

Журнал аналитической химии, 2019, T. 74, № 11, стр. 861-869
Проточные методы анализа как новый подход к осуществлению химико-аналитического и радиохимического контроля в атомной энергетике
Л. Н. Москвин a, *, И. В. Мирошниченко b
a Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
b Научно-исследовательский технологический институт им. А.П. Александрова
188540 Ленинградская обл., Сосновый, Бор, Копорское шоссе, 72, Россия
* E-mail: moskvinln@yandex.ru
Поступила в редакцию 25.03.2019
После доработки 19.04.2019
Принята к публикации 19.04.2019
Аннотация
Обсуждаются общие подходы к автоматизации химико-аналитического и радиохимического контроля в атомной энергетике. В случае радиоактивных сред с наибольшей удельной активностью обосновывается необходимость автоматизации контроля в режиме off-line, чтобы минимизировать объемы образующихся при этом радиоактивных сбросов. В качестве иллюстрации рассматриваются возможности автоматизации off-line анализа теплоносителей первых контуров ядерных энергетических установок при определении в них хлорид- и сульфат-ионов. На основании сравнения с результатами ионохроматографического анализа показано, что разработанные методики обладают сопоставимыми аналитическими характеристиками. Для иллюстрации возможностей радиохимического анализа приведена методика определения урана на принципах циклического инжекционного анализа, обеспечивающая более низкие пределы обнаружения по сравнению с радиометрическими методиками. В целом обосновывается тезис о целесообразности перехода к проточным методам анализа вместо применяемых в настоящее время статических методов.
Функционирование ядерных энергетических установок (ЯЭУ) можно рассматривать как технологический процесс, управление которым основано на данных, получаемых в результате измерения различных, в том числе и химических, показателей. В первую очередь это показатели, характеризующие технологические среды. В то же время одним из обязательных требований к любому технологическому процессу является обеспечение условий, гарантирующих отсутствие его отрицательного воздействия на окружающую среду. В связи с этим параметрами, позволяющими судить о нормальном протекании в ЯЭУ всех технологических процессов, являются показатели химического и радионуклидного состава как внутриобъектовых технологических сред, таких как водные теплоносители, так и жидких радиоактивных отходов (ЖРО), попадание которых в объекты окружающей среды нельзя полностью исключить.
Для надежного проведения химико-технологического контроля в том и другом случае важнейшим условием является автоматизация аналитических процедур. Она необходима в первую очередь для того, чтобы минимизировать время получения информации о химическом и радионуклидном составе контролируемых сред для оперативного вмешательства в контролируемый процесс. В случае аналитического контроля технологических процессов автоматизация дополнительно необходима для снижения дозовых нагрузок на персонал аналитических лабораторий, обеспечивающий химико-технологический контроль. Новые возможности для автоматизации химического анализа открылись с появлением проточных методов.
В случае объектов аналитического контроля с наибольшей удельной активностью, каковыми являются теплоносители первых контуров ЯЭУ и вода бассейнов выдержки отработанных тепловыделяющих сборок (ОТВС), главная цель автоматизации аналитического контроля – снижение дозовых нагрузок на персонал, выполняющий анализ соответствующих сред. При контроле ЖРО определяющей становится минимизация количеств потенциально опасных для окружающей среды сбросов. В обоих случаях необходима автоматизация химического анализа, выполняемого off-line, так как на объектах атомной энергетики создание систем аналитического контроля on-line во многих случаях невозможно в принципе, исходя из технических требований к контролируемым объектам, призванных обеспечить безопасность их эксплуатации.
Наряду с автоматизацией в методологии химического анализа наметилась общая тенденция его миниатюризации, направленная на сокращение расходов проб, реагентов и объемов образующихся отходов в плане реализации концепции “зеленой аналитической химии” [1]. Необходимость миниатюризации химического анализа с позиций “зеленой аналитической химии” в атомной энергетике возникает во всех случаях выполнения анализов радиоактивных сред, чтобы свести к минимуму попадание радионуклидов в окружающую среду. Общим решением одновременно проблем автоматизации и миниатюризации химического анализа является переход от статической схемы выполнения аналитических процедур к проточному анализу.
