомной структуры вокруг атомов 3d-металлов в аморфных матрицах представляет интерес для широкого круга областей (от геологии до катализа). Так, условия формирования тектитов определяют степень окисления и координацию атомов железа в диоксиде кремния, а реакция полимеризации этилена зависит от расположения одиночных атомов хрома на поверхности SiO2. Тектиты и импактиты представляют собой силикатные стeкла ударного происхождения [1]. Принято считать, что тектиты образуются из расплавленного высокоскоростного выброса на ранних стадиях формирования ударного кратера [2]. Химический состав тектитов включает значительные количества диоксида кремния (68–82%), высокое содержание глинозема, низкое содержание воды, микрополости, заполненные смесью газов, таких как диоксид углерода, водород, метан и благородные газы. Было известно, что такие параметры, как давление P, температура T, летучесть кислорода ${{f}_{{{{{\text{O}}}_{{\text{2}}}}}}},$ которые существовали во время процесса образования стекла, определяют координацию катионов. Одним из распространенных элементов в ударных стеклах является железо, поэтому железо потенциально может быть полезно в качестве датчика условий образования ударных пород. Например, зависимость окислительно-восстановительного состояния железа от P, T, ${{f}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ и состава обсуждалась для расплавов в [3]. О влиянии параметров P, T, ${{f}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ на соотношение Fe3+/Fe2+ в ударных расплавах сообщалось в [4]. Определение степени окисления Fe и локальной атомной структуры в тектитах и ударных стеклах является актуальной научной задачей с точки зрения понимания условий формирования этих объектов.
Спектроскопия рентгеновского поглощения является надежным инструментом для определения локальной атомной и электронной структуры вещества в конденсированном состоянии [5, 6]. Wilke и др. [7] показали, что предкраевая структура спектров поглощения за К-краем железа более эффективна для определения зарядового состояния атомов железа. Они продемонстрировали это на примере карты распределения интегральной интенсивности предкрая и его центра масс для соединений железа с различным координационным числом и валентностью. Так для тектитов и импактитов было показано [8–10], что атомы железа в аморфной матрице кремния могут занимать позиции с координационным числом между 4 и 6 и валентностью в широком интервале от Fe2+ до Fe3+. Сложности с количественным анализом спектров таких многокомпонентных систем заставляют искать новые подходы к анализу экспериментальных данных.
В последние годы машинное обучение стало мощным инструментом как для обработки получаемых в ходе экспериментов данных, так и для проведения теоретических исследований. Методы машинного обучения способны решать разнообразные задачи, такие как классификация и ранжирование объектов, построение аппроксимаций некоторых функций, нахождение скрытых зависимостей и скрытых признаков в данных и др. Методы машинного обучения уже использовали для анализа спектральных данных [11–13]. Трехмерная реконструкция локального окружения катиона железа в матрице SiO2 возможна на основе анализа спектра XANES за счет вклада эффектов многократного рассеяния при большой длине свободного пробега фотоэлектрона. Это делает возможным подтверждение структурной модели на основе сравнения экспериментального спектра XANES с теоретическим, построенным для гипотетической структуры [14–17]. Трудность заключается в том, что локальное окружение априори не известно, поэтому может потребоваться большое количество пробных структур [18] для достижения сходимости.
Как недавно было показано, методы машинного обучения (ML) были успешно применены для количественного анализа спектральных данных в целом [19–21] и рентгеновской спектроскопии ближнего края (XANES) в частности [22, 23]. В настоящей работе мы показываем применимость данных методов для извлечения структурных параметров соединений, таких как степень окисления и координационное число, по предкраевой области спектров XANES.
МЕТОДЫ
Предкраевая структура спектров рентгеновского поглощения была рассчитана в рамках мультиплетной теории поля лигандов [24] на основе орбиталей Ваннье (Gregory Wannier) [25–27] в программе XTLS [28, 29]. На первом этапе расчеты зонной структуры были выполнены с использованием подхода линеаризованного дополненного базиса плоских волн с полным потенциалом, как это реализовано в WIEN2k [30] для ячеек, которые содержат полный октаэдр (или тетраэдр) кислородов вокруг 3d-металла. Затем мы построили локальный базис максимально локализованных функций Ванье, взяв все зоны из определенного энергетического окна, образующего валентную полосу. Типичные значения энергии, ограничивающие область ванньеризации, составляют от –10 до 2 эВ относительно уровня Ферми. После построения низкоэнергетического базисного набора орбиталей Ванье мы получаем локальный низкоэнергетический гамильтониан, содержащий до 113 орбиталей Ванье. Однако систему такого размера нельзя использовать для многочастичных расчeтов, поскольку их сложность возрастает экспоненциально с размером базиса. Таким образом, мы должны ограничиться орбиталями, относящимися к низкоэнергетическим возбуждениям системы. Минимальная локальная модель, описывающая спектры за K-краем переходного металла, содержит пять 3d-подобных орбиталей железа и пять молекулярных орбиталей, построенных из суперпозиции орбиталей p-типа от кислорода. Сведение к пространству 10 орбиталей требует применения блочной процедуры Ланцоша к полной матрице орбиталей Ванье, которая преобразует ее в трехдиагональную форму, чтобы построить молекулярные орбитали с наибольшими хоппингами на 3d-орбиталях.
Для анализа предкраевой области рентгеновских спектров XANES использовались различные методы машинного обучения, такие как дерево принятия решений (Decision Tree), ансамблевый метод (ExtraTrees) [31], метод опорных векторов (SVM), логистическая регрессия, многослойный перцептрон (MLP). Данные методы использовались для предсказания координационного числа соединений и степени окисления атома железа в соединениях по их спектрам. В нашей работе мы использовали методы машинного обучения, реализованные в библиотеке scikit-learn для языка программирования Python. Для рассматриваемых методов машинного обучения использовались следующие параметры:
1. Decision Tree: максимальная глубина решающего дерева – 500; функция оценки качества разделения − информационный выигрыш (Information Gain).
2. ExtraTrees: количество деревьев – 300, функция оценки качества разделения – критерий Джини.
3. SVM: параметр регуляризации C = 1000.
4. Логистическая регрессия: использовалась L2-регуляризация с параметром C = 1000; алгоритм оптимизации – квазиньютоновский алгоритм BFGS с ограниченным использованием памяти (L-BFGS) [32]. Количество итераций – 4000.
5. MLP имеет три скрытых слоя размеров: 100, 50 и 10 с гиперболическим тангенсом в качестве функции активации (рис. 1). Сеть обучалась на протяжении 5000 эпох.
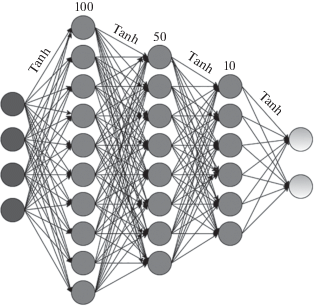
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Применение методов машинного обучения подразумевает наличие выборки данных для тренировки и тестирования алгоритмов, поэтому для проведения исследования необходимо было также подготовить и саму выборку. На первом этапе было необходимо создать структурные файлы соединений. Генерация структур производилась с помощью программного модуля molecule программного обеспечения Py FitIt [33]. Данный модуль позволяет конструировать молекулу из других соединений или отдельных атомов, задавая межатомные расстояния между составными частями или вращая эти части относительно некоторого центра на заданный угол. Были созданы структурные файлы соединений с различными координационными числами (рис. 2), у которых равномерно изменялось расстояние от центрального атома железа до ближайших атомов кислорода в диапазоне от 1.8 до 2.2 Å с шагом в 0.1 Å.
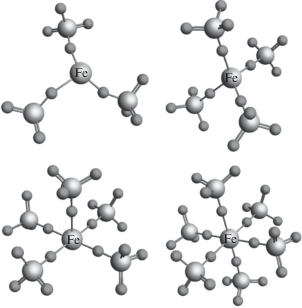
Для каждой структуры были найдены периодические волновые функции на основе решения уравнения Шрeдингера в теории функционала электронной плотности, проведена локализация по методу Ваннье и рассчитаны спектры за K-краем железа. Спектры рассчитывались для различных конфигураций d-оболочки на атомах железа, которые соответствовали валентности Fe2+ и Fe3+ (рис. 3a, 3б). При моделировании учитывались процессы переноса заряда с металла на лиганда. Были получены 60 спектров, которые и составили нашу обучающую выборку для применения методов машинного обучения.
Рис. 3.
Рассчитанные спектры предкраевой области XANES за K-краем железа для соединений с валентностью Fe2+ (а) и Fe3+ (б).
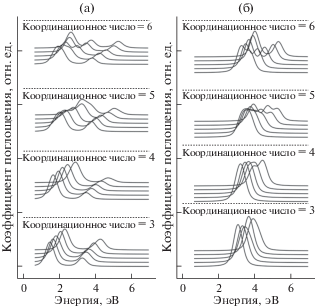
Из полученных графиков спектров (рис. 3) следует, что при изменении координационного числа заметно изменяется высота и ширина пиков на графике, а при изменении степени окисления атома железа происходит химический сдвиг и может изменяться и количество пиков, что объясняется различной мультиплетностью ионов железа 2+ и 3+. При увеличении межатомного расстояния между центральным атомом железа и ближайшими атомами кислорода уменьшается перекрытие d-орбиталей с орбиталями лигандов, а также параметр кристаллического поля. Это приводит к изменению энергетического положения спектральных особенностей, выраженного в виде расщепления или объединения пиков. Для оценки количества структурной информации, содержащейся в рассчитанных спектрах, было решено применить математические методы анализа данных.
Для успешного применения методов машинного обучения необходимо, чтобы все объекты обучающей выборки были представлены некоторым набором признаков, исчерпывающе описывающих каждый объект. Каждый спектр из нашей обучающей выборки представляет собой значения интенсивности коэффициента поглощения для различных значений энергии, причём большинство этих значений являются нулевыми или очень малыми. Более того, разные спектры определены на пересекающихся интервалах значений энергии, а не на одном и том же. По этим причинам тренировка методов машинного обучения по значениям интенсивности поглощения является малоэффективной. Для решения этой проблемы необходимо было выделить новые информативные признаки по имеющимся спектрам. Для этого был создан дескриптор, который строит по спектрам предкраевой области XANES новые признаки. В качестве признаков были выбраны положение и амплитуда пиков на графике спектров, площадь под кривым спектром, математическое ожидание и центральные моменты случайной величины до пятого порядка включительно, где в качестве случайной величины выступала интенсивность коэффициента рентгеновского поглощения. Последние характеристики описывают центр масс и моменты инерции соответственно. С помощью данного подхода мы снизили размерность пространства признаков с 1500 до 11, а также получили качественно лучшие признаки полученных спектров. Такая предобработка данных позволила значительно улучшить качество классификации целевых характеристик соединения.
В силу того, что наша выборка данных очень маленькая, то оценку качества моделей проводили методом скользящего контроля Leave-One-Out, так как данный метод использует все объекты выборки для контроля качества предсказаний обученной модели машинного обучения:
Предсказание степени окисления атома железа
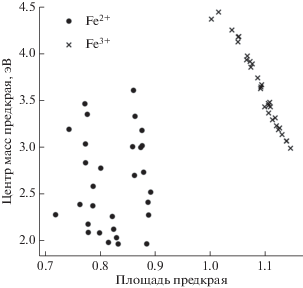
Задача предсказания степени окисления атома железа является задачей бинарной классификации. Перед решением данной задачи были построены графики рассеяния по всем парам признаков. Как следует из полученных графиков (рис. 4 ), классы не проникают друг в друга и линейно разделимы. Благодаря выделению новых признаков данная задача была решена всеми рассматриваемыми методами со стопроцентной точностью классификации.
Предсказание координационного числа соединений
Задача предсказания координационного числа представляет собой задачу многоклассовой классификации с шестью классами. Проведeнная нами предварительная обработка данных не гарантирует наилучшее выделение признаков. Для некоторых методов машинного обучения полученное пространство признаков может быть избыточным. Поэтому для возможного улучшения качества классификации был также использован метод главных компонент (PCA), который позволяет выделить признаки, по которым объекты обучающей выборки разделяются наилучшим образом, и отбросить лишние. В табл. 1 приведены точности результатов предсказания координационного числа рассмотренными методами машинного обучения по всем признакам и по признакам после применения метода главных компонент. В табл. 2 и 3 приведены точности предсказания координационного числа для спектров с одним и тем же значением степени окисления атома железа. При работе с такой выборкой для железа со степенью окисления 2+ качество предсказаний заметно выросло. Таким образом, для решения задачи анализа предкраевой области спектров XANES можно использовать каскад моделей для последовательного предсказания степени окисления атома железа, а затем координационного числа.
Таблица 1.
Точность классификации координационного числа всей выборки по полному набору параметров и после применения анализа основных компонент (PCA)
| Метод машинного обучения | Точность классификации по всем признакам | Точность классификации после применения PCA |
|---|---|---|
| Decision Tree | 0.58 | 0.61 |
| ExtraTrees | 0.55 | 0.65 |
| SVM | 0.75 | 0.66 |
| Logistic Regression | 0.60 | 0.61 |
| MLP | 0.66 | 0.75 |
Таблица 2.
Точность классификации координационного числа соединений со степенью окисления атома железа 2+ по полному набору параметров и после применения PCA
| Метод машинного обучения | Точность классификации по всем признакам | Точность классификации после применения PCA |
|---|---|---|
| Decision Tree | 0.66 | 0.70 |
| ExtraTrees | 0.86 | 0.96 |
| SVM | 0.83 | 0.80 |
| Logistic Regression | 0.86 | 0.86 |
| MLP | 0.90 | 0.93 |
Таблица 3.
Точность классификации координационного числа соединений со степенью окисления атома железа 3+ по полному набору параметров и после применения PCA
| Метод машинного обучения | Точность классификации по всем признакам | Точность классификации после применения PCA |
|---|---|---|
| Decision Tree | 0.33 | 0.60 |
| ExtraTrees | 0.16 | 0.56 |
| SVM | 0.60 | 0.40 |
| Logistic Regression | 0.36 | 0.43 |
| MLP | 0.60 | 0.68 |
Наилучшие результаты были показаны ансамблевым методом ExtraTrees и многослойным перцептроном. Стоит отметить тот факт, что не для всех методов машинного обучения метод главных компонент помог улучшить качество классификации. Метод опорных векторов (SVM) показал лучшие результаты при обучении по признакам без применения метода главных компонент.
ЗАКЛЮЧЕНИЕ
В данной работе были исследованы возможности методов машинного обучения для анализа предкраевой области спектров XANES. Для этого были построены 60 теоретических спектров в предкраевой области спектров рентгеновского поглощения в рамках ab initio-метода мультиплетной теории поля лиганд для соединений с различным координационным числом, степенью окисления и межатомным расстоянием между центральным атомом железа и ближайшими атомами кислорода. Были применены различные методы машинного обучения для решения задач определения степени окисления атома железа и определения координационного числа по полученным спектрам. При решении задачи определения степени окисления все методы показали стопроцентное качество классификации. При решении задачи определения координационного числа были рассмотрены 2 подхода:
1) задача решалась для полной выборки, получена точность классификации – 75%;
2) задача решалась отдельно для двух половин выборки с фиксированной степенью окисления атома железа, получена точность 96% для подвыборки с Fe2+ и 68% для подвыборки с Fe3+.
В дальнейшем планируется расчeт обучающей выборки большего размера для улучшения качества классификации и для возможности решения других задач, в частности, определения геометрических параметров соединения, таких как расстояние и углы между атомами в соединении, а также для использования данного подхода к другим классам соединений.
Список литературы
Koeberl C. // Large Meteorite Impacts and Planetary Evolution / Ed. Dressler B.O., Grieve R.A.F., Sharpton V.L. GSA Special Paper 293, 1994. P. 133–152.
Artemieva N. // Catastrophic Events Caused by Cosmic Objects / Ed. Adushkin V.V., Nemchinov I.V. Berlin: Springer, 2008. P. 267–289. https://doi.org/10.1007/978-1-4020-6452-4_8
Moretti R., Ottonello G. // J. Non-Cryst. Solids. 2003. V. 323. № 1−3. P. 111. https://doi.org/10.1016/S0022-3093(03)00297-7
Lukanin O.A., Kadik A.A. // Geochemistry International. 2007. V. 45. № 9. P. 857. https://doi.org/10.1134/S0016702907090029
Joly Y., Grenier S. // X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications / Ed. van Bokhoven J.A., Lamberti C. Chichester: John Wiley & Sons, 2016.
Bunker G. // Introduction to XAFS: a Practical Guide to X-ray Absorption Fine Structure Spectroscopy. Cambridge: Cambridge University Press, 2011.https://doi.org/10.1017/CBO9780511809194
Wilke M., Farges F., Petit P.-E., Brown Jr. G.E., Martin F. // American Mineralogist. 2001. V. 86. № 5−6. P. 714.https://doi.org/10.2138/am-2001-5-612
Giuli G., Pratesi G., Cipriani C., Paris E. // Geochimica et Cosmochimica Acta. 2002. V. 66. № 24. P. 4347.
Giuli G., Eeckhourt S.G., Paris E., Koeberl C., Pratesi G. // Meteoritics & Planetary Science. 2005. V. 40. № 11. P. 1575.
Kravtsova A.N., Guda L.V., Guda A.A., Trigub A.L., Badyukov D.D., Soldatov A.V. // Radiation Physics and Chemistry / Ed. Kozak M. et al. 2020. https://doi.org/10.1016/j.radphyschem.2018.12.017
Ghosh K., Stuke A., Todorović M., Jørgensen P.B., Schmidt M.N., Vehtari A., Rinke P. // Advanced Science. 2019. V. 6. № 9. P. 24. https://doi.org/10.1002/advs.201801367
Cianciosa M., Law K.J.H., Martin E.H., Green D.L. // Journal of Quantitative Spectroscopy and Radiative Transfer. 2020. V. 240. P. 37. https://doi.org/10.1016/j.jqsrt.2019.106671
Padarian J., Minasny B., McBratney A.B. // Geoderma Regional. 2019. V. 16. P. 56. https://doi.org/10.1016/j.geodrs.2018.e00198
Martini A., Borfecchia E., Lomachenko K.A., Pankin I.A., Negri C., Berlier G., Beato P., Falsig H., Bordiga S., Lamberti C. // Chem. Sci. 2017. V. 8. P. 6836. https://doi.org/10.1039/C7SC02266B
Braglia L., Borfecchia E., Maddalena L., Oien S., Lomachenko K.A., Bugaev A.L., Bordiga S., Soldatov A.V., Lillerud K.P., Lamberti C. // Catal. Today. 2017. V. 283. P. 89. https://doi.org/10.1016/j.cattod.2016.02.039
Tulchinsky Y., Hendon C.H., Lomachenko K.A., Borfecchia E., Melot B.C., Hudson M.R., Tarver J.D., Korzynski M.D., Stubbs A.W., Kagan J.J., Lamberti C., Brown C.M., Dinca M. // J. Am. Chem. Soc. 2017. V. 139. № 16. P. 5992.https://doi.org/10.1021/jacs.7b02161
Bugaev A.L., Guda A.A., Lazzarini A., Lomachenko K.A., Groppo E., Pellegrini R., Piovano A., Emerich H., Soldatov A.V., Bugaev L.A., Dmitriev V.P., van Bokhoven J.A., Lamberti C. // Catal. Today. 2017. V. 283. P. 119.https://doi.org/10.1016/j.cattod.2016.02.065
Bugaev A.L., Usoltsev O.A., Guda A.A., Lomachenko K.A., Pankin I.A., Rusalev Y.V., Emerich H., Groppo E., Pellegrini R., Soldatov A.V., van Bokhoven J.A., Lamberti C. // J. Phys. Chem. C. 2018. V. 122. № 22. P. 12 029.https://doi.org/10.1021/acs.jpcc.7b11473
Chatzidakis M., Botton G.A. // Towards calibration-invariant spectroscopy using deep learning, Scientific Reports. 2019. V. 9. № 2126. P. 1.https://doi.org/10.1038/s41598-019-38482-1
Ghosh K., Stuke A., Todorović M., Jørgensen P.B., Schmidt M.N. Vehtari A. Rinke P. // Advanced Science. 2019. V. 6. № 1801367. P. 1.https://doi.org/10.1002/advs.201801367
Torrione P., Collins L.M., Morton K.D. // Laser Spectroscopy for Sensing / Ed. Baudelet M. Woodhead Publishing, 2014. P. 125. https://doi.org/10.1533/9780857098733.1.125
Timoshenko J., Anspoks A., Cintins A., Kuzmin A., Purans J., Frenkel A.I. // Physical Review Letters. 2018. V. 120. P. 225502-1.https://doi.org/10.1103/PhysRevLett.120.225502
Zheng C., Mathew K., Chen C., Chen Y.M., Tang H.M., Dozier A., Kas J.J., Vila F.D., Rehr J.J., Piper L.F.J., Persson K.A., Ong S.P. // Comput. Mater. 2018. V. 4. № 12. P. 1.https://doi.org/10.1038/s41524-018-0067-x
Haverkort M.W., Zwierzycki M., Andersen O.K. // Physical Review. 2012. V. 85. P. 165113.https://doi.org/10.1103/PhysRevB.85.165113
Kunes J., Arita R., Wissgott P., Toschi A., Ikeda H., Held K. // Computer Physics Communications. 2010. V. 18. № 11. P. 1888.https://doi.org/10.1016/j.cpc.2010.08.005
Mostofi A.A., Yates J.R., Lee Y.S., Souza I., Vanderbilt D., Marzari N. // Computer Physics Communications. 2008. V. 178. № 9. P. 685.https://doi.org/10.1016/j.cpc.2007.11.016
Mostofi A.A., Yates J.R., Pizzi G., Lee Y.S., Souzo I., Vanderbilt D., Marzari N. // Computer Physics Communications. 2014. V. 185. № 8. P. 2309.https://doi.org/10.1016/j.cpc.2014.05.003
Kobayashi M., Niwa H., Takeda Y., Fujimori A., Senba Y., Ohashi H., Tanaka A., Ohya S., Hai P.N., Tanaka M., Harada Y., Oshima M. // Physical Review Letters. 2014. V. 112. P. 107 203.https://doi.org/10.1103/PhysRevLett.112.107203
Tanaka A., Jo T. // J. Physical Society of Japan. 1995. V. 64. № 6. P. 2248.
Schwarz K., Blaha P. // Computational Materials Science. 2003. V. 28. № 2. P. 259. https://doi.org/10.1016/s0927-0256(03)00112-5
Geurts P., Ernst D., Wehenkel L. // Mach. Learn. 2006. V. 63. P. 3.https://doi.org/10.1007/s10994-006-6226-1
Najafabadi M.M., Khoshgoftaar T.M., Villanustre F., Holt J. // J. Big Data. 2017. V. 4. P. 1.https://doi.org/10.1186/s40537-017-0084-5
Martini A., Guda S.A., Guda A.A., Smolentsev G., Algasov A., Usoltsev O., Soldatov M.A., Bugaev A.L., Rusalev Yu., Lamberti C., Soldatov A.V. // Computer Physics Communications. 2020. V. 250.https://doi.org/10.1016/j.cpc.2019.107064
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования