

Расплавы, 2022, № 5, стр. 521-536
Анализ процессов термической перестройки строения нанокластеров Ag75 и Ag101
Д. А. Рыжкова a, *, С. Л. Гафнер a, Ю. Я. Гафнер a, **
a Хакасский государственный университет им. Н.Ф. Катанова
Абакан, Россия
* E-mail: bashkova.daria@yandex.ru
** E-mail: ygafner@khsu.ru
Поступила в редакцию 10.05.2022
После доработки 24.06.2022
Принята к публикации 25.06.2022
- EDN: BDBTPT
- DOI: 10.31857/S0235010622050085
Аннотация
Методом молекулярной динамики с использованием модифицированного потенциала сильной связи TB-SMA проводилось исследование влияния декаэдрических “магических” чисел на формирование внутреннего строения наночастиц Ag (диаметром до 2.0 нм) для кластеров с ГЦК и аморфным начальным состоянием. Был сделан вывод, что в случае малых нанокластеров серебра даже четкое соответствие размера “магическому” числу той или иной структуры (ГЦК, ГПУ, Ih, Dh) не может гарантировать ее появление в ходе процесса термической эволюции. Если такие кластеры подвергнуть процедуре плавного нагрева путем постепенного подвода термической энергии, то протекающие при этом стохастические диффузионные процессы могут оказать очень сильное влияние на получаемые в конечном итоге структуры. Было отмечено наличие разнообразных возможных политипных переходов, чаще всего приводящих к образованию Ih- или Dh-модификаций. Несмотря на то, что полной фиксации структуры с осью симметрии пятого порядка при процессах плавного нагрева наночастиц различной исходной морфологии не происходило, показано, что использование кластеров с разрушенным начальным строением в большей степени позволяет решить проблему получения частиц с требуемой Ih- или Dh-фазой. ГЦК структура в кластерах такого малого размера не может считаться термически устойчивой и для ее стабилизации необходимо использовать наночастицы большего диаметра. Таким образом, если вид и качество строения имеет критическое значение для различных приложений, например, медицинских или плазмонных, то необходимо учитывать возможные процессы перестройки кластерной структуры, происходящие при реальных эксплуатационных условиях.
ВВЕДЕНИЕ
Сейчас широкое практическое применение наночастиц серебра (AgНЧ) в самых разных областях промышленности обеспечивается их уникальными оптическими, термодинамическими и электронными свойствами [1–6], а также большим разнообразием методов синтеза, которые позволяют получать частицы различных размеров и форм [7–10]. В последнее десятилетие мировое потребление наночастиц серебра неуклонно растет и к 2022 году уже достигло 200 т в год [4], причем использование AgНЧ в биомедицине составляет более трети от общего объема.
В зависимости от используемого метода синтеза наночастицы серебра могут быть изготовлены с различными морфологиями, такими как: сферические структуры, нитеобразные, треугольные пластины, шестиугольные пластины, пирамиды и кубы. В ходе проведенных опытов стало ясно, что уникальные химические и физические свойства AgНЧ определяются не только большим процентом атомов, расположенных на внешнем слое, но и кристаллографической ориентацией поверхности [1]. На количество поверхностных атомов влияет размер наночастицы, в то время как кристаллографическая ориентация зависит от ее формы. Поверхности с гранями {111}, {100} и, возможно, {110} в AgНЧ различаются не только плотностью поверхностных атомов, но также их электронной структурой, связью и химической реакционной способностью [1]. Данное наблюдение ставит перед учеными задачу синтеза AgНЧ со сферическим (квазисферическим), декаэдрическим или октоэдрическим строением, устойчивым в области комнатных температур или немного выше.
Другим интересным физическим явлением, связанным с использованием наночастиц серебра, стала поверхностно-усиленная Рамановская спектроскопия (surface-enhanced Raman Scattering – SERS). Данный метод продемонстрировал большой потенциал для быстрого обнаружения химических веществ в различных образцах и стал популярен в последние десятилетия в научном сообществе, благодаря большим перспективам применения. Однако, несмотря на многообещающие возможности SERS метода, оказалось, что для его практического использования необходимо решить множество теоретических и технических задач [11]. В частности, так как для улучшения рамановского сигнала используются, так называемые, SERS-активные подложки, представляющие собой неорганические или гибридные наноструктурированные материалы, то большое внимание должно быть уделено развитию методов формирования массивов металлических НЧ с контролируемыми параметрами, такими как размер, форма, межчастичное расстояние и степень упорядоченности.
Конструкция подложек SERS обычно направлена на максимальное увеличение плазмонного эффекта для усиления комбинационного рассеяния. Среди различных плазмонных материалов именно серебряные SERS-подложки обеспечивают наиболее сильное рамановское усиление и поэтому наиболее перспективны для широкого практического применения [11, 12]. В качестве усиливающей SERS-подложки также используются и наноколлоидное серебро, но и в этом случае явление SERS в значительной мере зависит от размера и формы AgНЧ. Хорошо известно, что сильное взаимодействие AgНЧ со светом приводит к эффекту поверхностного плазмонного резонанса в случае совпадения частоты падающего света с частотой колеблющихся электронов на поверхности. Поверхность AgНЧ не позволяет электронам проводимости покинуть частицу, создавая таким образом силу ответственную за частоту дипольного поверхностного плазмона [1], и меняя форму AgНЧ можно управлять колебаниями электронов.
Таким образом, в случае реального применения нанокластеров серебра в различных технических приложениях необходимо, прежде всего, найти условия устойчивости требуемого кристаллического строения и формы Ag наночастиц. Для решения такой задачи в работе будут рассмотрены и проанализированы возможные конфигурационные изменения в процессе термического воздействия на AgНЧ с числом атомов соответствующим геометрическим декаэдрическим “магическим” числам с целью определения размерных границ наночастиц, в которых может происходить политипный переход.
КОМПЬЮТЕРНАЯ МОДЕЛЬ
Для физически корректного решения поставленной задачи необходимо, прежде всего, определить наиболее адекватную методику исследования. Несомненно, результаты реальных экспериментов являются именно тем маркером, который позволяет проверить наши предположения и гипотезы. Но в случае анализа термической эволюции малых и очень малых наночастиц (нанокластеров) не все так однозначно. Так с точки зрения кристаллографии малым металлическим нанокластерам выгодно принять строение, соответствующее симметрии с осью пятого порядка, т.е. икосаэдрическое (Ih) или декаэдрическое (Dh), так как именно в этом случае достигается минимально возможное значение поверхностной энергии, играющей при таких размерах одну из основных ролей. Резонно предположить, что и результаты, полученные, например, при помощи электронной микроскопии, должны дать нам аналогичные данные. Однако в [13] было проведено подробное исследование электронным микроскопом высокого разрешения внутреннего строения наночастиц Cu3Au. В работе был сделан вывод о том, что морфология наночастиц существенно изменялась под действием электронного пучка микроскопа. То есть в [13] было показано, что в результате облучения частицы переходили в состояние жидких капель, а затем происходила их кристаллизация, в результате чего структура частиц с D = 2.0–20.0 нм была определена как ГЦК. В конечном итоге экспериментально было зафиксировано, что внутреннее упорядочение наночастиц даже при комнатной температуре является очень сложным процессом, зависящим от размера частиц и свойств окружающей среды.
Таким образом, можно прийти к выводу, что данные электронной микроскопии в случае металлических наночастиц очень малого размера (D < 2.0 нм) не всегда могут дать нам истинные результаты, и причиной является относительно высокая энергия электронов, приводящая к нежелательным процессам массированного термического воздействия. То есть, вместо анализа последствий плавного нагрева малых наночастиц в виде возможного изменения их внутреннего строения, мы можем получить несколько иную физическую реальность, а именно анализ результатов цикла плавление-кристаллизация. Формирование строения ряда металлических наночастиц в этом случае было изучено нами в ряде более ранних работ [14–18] и не является целью настоящего исследования.
На наш взгляд наиболее подходящим способом получения интересующей нас информации может стать компьютерное моделирование. Данная методика позволяет варьировать в самых широких пределах целый ряд как внутренних, так и внешних параметров нанокластеров (размер, форма, начальное строение, температура, темп подвода/отвода термической энергии и т.д.), что дает нам широкий простор для возможных модельных опытов, часто недостижимый при проведении реальных экспериментов. В качестве основы моделирования был выбран метод молекулярной динамики, который является достаточно адекватным с точки зрения определения структур металлических наночастиц с интересующим нас размером.
Другим важным моментом моделирования реальных систем является выбор правильной формы межатомного взаимодействия. Как показала практика, применение EAM потенциала (метод погруженного атома) при имитации малых металлических наночастиц не может считаться успешным в силу базовых особенностей построения такого рода взаимодействия. В случае достаточно больших частиц такой метод способен дать адекватные результаты, но при анализе изучаемых нами малых нанокластеров полученные на его основе данные перестают быть физически корректными. Так, в частности, при использовании EAM потенциала температуры плавления малых наночастиц перестают зависеть от размера кластера, то есть энергии связи атомов оказывается практически одинаковыми, несмотря на различное число координационных сфер в кластерах разного размера [19].
Поэтому в представленной работе компьютерное моделирование AgНЧ проводилось методом молекулярной динамики с использованием модифицированного потенциала сильной связи TB-SMA [20], хорошо зарекомендовавшего себя при исследовании внутреннего строения нанокластеров различных химических элементов. Метод основан на том, что большая группа свойств переходных металлов может быть полностью определена из плотности состояний внешних d-электронов. Второй момент плотности состояния есть μ2, и экспериментально установлено, что энергия когезии металлов пропорциональна ширине плотности состояний, которая в приближении вторых моментов есть просто $\sqrt {{{\mu }_{2}}} $. Если принимаются во внимание только ddσ- ddπ- и ddδ-орбитали, то, согласно данной модели, зонная энергия атома i может быть записана как
(1)
$E_{B}^{i} = - {{\left( {\sum\limits_j {\xi _{{\alpha \beta }}^{2}{{e}^{{ - 2{{q}_{{\alpha \beta }}}\left( {{{{{r}_{{ij}}}} \mathord{\left/ {\vphantom {{{{r}_{{ij}}}} {r_{0}^{{\alpha \beta }}}}} \right. \kern-0em} {r_{0}^{{\alpha \beta }}}}{\kern 1pt} - {\kern 1pt} 1} \right)}}}} } \right)}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}},$(2)
$E_{R}^{i} = \sum\limits_j {{{A}_{{\alpha \beta }}}{{e}^{{ - {{p}_{{\alpha \beta }}}\left( {{{{{r}_{{ij}}}} \mathord{\left/ {\vphantom {{{{r}_{{ij}}}} {r_{0}^{{\alpha \beta }}}}} \right. \kern-0em} {r_{0}^{{\alpha \beta }}}}{\kern 1pt} - {\kern 1pt} 1} \right)}}}} ,$Потенциалы, разработанные Клери и Розато [20], очень хорошо зарекомендовали себя при моделировании систем в кристаллическом состоянии и прошли подробную успешную проверку [20] по многим показателям. В частности, было произведено сравнение с экспериментальными данными ряда параметров точечных дефектов (вакансий, междоузлий и их малых комплексов), ряда термодинамических свойств металлов (температуры плавления, теплоты перехода, теплоемкости, коэффициента теплового расширения, константы Грюнайзена и т.д.), а также фононных спектров. Отметим, что рассчитанные в [20] характеристики в сравнении с экспериментальными результатами имеют расхождение не более 3–5%.
Температура системы определялась посредством средней кинетической энергии атомов, рассчитанной при помощи скоростного алгоритма Верле [21] с шагом по времени 1 фс. Начальные сферические кластеры были получены путем вырезания из идеальной ГЦК решетки с последующей термической релаксацией при 20 К. На этом этапе был сформирован первый ансамбль частиц. Затем AgНЧ были ступенчато нагреты с целью полного разрушения дальнего порядка в них и плавно охлаждены с высоким темпом отвода тепловой энергии до температуры 20 К. Из полученного массива наночастиц были отобраны наночастицы с аморфным строением для формирования второго ансамбля. Далее AgНЧ различной начальной морфологии ступенчато нагревались до температуры 1000 К с шагом 20 К, а в области структурных переходов с шагом 5 К. При каждом фиксированном значении частицы выдерживались в течение 1 нс.
Важным моментом моделирования является взаимодействие системы с тепловым резервуаром. Для метода молекулярной динамики естественным является микроканонический ансамбль, для которого постоянной величиной является энергия. Однако в большинстве случаев термодинамические свойства, рассчитанные при условии микроканонического ансамбля, не отражают реальность. При моделировании больший интерес представляет система не с фиксированным значением полной энергии Е, а с постоянным значением температуры Т, то есть уже канонический ансамбль. В такой ситуации система незамкнута, и ее можно представить находящейся в контакте с тепловым резервуаром. Для подобных целей было предложен и развит целый ряд специальных методик моделирования, и, в частности, термостат Нозе или его модификация термостат Нозе–Гувера (Nosé–Hoover), который мы и используем при вычислениях.
В этом случае общая энергия состоит из суммы кинетической и потенциальной энергии частиц и кинетической и потенциальной энергии теплового резервуара s
(4)
$H = \frac{1}{2}\sum\limits_{i{\kern 1pt} = {\kern 1pt} 1}^N {{{m}_{i}}\dot {r}_{i}^{2}} + \Phi ({{r}_{1}},{{r}_{2}},...,{{r}_{N}}) + {{\left( {\frac{{d{\kern 1pt} \ln {\kern 1pt} s}}{{dt}}} \right)}^{2}}\frac{Q}{2} + (3N + 1){{k}_{{\text{B}}}}{{T}_{0}}{\kern 1pt} \ln {\kern 1pt} s,$Моделирование проводилось с использованием компьютерной программы MDNTP, разработанной Dr. Ralf Meyer, University Duisburg Germany [22, 23]. Наличие структурных переходов определялось по калориметрическим кривым зависимости потенциальной энергии нанокластеров от температуры, функции радиального распределения и программам-визуализаторам OVITO и xmakemol. Наиболее информативным на первом этапе является зависимость потенциальной энергии кластера от температуры. Если она имеет непрерывный вид, без каких либо изломов, то это свидетельствует об отсутствии структурных переходов. Если же на данной кривой имеются отклонения от линейного характера зависимости Ер(Т), то данные участки исследуются более подробно с применением CNA (Common Neighbor Analysis) методики [24] на основе локальной парной корреляционной функции каждого атома кластера. Далее при помощи визуализаторов определяется реальный внешний вид и структура изучаемых наночастиц.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЯ
В наших более ранних работах по данной тематике [25, 26] уже проводился анализ термической стабильности нанокластеров серебра диаметром до 2 нм при условии их начального ГЦК строения. Было показано, что такой вид кристаллической структуры Ag кластеров оказывается термически стабильным для наночастиц диаметром D > 1.8 нм (170–180 атомов). Однако при меньших размерах сценарии термической эволюции внутреннего строения нанокластеров серебра значительно усложнялись. Причина состоит в огромном проценте поверхностных атомов, которые за счет уменьшения координационного числа, являются термически подвижными. Поэтому уже при комнатной температуре происходит явно выраженная диффузионная перестройка наночастиц с целью уменьшения их поверхностной энергии. Атомы верхнего слоя буквально скользят по поверхности, пытаясь занять энергетически более выгодные позиции. Результатом этого является формирование у нанокластера максимально возможной сферической формы, часто обладающей икосаэдрическим или декаэдрическим строением.
В работе [27] средствами компьютерного моделирования нами исследовалась структурная стабильность малых нанокластеров серебра с размерами, соответствующими ряду “магических” чисел ГЦК строения при условии исходной аморфной конфигурации. Полученные закономерности сравнивались с более ранними данными [25, 26] для аналогичного набора частиц с начальной ГЦК морфологией. Было показано, что характер термически индуцированных структурных переходов исследуемых нанокластеров резко отличается от ранее наблюдаемых: отсутствуют ГЦК и ГПУ конфигурации на фоне преобладания Ih модификаций. На основе полученных данных был сделан вывод, что использование разного начального строения малых нанокластеров серебра позволяет в ходе термической эволюции создать кластеры с требуемым внутренним строением, что может быть недостижимо при обычных химических или физических методах синтеза.
Результаты [27] по стабилизации икосаэдрического строения нанокластеров серебра диаметром до 2 нм показались нам очень многообещающими, поэтому в данной статье был проведен дальнейший анализ таких вопросов с учетом роли “магических” чисел уже декаэдрических структур. Хорошо известно, что кластеры с определенным числом атомов, отвечающим “магическим” числам, гораздо более стабильны по сравнению с кластерами иной величины. При этом степень дефектности поверхности кластеров с “магическими” числами атомов минимальна, в то время как у остальных она может быть весьма значительной. То есть в случае малых кластеров часто именно “магические” числа различной природы играют решающую роль в стабилизации того или иного внутреннего строения.
Так как основной целью представленной работы было определение влияния декаэдрических “магических” чисел на формирование внутреннего строения AgНЧ при условии термического воздействия, то для проведения компьютерных экспериментов были взяты частицы серебра диаметром до 2 нм. Выбор частиц столь малого размера обусловлен тем обстоятельством, что именно при таких величинах “магические” числа различной природы могут оказывать наиболее существенное воздействие на процессы атомного упорядочения.
Далее на примерах Ag75 и Ag101 изучим роль геометрических “магических” декаэдрических чисел при условии различной начальной морфологии (ГЦК или аморфная конфигурации), что позволяет более глубоко проследить природу возможных структурных переходов. На первом этапе в качестве начальных структур были использованы нанокластеры серебра, вырезанные из идеальной ГЦК-решетки (первый моделируемый ансамбль).
Рассмотрение начнем с меньшего по числу атомов кластера Ag75. Анализ полученных при моделировании данных показал неожиданно низкое влияние декаэдрических “магических” чисел для этого размера AgНЧ (рис. 1). Весь моделируемый ансамбль кластеров Ag75 вел себя при подводе тепловой энергии совершенно одинаково. А именно, исходное ГЦК-строение сохранялось на всем протяжении процесса подвода тепловой энергии вплоть до начала плавления, постепенно разрушаясь с поверхности. Только при температурах порядка Т = 560–600 К наблюдалось небольшое уменьшение потенциальной энергии, связанное с некоторой оптимизацией поверхности кластеров Ag75 в сторону более четко выраженной фасетной структуры, характерной при декаэдрическом строении. Далее при температурах 700–800 К происходили процессы, приводящие к попытке перестройки расположения атомов в направлении симметрии пятого порядка, предположительно икосаэдрического характера (рис. 1). Однако из-за слишком высокого значения термической энергии эта попытка оказывается неудачной и далее кластеры Ag75 переходят в расплавленное состояние.
Рис. 1.
Зависимость потенциальной энергии кластера Ag75 с начальным ГЦК-состоянием от температуры. Разный цвет атомов серебра определяется стандартной CNA-методикой (common neighbors analyze), анализирующей число связей каждого атома в 1–3 координационной сферах. Так атом красного цвета обозначает, что его координационное число в 1-ой сфере меньше 12, атом желтого цвета то, что его координационное число в 1-ой сфере равно 12, но во 2-ой меньше значения, характерного для плотноупакованных структур (ГЦК, ГПУ). Атомы синего цвета показывают, что их координационные числа в 1, 2 и 3-ей координационной сферах соответствуют ГЦК структуре, а зеленого – ГПУ.
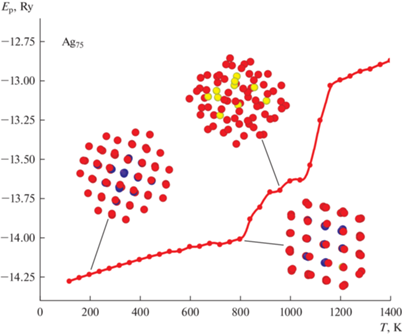
Таким образом, на основе полученных данных по зависимости внутреннего строения Ag75 от температуры, было установлено, что структурный переход для данного размера не свойственен и исходная ГЦК-фаза фактически является термически стабильной, по крайней мере до температуры Т = 800 К. Данный результат оказался достаточно неожиданным, так как при малых размерах кластеров влияние “магических” чисел обычно достаточно велико и часто приводит к перестройке начального расположения атомов. Попробуем объяснить причину полученного результата.
Как известно, при очень низких температурах Dh-кластер Ag75 обладает значением энергии, близким к энергии икосаэдра [28], которая, в свою очередь, имеет уровень несколько ниже энергии кластера с ГЦК-построением, причем такое малое различие по энергии сохраняется для Ih- и Dh-кластеров серебра, по всей видимости, практически до температуры плавления кластера. В случае “магического” Ih-размера кластера Ag55 [29] это приводит к очень быстрому преобразованию ГЦК-строения в икосаэдрическое, а для следующего по размеру “магического” Ih-кластера Ag147 в ходе нагрева примерно в одинаковой пропорции могла быть сформирована как икосаэдрическая, так и декаэдрическая модификация.
То есть, при подходящих условиях оси симметрии пятого порядка достаточно легко реализуется в кластерах серебра малого размера. Однако для ГЦК-кластера Ag75 такого явного перехода зафиксировано нами не было. Как было показано в работе [30], различие по энергиям между Dh-структурой и даже аморфной здесь может быть очень малым. Соответственно энергетический разрыв межу Dh-структурой и ГЦК будет еще меньше, из-за чего даже незначительные воздействия могут подавить ожидаемый в теории структурный переход ГЦК → Dh и таким воздействием, по нашему мнению, стало влияние геометрического ГЦК “магического” числа, наблюдаемого при N = 79 атомов. Действительно, как было показано нами ранее [26], кластер Ag79 сохранял неизменным свое исходное ГЦК-строение до температуры плавления и видимо наличие исходной ГЦК-структуры и близость к ГЦК “магическому” числу, явно стабилизирующему такой вид расположения атомов, не позволили кластеру Ag75 осуществить переход ГЦК → Dh.
Данное предположение находит свое подтверждение в случае следующего “магического” Dh-размера. Из полученных при моделировании данных для Ag101 можно сделать вывод, что уже на стадии предварительной термической релаксации, аналогично кластеру Ag55, происходил политипный переход ГЦК → Ih. Но если для Ag55 были получены идеальные икосаэдрические структуры, то в случае Ag101 сформированные на этапе релаксации структуры были икосаэдрического вида, но с большими нарушениями укладки атомов. С ростом температуры происходила коррекция Ih-структуры и при Т ≈ 470 К кластер Ag101 обладал уже достаточно правильным Ih-строением. Однако для полного завершения третей Ih-оболочки не хватает еще 46 атомов, что не дает возможности кластеру Ag101 сформировать глубокий минимум потенциальной энергии, гарантирующий стабильность имеющегося атомного строения. Поэтому уже при Т = 510 К Ag101 произвел перестройку атомов с образованием структуры близкой к декаэдрической, которая и была сохранена вплоть до температуры плавления (рис. 2). О низком качестве строения полученной Dh-структуры говорит тот факт, что, несмотря на большее число атомов, температура плавления Ag101 оказывается ниже, чем у Ag75. Данная особенность не стала для нас особенно удивительной, так как проведенный CNA анализ атомов, составляющих данные кластеры, показал значительное уменьшение межатомных связей в случае Ag101, особенно в области комнатных температур и выше.
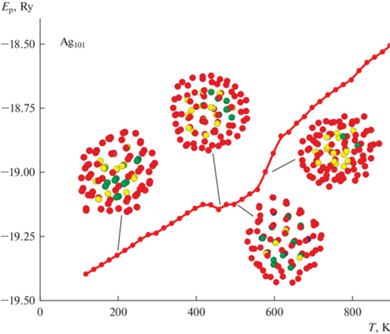
Следовательно, по результатам оценки термической стабильности ГЦК-кластера Ag101 можно сделать вывод о том, что, несмотря на строгое соответствие геометрическому “магическому” числу Dh-структуры, в ходе нагрева стохастически могла быть сформирована как икосаэдрическая, так и декаэдрическая модификация, что подтверждает близкие значения энергий данных структур с осью симметрии пятого порядка. Таким образом, в случае относительно высоких температур теоретическое энергетическое различие между Ih- и Dh-структурами может нивилироваться различными кинетическими факторами, в частности случайным характером диффузионных процессов и превышением величины тепловой энергии kT над энергетической щелью, разделяющей различные структурные модификации.
Перейдем теперь к рассмотрению результатов моделирования второго ансамбля частиц, т.е. кластеров Ag75 и Ag101 с первичной аморфной морфологией. Проанализируем полученные данные для набора наночастиц Ag75. Ожидаемо энергия начального состояния у таких кластеров была ощутимо выше, чем в случае исходного ГЦК-строения. Так при температуре T = 100 К различие по энергиям составило примерно 0.36 Ry или 0.065 эВ/атом. С ростом температуры различие резко падает и при Т = 300 К составляет уже 0.18 Ry или 0.0325 эВ/атом, немного уменьшаясь с дальнейшим ростом температуры. Причину такого сокращения значения ΔE можно легко определить по “мгновенным” снимкам кластера Ag75, показанным на рис. 3. Видно, что изначально аморфный кластер в ходе подвода тепловой энергии производит попытку перехода к икосаэдрическому строению. Однако сформированное расположение атомов оказывается не очень выгодным с энергетической точки зрения, так как не сопровождается существенным падением уровня потенциальной энергии. Как можно понять из рис. 3, с ростом температуры аморфное строение Ag75 начинает оптимизироваться, практически плавно переходя к некому варианту Ih-строению. Однако высокое значение потенциальной энергии, достаточно “рыхлый” внешний вид кластера и проведенный CNA анализ атомов Ag75 говорят о малом числе сформированных здесь межатомных связей. То есть в отличие от первого набора частиц с начальным ГЦК-строением аморфная морфология, часто получаемая при химических методах синтеза [13], может существенно облегчить получение наночастиц с осью симметрии пятого порядка. Однако в нашем случае качество такой структуры получилось достаточно низким и с дальнейшим ростом температуры AgНЧ просто вновь перешла в аморфное состояние. Плавный рост потенциальной энергии в данном случае обеспечивался явным увеличением площади поверхности кластера, который фактически начал растекаться с ростом температуры. Данный сценарий процесса термической эволюции кластеров Ag75 наблюдался нами примерно в 70% проведенных опытов.
Рис. 3.
Зависимость потенциальной энергии кластера Ag75 с начальным аморфным состоянием от температуры. Показан наиболее вероятный вариант термической эволюции.
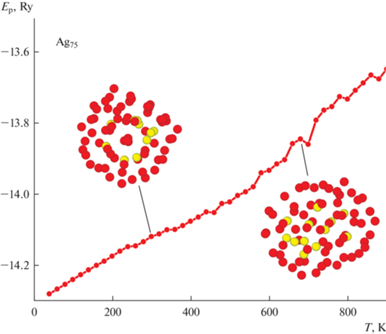
Полученный результат оказался достаточно неожиданным, так как изначально предполагалось, что наличие декаэдрического “магического” размера приведет к фиксации какой-либо ярко выраженной структуры с осью симметрии пятого порядка примерно в области комнатных температур, но этого не произошло. Попробуем понять такие данные компьютерного моделирования. Прежде всего, отметим достаточно высокое значение разности потенциальной энергии 0.065 эВ/атом на начальном этапе моделирования уже после проведения процедуры оптимизации строения. В работе [30] было проведено ab-initio исследование относительной стабильности кластеров золота с кристаллическим и аморфным построением аналогичного размера. Было определено, что различие по энергии связи на атом между наиболее стабильной аморфной и кристаллическими модификациями, составляет менее 0.01 эВ/атом. Примерно такой же результат был зафиксирован и нами для AgНЧ при использовании того же TB-SMA потенциала, но для других “магических” чисел [27, 29]. То есть причину такой большой величины энергии взаимодействия нельзя объяснить некорректно работающим потенциалом TB-SMA.
Причина неполной коррекции строения Ag75 после процедуры термической релаксации может заключаться в следующем. Как известно, при таких малых размерах AgНЧ именно Ih-строение, в целом, является энергетически более выгодным. Но такое строение формируется методом оптимизации поверхностной энергии, из-за чего поверхность идеального икосаэдра представляет собой 20 равносторонних треугольников (икосаэдрон Маркса). Только в этом случае можно получить достаточно ощутимый выигрыш в энергии. Поэтому можно предположить, что на начальном этапе нагрева рассматриваемых нами НЧ, Ag75 (рис. 3) действительно начинает формировать Ih-строение. Но размер кластера (75 атомов) не позволяет этого сделать полноценно. Ближайший идеальный Ih-размер (“магическое” число) 55 атомов, следующее – 147. Таким образом, у Ag75 имеется либо 20 избыточных атомов, либо, наоборот, для образования правильного Ih-строения не хватает примерно половины атомов. Следовательно, в ходе подвода тепловой энергии кластер Ag75 может начинать образовывать достаточно правильную первую и вторую Ih-оболочки. Но далее у него остается 20 “лишних” атомов, которые хаотично перемещаются по его поверхности пытаясь сформировать некое подобие третьей Ih-оболочки. На наш взгляд, именно хаотическое диффузионное перемещение этих атомов и отвечает за образование “рыхлого” внешнего вида кластерAg75 с повышенным значением потенциальной энергии.
Однако отсутствие декаэдрического строения в рассмотренных 70% модельных случаев понять сложнее. Известно, что энергетическая щель между Ih-, Dh- и ГЦК-структурами не велика, что ясно из результатов ab-initio расчетов. То есть все эти три политипные модификации, при определенных внешних условиях, могут стать устойчивыми и отсутствие здесь Dh-строения можно объяснить только очень близким ГЦК “магическим” числом (N = 79 атомов). На образование идеальных, или почти идеальных Dh- и ГЦК-структур не накладывается столь жестких ограничений, как при формировании идеального Ih-строения, разница по энергиям между данными структурами тоже мала и поэтому при моделируемом нами “магическом” размере N = 75 может возникнуть ситуация своеобразной логической неопределенности, приводящая, как мы видим, к попытке формирования уже не ГЦК, и не Dh-модификации, а не столь свойственной для этого размера кластера икосаэдрической фазы. Именно такое Ih-строение можно считать универсальной заменой иным кристаллическим модификациям во всех сложных случаях перестройки атомной структуры малых кластеров серебра, что мы и видим на рис. 3.
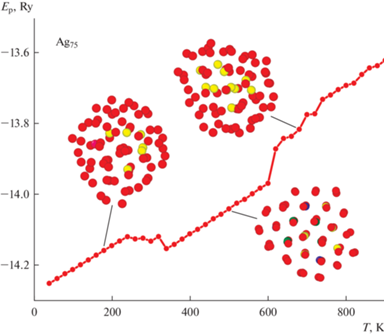
Однако в остальных 30% опытов были получены несколько иные результаты (рис. 4). Здесь мы наблюдаем достаточно стандартное поведение перестройки кластерной структуры под действием термического фактора. То есть изначально аморфный кластер Ag75 в области комнатной температуры образует устойчивое декаэдрическое строение с наличием резкого падения потенциальной энергии. Возникает вопрос, почему же в этих случаях декаэдр все же смог образоваться? Если сравнить рис. 3 и 4, то видно, что в последнем случае кластеры на начальном этапе обладали еще большим значением потенциальной энергии. На наш взгляд, именно это обстоятельство и играет решающую роль в характере происходящих процессов. Аморфное строение можно охарактеризовать как строение с максимально возможной свободой перемещения атомов из-за чего аморфные кластеры могут обладать достаточно разнообразной формой и расположением атомов. Именно из-за этой свободы энергия таких AgНЧ и может варьироваться в некоторых пределах. Если значение энергии станет слишком большим, то для AgНЧ станет энергетически выгодным не плавная оптимизация строения (рис. 3), а переход к наиболее устойчивой при данной температуре структурной модификации. При выбранных нами условиях моделирования это оказывается именно декаэдрическое строение.
Рис. 4.
Зависимость потенциальной энергии кластера Ag75 с начальным аморфным состоянием от температуры при наличии структурного перехода к Dh-модификации.
Далее перейдем к рассмотрению данных, полученных для ансамбля кластеров Ag101 с исходным аморфным строением, и вначале изучим процесс термической эволюции структуры для наиболее вероятного случая (рис. 5), реализующегося примерно в 70–80% модельных опытов. По сравнению с ранее описанным случаем кластера Ag101 с ГЦК-строением (рис. 2), здесь также есть свои особенности поведения. Так, если на рис. 2 уже на стадии предварительной термической релаксации наблюдался политипный переход ГЦК → Ih, то при условии начального аморфного строения кластер Ag101 сохранял исходное строение вплоть до температуры, примерно 450 К. Полученная температура политипного перехода четко коррелирует с Т ≈ 470 К, при которой кластер Ag101 (ГЦК-строение) обладал уже достаточно правильным Ih-строением. То есть независимо от начального строения при температурах порядка Т = 450–480 К в кластерах Ag101 происходит переход к икасаэдрической фазе. Другим моментом, сближающим вид термической эволюции на рис. 2 и рис. 5, является их энергетическая составляющая. Оказалось, что, несмотря на разное начальное структурное состояние, выигрыш в энергии оказался на удивление небольшим, всего порядка 0.005 эВ/атом вплоть до температур структурного перехода, после которого стало возможным оценивать только качество возникающих икосаэдрических кластеров Ag101.
Рис. 5.
Зависимость потенциальной энергии кластера Ag101 с начальным аморфным состоянием от температуры. Показан наиболее вероятный вариант термической эволюции.
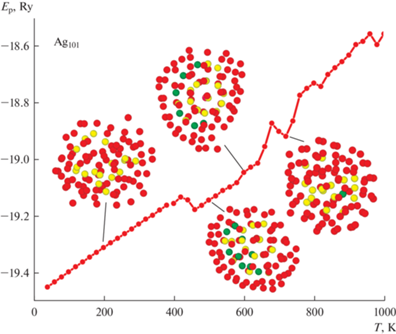
На наш взгляд, полученный результат является достаточно правдоподобным. Как уже говорилось выше, в ходе ряда компьютерных экспериментов было определено, что различие по энергии связи между наиболее стабильной аморфной и кристаллической модификациями, составляет менее 0.01 эВ/атом, и то в области низких температур. С ростом же температуры аморфный кластер, благодаря большой подвижности атомов, может сформировать практически сферическую форму без наличия фасеточного строения, характерного для кристаллических структур. Именно возможный выигрыш в поверхностной энергии может стать ключом к пониманию стабильности аморфной фазы при данных размерах кластеров и средних температурах. Кроме этого, в случае Ag101 (ГЦК-фаза) сформированные на этапе релаксации структуры хоть и были икосаэдрического вида, но с большими нарушениями укладки атомов, что также нивелировало энергетический разрыв между структурами. Далее, при температурах порядка 450–500 К термической энергии становится достаточно уже для более глубокой перестройки структуры, из-за чего и появляется возможность образования строения с осью симметрии пятого порядка (рис. 5).
Остальные полученные в ходе проведения компьютерных экспериментов варианты термической эволюции Ag101 представляли собой нагрев первичной аморфной структуры без каких-либо политипных переходов. Потенциальная энергия таких кластеров плавно увеличивалась в ходе подвода тепловой энергии, а в области температур, при которых в иных случаях (рис. 5) наблюдался переход к жидкому состоянию, фиксировалось некое “растекание” кластера с увеличением его площади поверхности и потерей сферической формы. Возможные причины устойчивости аморфного кластерного строения были обсуждены выше. Помимо этого, можно добавить, что с ростом размера кластера вероятность реализации строения с осью симметрии пятого порядка достаточно существенно уменьшается, из-за чего в ряде модельных опытов с кластерами Ag101 мы вообще не смогли ее получить, в отличие от более мелкого кластера Ag75.
ЗАКЛЮЧЕНИЕ
Таким образом, на основе проведенных в данной работе компьютерных экспериментах, так и основываясь на наших более ранних работах [24, 26, 28], можно сделать вывод о том, что в случае малых нанокластеров серебра даже четкое соответствие размера “магическому” числу той или иной структуры (ГЦК, ГПУ, Ih, Dh) далеко не всегда гарантирует ее появление в ходе процесса термической эволюции. Конечно с энергетической точки зрения структура кластеров с соответствующим им “магическим” размером является наиболее устойчивой и эти данные ab-initio вычислений мы не опровергаем ввиду их очевидности. Однако данный вывод был получен и справедлив для случая очень низких температур и оптимизированных кластеров правильного строения. Но если такие кластеры подвергнуть процедуре плавного нагрева путем постепенного подвода термической энергии, то протекающие при этом стохастические диффузионные процессы могут оказать очень сильное влияние на получаемые в конечном итоге структуры.
Данный вывод был проверен нами для двух возможных начальных состояниях Ag кластеров (ГЦК и аморфное) и было отмечено наличие разнообразных политипных переходов, чаще всего приводящих к образованию Ih- или Dh-модификаций. Несмотря на то, что полной фиксации структуры с осью симметрии пятого порядка при процессах плавного нагрева наночастиц различной исходной морфологии все же не происходило, несомненно то, что использование кластеров серебра с разрушенным начальным строением в большей степени позволяет решить проблему получения Ag наночастиц с требуемой Ih- или Dh-фазой. ГЦК же структура в кластерах такого малого размера (D < 2 нм) все же не может считаться термически устойчивой и для ее стабилизации необходимо использовать наночастицы большего размера. Таким образом, если в ходе реального применения наночастиц серебра в различных медицинских или плазмонных приложениях вид и качество строения имеет критическое значение, то необходимо учитывать возможные процессы перестройки кластерной структуры, происходящие при реальных эксплуатационных условиях применения.
Список литературы
Misirli G.M., Sridharan K., Abrantes S.M.P. // Beilstein J. Nanotechnol. 2021. 12. P. 440–461. https://doi.org/10.3762/bjnano.12.36
Adibkia K., Ehsani A., Jodaei A., et al. // Beilstein J. Nanotechnol. 2021. 12. P. 786–797. https://doi.org/10.3762/bjnano.12.62
Matos Y.B, Romanus R.S., Torquato M., et al. // Beilstein J. Nanotechnol. 2021. 12. P. 798–807. https://doi.org/10.3762/bjnano.12.63
Pem B., Ćurlin M., Jurašin D.D. Vrček V., et al. // Beilstein J. Nanotechnol. 2021. 12. P. 665–679. https://doi.org/10.3762/bjnano.12.53
Kiss F.D., Miotto R., Ferraz A. // Nanotechnology. 2011. 22. 275708. https://doi.org/10.1088/0957-4484/22/27/275708
Luo W., Hu W., Xiao S. // J. Phys. Chem. C. 2008. 112. № 7. P. 2359–2369. https://doi.org/10.1021/jp0770155
Syafiuddin A., Salmiati, Salim M.R., et al. // J. Chin. Chem. Soc. 2017. 64. P. 732–756. https://doi.org/10.1002/jccs.201700067
Beyene H.D., Werkneh A.A., Bezabh H.K., et al. // SM&T. 2017. 13. P. 18–23. https://doi.org/10.1016/j.susmat.2017.08.001
Zhang Z., Shen W., Xue J., et al. // Nanoscale Res. Lett. 2018. 13. P. 54. https://doi.org/10.1186/s11671-018-2450-4
Naz G., Asghar H., Ramzan M., et al. // Beilstein J. Nanotechnol. 2021. 12. P. 624–632. https://doi.org/10.3762/bjnano.12.51
Ranishenka B.V., Panarin A.Yu., Chelnokova I.A., et al. // Beilstein J. Nanotechnol. 2021. 12. P. 902–912. https://doi.org/10.3762/bjnano.12.67
Filipczak P., Pastorczak M., Kardaś T., et al. // J. Phys. Chem. C. 2021. 125. P. 1999–2004. https://doi.org/10.1021/acs.jpcc.0c06937
Tran D.T., Jones I.P., Johnston R.L., et al. // J. Phys. Conf. Ser. 2010. 241. 012086. https://doi.org/10.1088/1742-6596/241/1/012086
Gafner S.L., Gafner J.J., Kosterin S.V. Formation of an icosahedral structure during crystallization of copper nanoclusters. In book: Copper. Better properties for innovative products. Weinheim: Wiley-VCH, 2006.
Гафнер С.Л., Редель Л.В., Гафнер Ю.Я. К вопросу о формировании структурных модификаций в нанокластерах Ni // ФММ. 2007. 104. № 2. С. 189–195.
Самсонов В.М., Харечкин С.С., Гафнер С.Л., и др. Молекулярно-динамическое исследование плавления и кристаллизации наночастиц // Кристаллография. 2009. 54. № 3. С. 530–536.
Гафнер С.Л., Редель Л.В., Гафнер Ю.Я. Моделирование процессов структурообразования нанокластеров меди в рамках потенциала сильной связи // ЖЭТФ. 2009. 135. № 5. С. 899–916.
Гафнер Ю.Я., Головенько Ж.В., Гафнер С.Л. Формирование структуры нанокластеров золота при процессах кристаллизации // ЖЭТФ. 2013. 143. № 2. С. 288–305.
Samsonov V.M., Vasilyev S.A., Nebyvalova K.K., et al. // J. Nanopart. Res. 2020. 22. 247. https://doi.org/10.1007/s11051-020-04923-6
Cleri F., Rosato V. // Phys. Rev. B. 1993. 48. I. 1. P. 22. https://doi.org/10.1103/PhysRevB.48.22
Verlet L. // Phys Rev. 1967. 159. № 1. P. 98. https://doi.org/10.1103/PhysRev.159.98
Kadau K., Germann T.C., Lomdahl P.S., Holian B.L. // Science 2002. 296. I. № 5573. P. 1681–1684. https://doi.org/10.1126/science.1070375
Meyer R., Gafner J.J., Gafner S.L., Stappert S., Rellinghaus B. and Entel P. // Phase Transitions 2005. 78. № 1–3. P. 35–46. https://doi.org/10.1080/0141590412331316753
Honeycutt J.D., Andersen H.C. // J. Phys. Chem. 1987. 91. № 19. P. 4950–4963. https://doi.org/10.1021/j100303a014
Редель Л.В., Гафнер Ю.Я., Гафнер С.Л. Роль “магических” чисел при формировании структуры в малых нанокластерах серебра // ФТТ. 2015. 57. С. 2061–2070.
Gafner Y., Gafner S., Bashkova D. // J. Nanopart. Res. 2019. 21. P. 243–248. https://doi.org/10.1007/s11051-019-4691-2
Рыжкова Д.А., Гафнер С.Л., Гафнер Ю.Я. // Письма в ЖЭТФ. 2021. 113. № 10. С. 668–677. https://doi.org/10.31857/S1234567821100062
Liu D., Wen Z., Jiang Q. // CurrNanosci. 2011. 7. P. 463–470. https://doi.org/10.2174/157341311795542598
Рыжкова Д.А., Гафнер С.Л., Гафнер Ю.Я. // ФТТ. 2022. 64. № 3. С. 313–318.
Garzón I.L., Michaelian K., Beltrán M.R., et al. // Phys. Rev. Lett. 1998. 81. P. 1600. https://doi.org/10.1103/PhysRevLett.81.1600
Дополнительные материалы отсутствуют.