К настоящему времени предложен целый ряд проточных методов, позволяющих найти решение проблем автоматизации и миниатюризации химического анализа. Их объединяет общая идея “объединения всех стадий химического анализа в единую поточную линию – своего рода конвейер, на котором последовательно выполняются все операции, предусмотренные методикой анализа. Осуществляется отбор аликвоты пробы, выполняются операции пробоподготовки, например, если в этом есть необходимость, концентрирование аналитов из пробы анализируемой среды, проводятся химические реакции с целью получения “аналитических форм” последних и, наконец, измеряется аналитический сигнал” [2].
Чтобы организовать подобную поточную линию, создается поток или самой анализируемой среды, или специально подаваемой в проточный анализатор жидкости-носителя, в которую вводятся аликвоты проб. Этот поток проходит через систему коммуникаций, обеспечивающих последовательное воспроизведение через определенные промежутки времени всех перечисленных выше процедур, входящих в общую схему выполнения анализа. Подобные системы коммуникаций, называемые “гидравлическими схемами” или “потоко-распределительными системами” [3] имеют свою специфику в каждом проточном методе. Проведено сравнение различных проточных методов в плане применимости для решения задач автоматизации и миниатюризации химического анализа [4].
Основной разграничительный признак между проточными методами анализа – это условия протекания реакций образования “аналитических форм” аналитов. По этому признаку все проточные методы можно условно разделить на “неравновесные” и “равновесные”. В первых по реализуемой схеме объединения потоков растворов проб и реагентов не гарантируется достижение равновесия в реакциях образования “аналитических форм” к моменту регистрации аналитического сигнала, так как смешение пробы и растворов реагентов происходит за счет взаимодиффузии. Соответственно аналитический сигнал не достигает своей максимально возможной величины. В “равновесных” методах в гидравлических схемах создаются условия для принудительного смешения пробы и растворов реагентов, что позволяет достичь максимальной для данного конкретного случая величины аналитического сигнала.
Наибольшее распространение нашел проточно-инжекционный анализ (ПИА) [5], относящийся к первой группе. Популяризации ПИА мы обязаны не только авторам метода [6–8], но и их многочисленным последователям. Наконец, этому наиболее популярному из проточных методов посвящен ряд глав в уже упоминавшейся коллективной монографии [2]. Автор метода Ружичка уже в ранних публикациях, посвященных осмыслению потенциальных возможностей своего открытия, видит ему место как в науке, так и в промышленности для решения задач технологического контроля [9]. Концепция выполнения анализа в ПИА предусматривает периодическое введение дискретных порций пробы и растворов реагентов в непрерывный ламинарный поток носителя. Различные варианты схемы ПИА допускают включение реагента в состав раствора-носителя или использование в качестве последнего непосредственно пробы с дискретным вводом в нее растворов реагентов. Взаимное смешение пробы и растворов реагентов происходит при этом в смесительных спиралях, включенных в гидравлические схемы, под действием конвекции и диффузии. Строго воспроизводимые условия по времени подачи пробы и растворов реагентов и скорости всех потоков позволяют осуществлять измерение аналитического сигнала в ПИА независимо от времени достижения равновесия в системе, что обеспечивает высокую производительность (до нескольких сотен проб в час) [10] и за счет некоторых потерь в чувствительности гарантирует высокую воспроизводимость результатов анализа. Относительное стандартное отклонение, как правило, не превышает 0.02. Однако схема так называемого прямого ПИА, предполагающая непрерывную подачу растворов реагентов в поток носителя с пробой, не позволяет оптимизировать аналитический процесс в направлении минимизации их расходов. В результате общий расход реагентов в схеме ПИА может достигать десятков мл/мин. Поэтому ПИА в описанном выше традиционном (прямом) варианте, решая проблему автоматизации методик химического анализа, не решает проблему его миниатюризации. При этом автоматизация преимущественно осуществляется в варианте анализа off-line. Для анализа on-line более предпочтителен вариант смешанного проточного метода: ПИА и непрерывный проточный анализ (НПА) [11]. Подобный вариант проточного метода позволяет осуществлять анализ контролируемой среды on-line с периодической проверкой правильности показаний за счет периодического ввода в поток пробы стандартных добавок.
Идеи “неравновесных” проточных методов воплотились в целом ряде их вариантов. В первую очередь это метод последовательного инжекционного анализа (SIA) [11], обеспечивший унификацию гидравлических схем и в какой-то степени снижение расходов реагентов по сравнению с ПИА.
Главным достоинством неравновесных проточных методов является высокая производительность, достигаемая за счет некоторого проигрыша в чувствительности по сравнению с неавтоматизированными статическими аналогами методик. Но, учитывая то, что при разработке автоматизированных методик анализа производительность далеко не всегда является основным из предъявляемых к ним требований, в последние годы почти одновременно предложено несколько вариантов проточных методов, обеспечивающих максимальную чувствительность при минимальном расходе реагентов за счет принудительного смешения заданных объемов пробы и раствора реагента. Для этой группы методов характерно включение в их гидравлические схемы смесительных реакционных емкостей, в которые подаются потоки растворов пробы и реагентов и происходит их смешение, и могут быть созданы условия для достижения равновесия в реакциях образования аналитических форм. К этим методам относятся зонный флюидный проточный анализ (ZF) [12], проточно-порционный анализа (FBA) [13] и циклический инжекционный анализ (ЦИА) [14].
Среди них наиболее универсальным решением при воплощении идеи равновесных методов явился ЦИА, где перемешивание осуществляется барботированием газа, в простейшем случае воздуха. Для этого в аэрогидравлическую схему проточного аналитического процесса дополнительно включен канал для подачи газа, предназначенного для перемешивания пробы с растворами реагентов и для интенсификации растворения твердофазных проб или извлечения из них аналитов [15]. Кроме того, добавление в схему “газовой” линии расширяет возможности метода, включая решение задач анализа газообразных сред.
При поиске альтернативы используемым фотометрическим методикам анализа дополнительно было необходимо учитывать специфику приборной оснащенности промышленных аналитических лабораторий на объектах атомной энергетики и уровень квалификации работающего в них персонала, который хорошо владеет всем арсеналом фотометрических методик и адекватным им инструментарием. Помимо “человеческого фактора” существенное значение имеет и экоаналитический. Наиболее привлекательным решением является автоматизация существующих фотометрических методик на принципах проточных методов анализа с минимальной модернизацией имеющегося приборного оснащения базовых и заводских лабораторий.
С учетом сформулированных подходов к решению поставленных задач в качестве инструментального средства достижения поставленной цели разработана автоматизированная проточно-инжекционная приставка к серийно выпускаемым фотометрам КФК-3, являющимся обязательным атрибутом приборного оснащения базовых и заводских лабораторий, обеспечивающих аналитический контроль жидких радиоактивных сред. Для компоновки гидравлических схем под конкретные методики анализа приставка, блок-схема которой представлена на рис. 1, включает переключатель потоков на 4 входа и 2 выхода, трехканальный перистальтический насос с регулируемым расходом от 0.5 до 10 мл/мин и проточную кювету, встроенную в кюветное отделение фотометра. Объем проточной кюветы составляет 50 мкл с длиной оптического пути 30 мм. Коммутация элементов гидравлической схемы осуществляется с помощью полиэтиленовых трубок внутренним диаметром 1.0 мм. Для управления приставкой и обработки результатов используется внешний компьютер или встроенный в приставку микропроцессор. Детектирование аналитического сигнала выполняется на фотоприемнике измерительной схемы фотометра. С учетом возможностей используемого фотометра в зависимости от конкретной методики анализа с его помощью обеспечивается детектирование в спектральном диапазоне 320−900 нм.
Рис. 1.
Блок-схема ПИА-приставки к КФК-3. Линии ввода в анализатор: I – проба/стандартный раствор, II – дистиллированная вода, III–V – растворы реагентов: 1 – кран-переключатель потоков, 2 – перистальтический насос, 3 – реакционная спираль, 4 – фотометр с проточной кюветой.
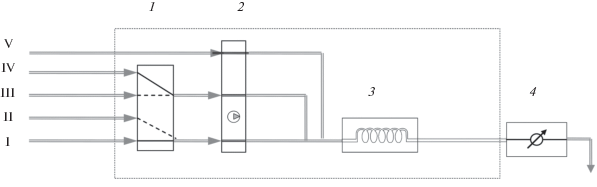
ПИА-приставка, блок-схема которой представлена на рис. 1, в равной степени обеспечивает возможность автоматизации фотометрических методик анализа теплоносителей первых контуров ЯЭУ и ЖРО. Различия проявляются только в средствах дополнительного оснащения приставок в том и в другом случаях. В случае теплоносителей это сорбционные колонки с ионитами для предварительного концентрирования микроконцетраций аналитов, таких как хлорид- и сульфат-ионы. При анализе ЖРО, учитывая их специфику как объектов анализа, проявляющуюся в наличии в них нефтепродуктов, поверхностно-активных веществ и взвесей продуктов коррозии, требуется более сложная пробоподготовка, нивелирующая мешающее влияние примесей, находящихся в форме коллоидов и взвесей. Для устранения мешающего влияния коллоидов и взвесей при выполнении проточного анализа разработаны специальный сменный фильтр-патрон и диализная ячейка, устанавливаемые в гидравлических трассах последовательно. В конструкцию сменного фильтр-патрона, устанавливаемого в приставке на линии подачи пробы, входит предколонка со слоем титанового порошка, удаляющая крупные частицы размером более 0.1 мм. Очистка пробы от мелкодисперсных частиц размером до 10 мкм происходит на фильтре, изготовленном из пористого фторопласта.
Проблема очистки пробы от коллоидных частиц решается методом диализа через целлофановые мембраны. Проточная диализная ячейка лабиринтного типа объемом 10 см3 с противоточной подачей отдающего и принимающего растворов устанавливается на линии подачи пробы непосредственно после двухступенчатого фильтра. Принципиальная схема проточной диализной ячейки представлена на рис. 2.
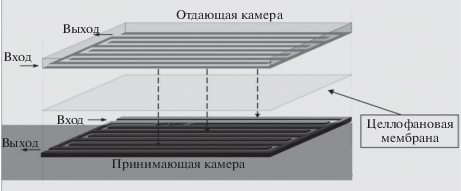
Эффективная площадь мембраны в ячейке, составляющая 30 см2, выбрана на основании результатов предварительных исследований эффективности массообмена между ее отдающей и принимающей камерами. Влияние гидродинамических условий процесса диализа на эффективность массопереноса контролируемых примесей через мембраны исследовано в диапазоне объемных скоростей растворов от 0.5 до 10 мл/мин. Установлено, что при включении проточного диализатора в гидравлическую схему проточно-инжекционного анализатора удается обеспечить содержание ионных форм аналитов в принимающей камере на уровне от 10 до 20% от их начальной концентрации в пробе ЖРО. С целью оперативной проверки эффективности переноса контролируемых примесей в отдающую камеру диализной ячейки периодически подаются стандартные растворы, и соответствующая поправка вводится в результаты анализа.
Разработанная приставка к ФЭКу и вспомогательное оснащение к ней обеспечили возможность автоматизации фотометрических методик анализа ЖРО и природных вод. Та же приставка обеспечивает возможность автоматизации методик анализа теплоносителей ЯЭУ с адекватными изменениями в схеме пробоподготовки, которые включают замену диализатора колонкой для концентрирования аналитов.
Для заполнения ионообменных колонок в этом случае использовали сильноосновный анионит АВ-17 с размером частиц 50–100 мкм. Для уменьшения гидравлического сопротивления колонки ионит перед ее заполнением смешивали с гранулированным пористым политетрафторэтиленом (фракция частиц 0.25–0.5 мм). Размер колонки составил 4 × 20 мм (внутренний объем 0.25 мл). Параметры колонки обеспечивают обменную емкость загрузки смолы 0.1 мг/моль, что перекрывает диапазон возможных содержаний определяемых анионов в пробах водных теплоносителей ЯЭУ. Максимально допустимая скорость фильтрации пробы через колонку составила 5 мл/мин при эффективности выделения на ионообменной смоле не менее 95% от содержания аналитов в пробе независимо от ее объема в пределах до 100 мл. Допустимая скорость элюирования определяемых анионов без существенного размытия их зон в элюате обеспечивается при объемной скорости элюента не более 0.5 мл/мин. В качестве элюента для хлорид- и сульфат-ионов выбрана 0.1 М HNO3, что одновременно обеспечивает необходимые условия для образования аналитических форм обоих аналитов.
С целью унификации гидравлических схем ПИ-определения хлорид- и сульфат-ионов в качестве базовых стационарных методик их определения были выбраны турбидиметрические методики. Использование в проточном анализе турбидиметрического метода определения хлорид- и сульфат-ионов осложняется рядом факторов. В частности, помутнение раствора, обусловленное образованием коллоидных взвесей AgCl и BaSO4, происходит сравнительно медленно. С целью адаптации турбидиметрических методик к условиям ПИА выполнен цикл специальных исследований, направленных на поиск условий повышения и стабилизации величины аналитического сигнала при турбидиметрическом определении аналитов. Кроме того, найдены условия для ускорения стабилизации оптической плотности растворов.
Для унификации аналитических процедур при выполнении анализов предложена адекватная разработанным методикам, включающим стадию предварительного концентрирования аналитов, схема градуировки, которая может рассматриваться как общеметодическое решение проблемы определения микроконцентраций аналитов, когда отсутствует принципиальная возможность приготовления их адекватных стандартных растворов. Для методик анализа с предварительным концентрированием аналитов в условиях, гарантирующих полноту их выделения, построение градуировочного графика предложено проводить в координатах: аналитический сигнал−общее количество аналита в аликвоте пробы, вводимой в анализатор. В этом случае один и тот же градуировочный график может быть использован для разных объемов пробы с различной концентрацией аналитов. Определяемым параметром в этом случае является общее содержание аналита в пробе, а концентрация в ней вычисляется как отношение найденного количества аналита к объему пробы.
При построении градуировочной зависимости для проточно-инжекционного определения хлорид- и сульфат-ионов с предварительным концентрированием использовали стандартные растворы с концентрациями ${\text{SO}}_{4}^{{2 - }}$ и Cl– 100 мкг/л и объемом пробы, наносимой на концентрирующую колонку, от 2.5 до 20 мл. Использование относительно концентрированных стандартных растворов является единственно приемлемым способом градуировки в условиях технической невозможности приготовления адекватных стандартных образцов, так как диапазон определяемых концентраций обоих анионов находится на уровне их фоновых содержаний в воде высокой чистоты, получаемой любым из методов препаративного получения и, в частности, общепринятым методом ионообменной деионизации.
Для проверки правильности результатов анализа, получаемых по разработанной методике, последовательно использовали методы добавок и сравнения с результатами, получаемыми реперным методом ионной хроматографии. Для проверки правильности использовали реальные пробы водных теплоносителей. Параллельно выполняли серии анализов технологических проб как при бескоррекционном (второй контур), так и при аммиачном (первый контур) водных режимах реакторной установки с использованием метода добавок. Из отобранных проб теплоносителей отбирали две равные аликвоты. В первой из них определяли исходное содержание хлорид- и сульфат-ионов. В другую часть пробы вводили добавки определяемых анионов, приготовленные на основе ГСО 7262 и ГСО 7253 (величина добавки составляла 50−100% от среднестатистического содержания хлоридов и сульфатов в исходной пробе), и анализировали вторую аликвоту пробы с добавкой. Деионированная вода, используемая для приготовления растворов добавок, имела удельную электропроводимость менее 0.1 мкСм/см, что гарантирует содержание в ней определяемых анионов на уровне менее 10 мкг/л. Результаты определения хлорид- и сульфат-ионов в водных теплоносителях методом ПИА по разработанным методикам (n = 5, Р = 0.95) приведены в табл. 1. Показатель точности оценивали в соответствии с алгоритмом оценивания характеристик погрешности результатов анализа с использованием метода добавок.
Таблица 1.
Результаты проточно-инжекционного турбидиметрического определения хлорид- и сульфат-ионов в водных теплоносителях ядерных энергетических установок с использованием метода добавок
| Образец для контроля | Содержание хлорид-ионов, мкг/л | Величина добавки, мкг/л | Содержание сульфат- ионов, мкг/л |
Величина добавки, мкг/л | ||
|---|---|---|---|---|---|---|
| проба без добавки | проба с добавкой | проба без добавки | проба с добавкой | |||
| Проба № 1 (первый контур) | 17 ± 6 | 24 ± 6 | 10 ± 3 | 25 ± 12 | 41 ± 19 | 20 ± 5 |
| Показатель точности, % | 35 | 25 | 48 | 46 | ||
| Проба № 2 (второй контур) | 9 ± 4 | 21 ± 6 | 10 ± 3 | 19 ± 10 | 44 ± 19 | 20 ± 5 |
| Показатель точности, % | 38 | 29 | 52 | 43 | ||
Результаты сравнения разработанной и ионохроматической методик приведены в табл. 2.
Таблица 2.
Результаты (мкг/л) определения содержания хлорид-ионов в теплоносителе
| Метод проточно-инжекционного анализа | Метод ионной хроматографии | ||
|---|---|---|---|
| с1 | ±Δ | с2 | ±Δ |
| 3.0 | 1.1 | 4.1 | 2.0 |
| 4.2 | 2.6 | 3.8 | 1.9 |
| 4.1 | 1.5 | 4.0 | 2.0 |
| 5.0 | 1.9 | 6.4 | 3.2 |
| 3.2 | 1.2 | 2.2 | 1.1 |
| 4.8 | 1.8 | 3.4 | 1.7 |
| 21.0 | 5.7 | 20.3 | 5.8 |
| 3.0 | 1.1 | 2.1 | 1.1 |
| 3.3 | 1.2 | 3.6 | 1.8 |
| 4.0 | 1.5 | 3.0 | 1.5 |
| 4.5 | 1.7 | 3.3 | 1.6 |
| 6.2 | 2.4 | 4.8 | 2.4 |
| 10.5 | 3.8 | 11.6 | 5.8 |
| 12.0 | 4.6 | 10.0 | 5.0 |
| 40.0 | 11.0 | 43.0 | 12.0 |
| 3.8 | 1.4 | 3.9 | 2 |
| 3.3 | 1.2 | 4.3 | 2.2 |
| 5.5 | 2.0 | 5.5 | 2.7 |
Поскольку методики определения хлорид-ионов сравнивали в области следовых концентраций (менее 50 мкг/л), принимали специальные меры для исключения неконтролируемых грубых промахов, связанных с загрязнениями посуды. Пробы отбирали в одну специально подготовленную пробоотборную емкость, из которой отбирали аликвоты необходимого объема для каждой методики измерений в тщательно отмытую деионированной водой и сполоснутую отбираемым на анализ теплоносителем полиэтиленовую посуду.
Результаты статистической обработки данных, полученных обоими методами, приведены в табл. 3. Корреляционный анализа данных, полученных обоими методами, позволил сделать вывод о наличии корреляций между результатами определения обоих анионов сравниваемыми методами. При сопоставлении параллельных рядов результатов определений проточно-инжекционным и ионохроматографическими методами анализа коэффициент парных корреляций R составил 0.98.
Таблица 3.
Расчетные значения статистических параметров результатов определения концентрации хлоридов с использованием различных методик анализа
| Параметр | Ионная хроматография Цвет-3006М |
Проточно-инжекционный анализ (ПИА-приставка) |
|---|---|---|
| Среднее, мкг/л | 3.9 | 4.1 |
| Медиана, мкг/л | 3.8 | 4.0 |
| Стандартное отклонение, мкг/л | 1.1 | 0.98 |
| Доверительный интервал ±Δх | 0.7 | 0.6 |
Проведенные испытания показали, что абсолютная величина случайной вариации результатов определения содержания хлорид- и сульфат-ионов в теплоносителе сопоставима для сравниваемых методик анализа, что явилось основанием для аттестации разработанных методик в УНИИМ в соответствии с требованиями ГОСТ 8.563 и рекомендацией возможности их практического применения на объектах атомной энергетики.
Для иллюстрации предлагаемого подхода к автоматизации методик радиохимического анализа выбрана актуальная для радиохимического контроля на ЯЭУ задача определения концентрации урана в теплоносителях первых контуров и в воде бассейнов выдержки ОТВС. В качестве автоматизируемой выбрана фотометрическая методика определения урана с арсеназо III. Чтобы обеспечить получение рекордно низких пределов обнаружения в методику включили стадию концентрирования аналита. При этом исходили из следующих предпосылок. Во-первых, фиксируемые значения концентрации урана в теплоносителе находятся в диапазоне от 0.01 до 10 мкг/л [15]. Во-вторых, указанная нижняя граница концентрации урана в теплоносителе соответствует величине его объемной радиоактивности ~3 × 10–14 Ки/л (1 Ки 238U = 3 × 106 г), что на три−четыре порядка ниже предела обнаружения существующих методик альфа-спектрометрического анализа для радиометрического определения урана. В связи с этим для определения микроконцентрации урана в водных средах предпочтительными оказываются не ядерно-физические, а проточные химические методы анализа, обладающие более высокой чувствительностью и экспрессностью.
Чтобы обеспечить минимальные пределы обнаружения, при разработке методики определения микроконцентраций урана в водных теплоносителях потребовалось включение в схему анализа нестандартной методики предварительного концентрирования аналита на диоксиде марганца, синтезируемом непосредственно в процессе выполнения анализа. Исходили из того, что для автоматизации методик, включающих нетрадиционные операции пробоподготовки, наиболее привлекательны “равновесные” проточные методы и, в частности, циклический инжекционный анализ (ЦИА). В этом случае использовали универсальный проточный анализатор “ПИАКОН-С” производства ООО “Росаналит” с фотометрической ячейкой. Объем проточной кюветы составлял 300 мкл при длине поглощающего слоя 20 мм. Предел абсолютной погрешности анализатора при измерении коэффициента пропускания равен 1.5%.
В проточной ячейке для концентрирования on-line обеспечивается синтез в потоке диоксида марганца и определение на пористой мембране из пористого политетрафторэтилена, которая обеспечивает минимальное гидравлическое сопротивление сформировавшегося слоя сорбента.
Для повышения чувствительности определения урана по фотометрической реакции с арсеназо III в гидравлическую схему дополнительно был включен редуктор Джонса с гранулированным цинком для восстановления U(VI), обычно присутствующего в теплоносителях первых контуров, до U(IV). В окончательном варианте аэрогидравлическая схема для выполнения приведена на рис. 3.
Рис. 3.
Аэрогидравлическая схема ЦИА для проточного определения урана: 1–6 – соответственно линии подачи пробы, воды, и последовательно растворов реагентов: 0.01%-ного раствора арсеназо III, 3 М HCl, 10–3 М раствора аскорбиновой кислоты, 10−3 М раствора перманганата калия. Обозначения: МК – многоходовой переключатель; Р – перистальтический насос; Я – проточная ячейка для концентрирования урана; РЕ, ВЕ – реакционная и вспомогательная емкости соответственно; R – редуктор Джонса; D – проточный фотометрический детектор; W – линия сброса отработанного аналита.
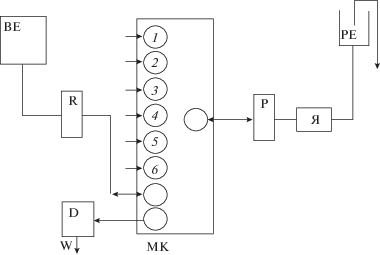
Согласно приведенной схеме в реакционную емкость (РЕ) ЦИ-анализатора поочередно дозировали по 3 мл 10–3 М растворов перманганата калия и аскорбиновой кислоты. Образовавшаяся в реакционной емкости суспензия MnO2 ⋅ nH2O путем реверса насоса направлялась в ячейку для концентрирования. Затем пробу, содержащую уран(VI), объемом от 100 до 200 мл пропускали через ячейку для концентрирования при расходе пробы до 10 мл/мин.
После завершения этапа концентрирования урана через ячейку пропускали 2 мл 10–3 М раствора аскорбиновой кислоты со скоростью 0.5 мл/мин. В предварительном эксперименте по результатам люминесцентного определения урана в растворе на выходе из ячейки установлено, что выбранные условия обеспечивают полноту растворения находящегося в ячейке сорбента и перевод в раствор сорбированного на нем урана. Раствор из ячейки для концентрирования направлялся в реакционную емкость. Далее в реакционную емкость пода-вали 0.5 мл 3 М HCl и полученный раствор с помощью реверса насоса направлялся через редуктор во вспомогательную емкость (ВЕ). Полученный раствор, содержащий уран(IV), вновь направляли в реакционную емкость, в которую далее водили 2 мл 0.01%-ного раствора арсеназо III в 1 M НСl. Перед перекачкой раствора из ВЕ в РЕ последнюю промывали 1 М HCl, подаваемой через многоходовой кран по отдельной линии. Для завершения реакции образования комплекса урана(IV) с арсеназо III раствор в РЕ перед его перекачкой в фотометрическую ячейку выдерживали в течение 3 мин.
При проверке разработанной методики на модельных растворах урана(VI) установлено, что она обеспечивает нижнюю границу диапазона определяемых концентраций 2 мкг/л при объеме пробы 200 мл и времени выполнения одного анализа 15 мин.
Приведенные методики убедительно иллюстрируют целесообразность изменения подходов к выборам методик химического анализа жидких сред в атомной энергетике. На смену статическому анализу целесообразен переход к методикам проточного анализа.
Список литературы
Armenta S., Gariguas M. de la Gnardia. Green analytical chemistry // Trends Anal. Chem. 2008. V. 27. № 6. P. 497.
Проточный химический анализ / Проблемы аналитической химии. Т. 17 / Под ред. Золотова Ю.А. М.: Наука, 2014. 428 с.
Шпигун Л.К., Золотов Ю.А. Проточно-инжекционный анализ. М.: Знание, 2004. 477 с.
Vakh C., Falkova M., Timofeeva I., Moskvin A., Moskvin L., Bulatov A. Flow analysis: A novel approach for classification // Crit. Rev. Anal. Chem. 2015. https://doi.org/10.1080/10408347.2015.1087301
Ruzicka J., Hansen E.H. Flow injection analysis // Anal. Chim. Acta. 1975. V. 78. P. 145.
Ruzicka J., Hansen E.H. The first decade of flow injection analysis from serial assay to diagnostic tool // Anal. Chim. Acta. 1986. V. 179. P. 1.
Ruzicka J., Hansen E.H. Flow injection analysis. 2nd ed. / Ser. “Chemical Analysis”. V. 62. N.Y.: John Wiley & Sons, 1988. 498 p.
Кузнецов В.В. Проточно-инжекционный анализа // Соровский образовательный журн. 2010. Т. 11. С. 56.
Ruzicka J. Flow analysis – a servey of its potential as solution handeing and data gathering technique in chemical research and industry // Frez’s J. Anal. Chim. 1988. V. 329. № 6. P. 653.
Москвин Л.Н., Москвин А.Л. Вода и водные среды: химический анализ “on line”, проблемы и решения // Успехи химии. 2005. № 2. С. 155.
Ruzicka J., Marshall G.D. Sequential injection: A new concept for chemical sensors, process analysis and laboratory assays // Anal. Chim. Acta. 1990. V. 237. P. 329.
Marshall G., Wolcott D., Olson D. Zone fendics in flow analysis: Potentialities and application // Anal. Chem. Acta. 2003. V. 499. P. 29.
Diniz P.D., Almeida L.F., Harding D.P., Araujo M.C.U. Flow-batch analysis // Trends Anal. Chem. 2012. V. 35. P. 39.
Мозжухин А.В., Москвин А.Л., Москвин Л.Н. Циклический инжекционный анализ как новый метод проточного анализа // Журн. аналит. химии. 2007. Т. 62. С. 527.
Falkova M.T., Pushina M.O., Bulatov A.V., Alekseeva G.M., Moskvin L.N. Stepwise injection spectrophotometric determination of flavonoids in medicinal plants // Anal. Lett. 2014. V. 47. P. 970.
Дьяков А.А., Менькин Л.И., Карпенко С.Г. Определение концентрации 235-U в водном теплоносителе первого контура ядерного реактора // Атомная энергия. 1986. Т. 61. № 5. С. 334.
Епимахов В.Н., Глушков С.В. Определение альфа-излучающих радионуклидов в водном теплоносителе АЭУ с использованием мембран, импрегнированных гидратированным диоксидом марганца // Радиохимия. 1994. Т. 36. № 6. С. 514.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии