

Журнал аналитической химии, 2022, T. 77, № 10, стр. 938-946
Быстрый гидролиз и дериватизация метаболитов нитрофуранов с новым дериватизирующим агентом 5-нитро-2-фуральдегидом при их ВЭЖХ-МС/МС-определении в курином мясе
А. О. Мелехин a, b, В. В. Толмачева a, *, Ю. Н. Холявская b, Е. С. Седых b, С. Г. Дмитриенко a, В. В. Апяри a, А. Л. Баиров b
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, ГСП-1, Россия
b Центральная научно-методическая ветеринарная лаборатория
111622 Москва, ул. Оранжерейная, 23, Россия
* E-mail: nikatolm@mail.ru
Поступила в редакцию 21.02.2022
После доработки 23.03.2022
Принята к публикации 24.03.2022
- EDN: PZZFQO
- DOI: 10.31857/S0044450222100115
Аннотация
С использованием 5-нитро-2-фуральдегида в качестве нового дериватизирующего агента предложен способ быстрой пробоподготовки образцов пищевых продуктов при определении в них метаболитов нитрофуранов методом ВЭЖХ-МС/МС. На примере образцов мяса кур-бройлеров, получавших в течение 10 дней нитрофураны, показано, что в термостатированной ультразвуковой бане время гидролиза-дериватизации можно значительно уменьшить за счет повышения температуры до 80°C и концентрации соляной кислоты до 0.5 М. Проведено определение метаболитов фуралтадона, фуразолидона, нитрофурантоина и нитрофуразона в курином мясе, печени, желудках и сердце кур с использованием разработанного способа и официальной методики. Предложенный способ обеспечивает количественное выделение метаболитов.
Проблеме безопасности пищевых продуктов в настоящее время уделяется большое внимание [1]. Среди потенциально вредных веществ, которые необходимо контролировать в пищевых продуктах, значительное место занимают ветеринарные препараты [2, 3]. Употребление в пищу продуктов, содержащих остатки таких препаратов, связано с риском для здоровья потребителей, поскольку эти продукты могут вызывать аллергические реакции, индуцировать устойчивость патогенов к антибиотикам, оказывать токсическое микробиологическое действие, вызывать канцерогенные или тератогенные эффекты [4, 5].
Перечень ветеринарных препаратов, которые необходимо контролировать в продуктах животного происхождения, включает, наряду с сульфаниламидами, тетрациклинами, фторхинолонами, амфениколами и некоторыми другими антибиотиками, также нитрофураны [5]. Нитрофураны – класс синтетических антибактериальных и противопаразитарных препаратов широкого спектра действия, которые до 1994 г. широко применялись в качестве терапевтических и ветеринарных препаратов, а также кормовых добавок в ветеринарной практике, животноводстве и аквакультуре [6]. В отличие от большинства других ветеринарных препаратов, при попадании в организм животных нитрофураны в течение нескольких часов быстро метаболизируются. Метаболиты нитрофуранов остаются в продуктах животного происхождения в течение длительного времени в виде связанных с белками соединений [7]. После того как было установлено, что нитрофураны и их метаболиты обладают канцерогенным и мутагенным действием [8], их использование в животноводстве было запрещено в большинстве стран, включая Россию [9, 10]. Однако данные мониторинга качества пищевых продуктов свидетельствуют о том, что несмотря на установленные законодательством запреты нитрофураны продолжают использовать не только в развивающихся странах, но и в Европейском Союзе, что обусловлено их высокой антибактериальной активностью, низкой стоимостью и доступностью [11, 12]. Согласно базе данных Системы быстрого оповещения для пищевых продуктов и кормов (RASFF) в течение 2002–2017 гг. зарегистрировано 854 случая загрязнения пищевых продуктов нитрофуранами [13].
В рамках финансируемого ЕС проекта FoodBRAND (обнаружение связанных остатков и нитрофуранов) разработан подтверждающий метод ВЭЖХ-МС/МС для определения в пище четырех запрещенных нитрофуранов, а именно: фуралтадона, фуразолидона, нитрофурантоина и нитрофуразона, в форме их соответствующих метаболитов: аминоморфолинометил-2-оксазолидинона, 3‑амино-2-оксазолидинона, 1-аминогидантоина и семикарбазида [14]. Контроль содержания метаболитов нитрофуранов в пищевых продуктах существенно не изменился с момента внедрения методологии FoodBRAND; во многих странах эта методика с небольшими модификациями используется в качестве официальной [15–19]. Из-за высокой полярности, низкой молекулярной массы и отсутствия хромофоров определение метаболитов нитрофуранов в пищевых продуктах проводят после длительной пробоподготовки, включающей кислотный гидролиз и дериватизацию с использованием 2-нитробензальдегида (0.1–0.2 М HCl, 16 ч при 37‒40°С), а также очистку гидролизатов методом жидкостной или твердофазной (ТФЭ) экстракции. Пробоподготовка является самым длительным и трудоемким этапом анализа, поэтому неудивительно, что в последнее десятилетие были предложены альтернативные подходы к оптимизации условий кислотного гидролиза-дериватизации за счет повышения температуры [20–22], изменения концентрации HCl [23], применения ультразвукового [24, 25] или микроволнового [26–30] излучения.
Ранее [31, 32] для дериватизации метаболитов нитрофуранов при их определении методом ВЭЖХ-МС/МС в куриных яйцах [31] и меде [32] мы предложили использовать новый дериватизирующий агент 5-нитро-2-фуральдегид (5-НФА), а для очистки гидролизата – сверхшитый полистирол (ССПС) [31] и магнитный ССПС [32], которые до этого для этих целей не применяли. Дериватизацию метаболитов проводили в соответствии с официальной методикой в 0.1 М соляной кислоте при 37–40°C течение 16 ч.
Цель настоящей работы состояла в изучении возможности сокращения времени кислотного гидролиза и дериватизации за счет изменения температуры, концентрации соляной кислоты и применения ультразвука. Для достижения поставленной цели нам понадобились образцы, содержащие метаболиты нитрофуранов. В качестве объектов исследования использовали куриное мясо и субпродукты кур-бройлеров, которые в течение 10 дней получали нитрофураны. Очистку гидролизатов перед ВЭЖХ-МС/МС-определением проводили методом ТФЭ с использованием ССПС.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы. Использовали хроматографически чистые метанол, ацетонитрил и муравьиную кислоту (Fisher Scientific Inc., США), н-гексан (Fisher Scientific Inc., США), 5-нитро-2-фуральдегид (Sigma-Aldrich, США), 2-нитро-бензальдегид (2-НБА), трихлоруксусную кислоту (Fisher Scientific Inc., США), гидрофосфат калия (Fisher Scientific Inc., США), соляную кислоту х. ч., сверхсшитый полистирол Диапак П-3 (Био-ХимМак, Россия). ССПС перед использованием активировали ацетонитрилом. Очищенную воду получали с помощью системы Milli-Q Synthesis (Millipore, США).
В качестве стандартных образцов использовали фуралтадон (ФД), фуразолидон (ФТ), нитрофурантоин (НФТ), нитрофуразон (НФ), 3-амино-5-метилморфолино-2-оксазолидинон (AМОЗ), 3-амино-2-оксазолидинон (АОЗ), 1-аминогидантоина (АГД) гидрохлорид, семикарбазида (СЕМ) гидрохлорид с содержанием основного вещества не менее 95.0% (Sigma-Aldrich, США). В качестве внутренних стандартов метаболитов нитрофуранов использовали d5-3-амино-5-метилморфолино-2-оксазолидинон (AМОЗ-D5), 13С3-аминогидантоин (АГД-13C3), d4-3-амино-2-оксазолидинон (AОЗ-D4), 15N2,13C-семикарбазид (СЕМ-15N213C) с содержанием основного вещества не менее 99.0% (Witega, Германия).
Исходные растворы нитрофуранов, их метаболитов и изотопно-меченных метаболитов с концентрацией 200 мкг/мл готовили растворением соответствующей навески в ацетонитриле. Растворы хранили при ‒20°С не более шести месяцев. Растворы смеси нитрофуранов, смеси метаболитов нитрофуранов с концентрацией 1.0 мкг/мл готовили путем разбавления исходных в метаноле. Аналогичным образом готовили раствор смеси внутренних стандартов с концентрацией 1.0 мкг/мл. Срок хранения смесей составлял 1 мес. Рабочие растворы готовили разбавлением исходных метанолом в день использования.
Анализируемые образцы. Для построения градуировочных зависимостей использовали образцы “чистого” куриного мяса, собранные Центральной научно-методической ветеринарной лабораторией (Москва, Россия) в 2021 г. в процессе государственного мониторинга пищевой продукции. Образцы хранили при –20°С в холодильнике. Для изучения влияния различных факторов на гидролиз и дериватизацию связанных с белками метаболитов нитрофуранов использовали образцы от кур-бройлеров, которые в течение 10 дней в виде инъекций получали нитрофураны. Общее количество нитрофуранов, которые получили бройлеры, составило 345, 125, 267 и 222 мкг/кг живого веса бройлеров для фуралтадона, фуразолидона, нитрофурантоина и нитрофуразона соответственно. Забой проводили спустя день после введения последней дозы. Образцы мышц, печени, желудков и сердца от каждого животного собирали отдельно, замораживали и хранили при –20°С в холодильнике.
Аппаратура. Использовали высокоэффективный жидкостной хроматограф ExionLC (Shimadzu, Япония) в сочетании с тройным квадрупольным масс-спектрометрическим детектором SCIEX Triple QuadTM 5500 (AB Sciex, Сингапур), оснащенный бинарным насосом и автосамплером. Масс-спектрометр был настроен на сбор данных в режиме мониторинга множественных реакций (ММР). Установлены следующие оптимальные значения параметров: напряжение на распыляющем капилляре – 4500 В; температура испарителя – 550°C; в качестве газа завесы и газа в ячейке использовали азот; давление газа соударений – 10 фунтов на квадратный дюйм (psi); давление газа завесы – 35 psi; давление осушающего газа и давление распыляющего газа – 50 psi; входной потенциал – 10 В. Применяли аналитические весы Sartorius AC 121S (Sartorius, Германия), систему подготовки деионизованной воды Milli-Q Synthesis (Millipore, США), ультразвуковую баню S70H, ElmaSonic (Германия) с частотой ультразвука 37 кГц и эффективной мощностью 150 Вт, центрифугу лабораторную Thermo Scientific SL40R (Thermo Scientific, США), систему упаривания закрытого типа TurboVapII CaliperLifeSciences (Caliper Life Sciences, США), вакуумную установку для ТФЭ М6 (Манифолд, Россия), шейкер для пробирок MultiReax (Heidolph, Германия).
Условия хроматографического разделения и детектирования. Разделение проводили на колонке Acclaim™ 120 C18 (100 × 2.1 мм) c диаметром зерна сорбента 3.0 мкм (Thermo Scientific, США) в режиме градиентного элюирования. Использовали подвижные фазы, состоящие из 0.5%-ной муравьиной кислоты в воде (А) и 0.5%-ной муравьиной кислоты в смеси ацетонитрила и метанола (50 : 50) (Б). Разделение проводили, применяя следующую программу градиентного элюирования: 20–80% B (0–7 мин), 80% B (7–7.5 мин), 80–20% B (7.5–8 мин). Скорость потока составляла 0.3 мл/мин. Температуры колонки и автосамплера поддерживали во время работы на уровнях 40 и 15°C соответственно, объем вводимой пробы составлял 10 мкл.
Идентификация и определение. Продукты дериватизации метаболитов нитрофуранов с 5-НФА (соответствующие исходные нитрофураны) идентифицировали по полученным хроматограммам с использованием метода MМР. Параметры MМР для нитрофуранов и внутренних стандартов приведены в табл. 1. Неизвестную концентрацию аналита в пробе определяли методом градуировочного графика (матричная градуировка). Аналитическим сигналом служило отношение площади пика аналита к площади пика соответствующего внутреннего стандарта. Линейность градуировочных графиков оценивали на модельных “чистых” образцах куриного мяса, не содержащих остаточных количеств определяемых аналитов, с добавками метаболитов нитрофуранов на уровнях концентраций 1, 10, 50, 100 и 250 мкг/кг и добавкой смеси внутренних стандартов c концентрацией 100 мкг/кг. Коэффициенты линейной зависимости площадей хроматографических пиков препаратов от их концентрации в анализируемом образце составили не менее 0.99.
Таблица 1.
Основные характеристики продуктов дериватизации метаболитов нитрофуранов с 5-нитро-2-фуральдегидом при определении методом ВЭЖХ-МС/МС в режиме мониторинга множественных реакций
| Метаболит | Нитрофуран (продукт дериватизации) |
tR, мин | Q1, m/z | Q3, m/z | ПД*, B | ЭС**, эВ |
|---|---|---|---|---|---|---|
| AМОЗ | ФД | 2.18 | 325.0(+) | 252.0/281.0 | 60/60 | 25/19 |
| AОЗ | ФТ | 3.67 | 226.0(+) | 122.0/113.0 | 60/60 | 18/19 |
| АГД | НФТ | 3.43 | 236.8(–) | 151.9/123.8 | –100/–100 | –17/–21 |
| СЕМ | НФ | 3.32 | 196.8(–) | 149.9/123.8 | –100/–100 | –13/–14 |
| AМОЗ-D5 | ФД-D5 | 2.18 | 330.0(+) | 286.0 | 60 | 19 |
| AОЗ-D4 | ФТ-D4 | 3.67 | 230.0(+) | 117.0 | 60 | 19 |
| АГД-13C3 | НФТ-13C3 | 3.43 | 239.8(–) | 151.9 | –100 | –17 |
| СЕМ-15N213C | НФ-15N213C | 3.32 | 199.8(–) | 152.9 | –100 | –13 |
Пробоподготовка. В центрифужную пробирку емк. 50 мл вносили 1.00 ± 0.01 г тщательно измельченной пробы куриного мяса и промывали два раза по 5 мл этилацетата для удаления не связанных с белками метаболитов. После центрифугирования и удаления этилацетатной фазы к пробе добавляли 100 мкл раствора внутреннего стандарта (1000 нг/мл), 100 мкл метанола, 5 мл 0.1, 0.2 или 0.5 М соляной кислоты и 400 мкл 0.2 М 5-НФА в метаноле для проведения дериватизации. Содержимое пробирки перемешивали на шейкере в течение 20 мин и помещали в ультразвуковую баню на 10, 20, 30, 40, 60, 90 или 120 мин или в термостат на 16 ч при 37°C. После проведения кислотного гидролиза и дериватизации образцы охлаждали до комнатной температуры, добавляли 1 мл 20%-ного раствора трихлоруксусной кислоты для осаждения белков, перемешивали на шейкере в течение 10 мин. Затем добавляли 10 мл деионизованной воды и доводили рН до 4 раствором К2НРО4 (1 М раствор). Для обезжиривания пробы к смеси добавляли 3 мл гексана, перемешивали на шейкере в течение 10 мин и центрифугировали при 4000 об/мин в течение 10 мин. Гексан удаляли, а водный слой очищали методом ТФЭ с помощью картриджа шприцевого типа, заполненного 30 мг ССПС (30 × 10 мм), по методике, разработанной нами ранее [31]. Твердофазную экстракцию проводили на вакуумной установке для ТФЭ (Манифолд М6, Россия). Картридж кондиционировали 2 мл ацетонитрила и 3 мл деионизованной воды. Перед элюированием картридж промывали 3 мл деионизованной воды, а затем элюировали аналиты 2 мл ацетонитрила. Элюаты упаривали в атмосфере азота при 40°C досуха, вновь растворяли в 0.5 мл смеси подвижных фаз A и Б (80 : 20) и использовали для дальнейшего ВЭЖХ-МС/МС-анализа. Пробоподготовку куриных печени, сердца и желудков проводили аналогичным образом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Традиционную длительную пробоподготовку образцов пищевых продуктов при определении в них метаболитов нитрофуранов (инкубация в водяной бане 16 ч, 37°C), как показано в нескольких работах, можно сократить до 4 ч [20] и 2 ч [21, 22] за счет повышения температуры до 55 и 60°C соответственно. При этом, как отмечается в работах [21, 22], повышение температуры приводит не только к сокращению времени, но и к повышению выхода продуктов дериватизации [21, 22]. Еще более эффективным оказалось одновременное воздействие температуры и ультразвукового или микроволнового излучения. При проведении термостатического ультразвукового гидролиза-дериватизации это время удалось снизить с 16 до 2 ч (45 кГц, 60°C) [24] и даже до 20 мин (45 кГц, 50°C) [25]. При проведении процедуры в микроволновой печи это время составляет 4 (65°C) [26], 6 (95°C) [27, 28], 7.2 (90°C) [29, 25 ] и 20 (70°C) мин [30, 24 ], но при этом часто уменьшается выход продуктов дериватизации за счет разложения нитрофенильных производных некоторых нитрофуранов [26]. Напротив, в работе [23] сообщается о значительном снижении времени гидролиза-дериватизации (до 5 мин) при комнатной температуре за счет повышения концентрации соляной кислоты и количества метанола в реакционной смеси.
Столь неоднозначные и зачастую противоречивые сведения о влиянии температуры на процесс гидролиза-дериватизации, по-видимому, связаны с тем, что при выборе условий не всегда использовали образцы пищевых продуктов, полученные после воздействия на животных нитрофуранами, как это было сделано в работах [20, 22, 24, 26], а использовали модельные образцы, приготовленные путем добавления метаболитов нитрофуранов к анализируемым пищевым продуктам [20, 21, 23, 25, 27–30]. Очевидно, что гидролиз метаболитов нитрофуранов из образцов, полученных естественным путем в процессе воздействия на животных, будет отличаться от гидролиза в модельных образцах. Кроме того, в качестве дериватизирующего агента в цитируемых выше работах использовали не только 2-НБА [20–24, 26–28], но и другие дериватизирующие агенты [25, 29, 30], что также могло повлиять на время дериватизации, при этом исследования проводили при разных концентрациях соляной кислоты.
В настоящей работе с целью снижения времени анализа изучено влияние температуры и концентрации соляной кислоты на ВЭЖХ-МС/МС-определение метаболитов нитрофуранов в курином мясе с использованием 5-нитро-2-фуральдегида при проведении пробоподготовки в термостатированной ультразвуковой ванне. Ранее нами доказано, что продуктами дериватизации метаболитов нитрофуранов с этим новым дериватизирующим агентом являются соответствующие исходные нитрофураны [31, 32].
Предварительные испытания. Для оценки содержания метаболитов нитрофуранов в образцах мяса кур-бройлеров, получавших в течение 10 дней нитрофураны, провели их определение в виде нитрофенилпроизводных по официальной методике, регламентированной действующим ГОСТ 32014-2012 [33], с использованием в качестве дериватизирующего агента 2-НБА и по разработанной нами ранее методике [31] в виде исходных нитрофуранов с использованием в качестве дериватизирующего агента 5-НФА. Гидролиз и дериватизацию в этой серии экспериментов проводили одновременно в соответствии с официальной методикой путем нагревания образцов на водяной бане в присутствии 0.1 М HCl в течение 16 ч. Установлено, что результаты, полученные с использованием разных дериватизирующих агентов (табл. 2), не различаются между собой. Следует отметить, что найденные количества метаболитов нитрофуранов не коррелируют с введеным бройлерам количеством, что может указывать на разные пути метаболизма нитрофуранов в организме животных, о чем сообщалось ранее [34, 35].
Таблица 2.
Результаты (мкг/кг) определения метаболитов нитрофуранов в образцах куриного мяса кур-бройлеров, получавших в течение 10 дней нитрофураны (n = 3, P = 0.95)
| Дериватизирующий агент, условия проведения гидролиза и дериватизации | AМОЗ | AОЗ | АГД | СЕМ |
|---|---|---|---|---|
| 2-Нитробензальдегид, 0.1 М HCl, 40°С, водяная баня, 16 ч (ГОСТ 32014-2012) | 40 ± 6 (0.06)* | 31 ± 4 (0.05) | 88 ± 11 (0.05) | 110 ± 10 (0.04) |
| 5-Нитро-2-фуральдегид, 0.1 М HCl, 40°С, водяная баня, 16 ч | 41 ± 5 (0.05) | 30 ± 3 (0.04) | 91 ± 9 (0.04) | 105 ± 13 (0.05) |
Кроме того, с учетом дальнейших исследований оценили термостабильность 5-НФА и четырех нитрофуранов в 0.1, 0.2 и 0.5 М растворах соляной кислоты и установили, что повышение температуры от 40 до 80°C при нагревании образцов в термостатированной ультразвуковой бане не приводит к разрушению этих соединений.
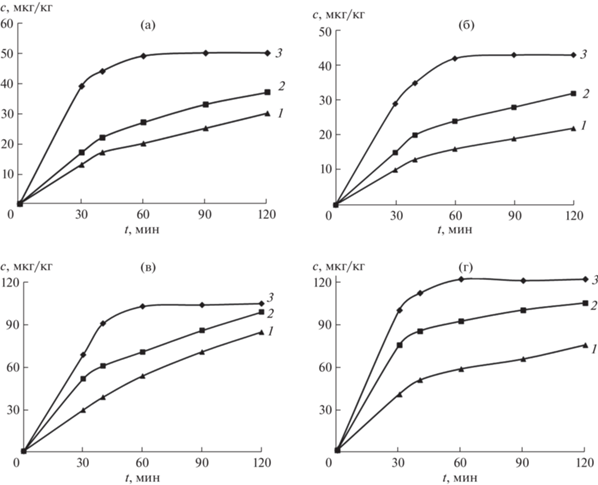
Влияние температуры и времени изучали на примере четырех образцов куриного мяса, содержащих известные количества метаболитов AОЗ, AМОЗ, АГД и СЕМ. В этой серии экспериментов гидролиз-дериватизацию проводили в термостатированной ультразвуковой бане (37 кГц) в присутствии 0.1 М HCl при 40, 60 и 80°C в течение 30, 60, 90 и 120 мин. Как видно из рис. 1, максимальный выход продуктов дериватизации, коррелирующий с содержанием этих метаболитов в анализируемых образцах (табл. 2), достигается при 80°C. Полученные результаты указывают на то, что повышение температуры от 40 до 80°C позволяет снизить время гидролиза-дериватизации в 0.1 М HCl до 60 мин.
Рис. 1.
Влияние температуры на выход продуктов дериватизации метаболитов нитрофуранов АМОЗ (а), АОЗ (б), АГД (в) и СЕМ (г) с 5-нитро-2-фуральдегидом в зависимости от времени проведения гидролиза-дериватизации в термостатированной ультразвуковой бане. сHCl = 0.1 М; 40°C (1), 60°C (2), 80°C (3).
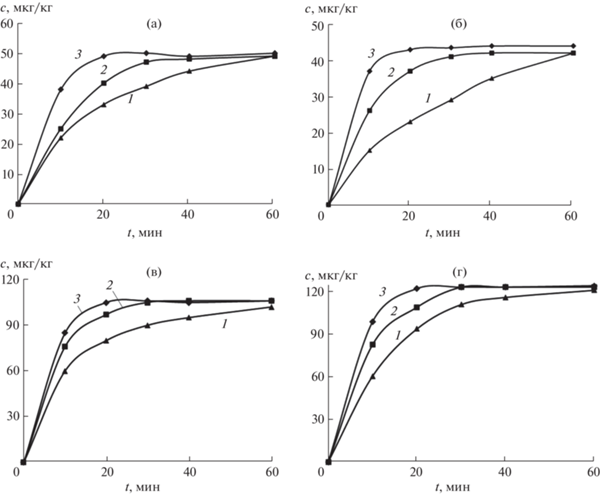
Влияние концентрации соляной кислоты и времени. В этой серии экспериментов гидролиз-дериватизацию проводили в термостатированной ультразвуковой бане (37 кГц) при 80°C в присутствии 0.1, 0.2 и 0.5 М HCl в течение 10, 20, 30, 40 и 60 мин. Данные рис. 2 указывают на то, что увеличение концентрации соляной кислоты от 0.1 до 0.2 и далее до 0.5 М приводит к сокращению времени от 60 до 30 и далее до 20 мин соответственно. Таким образом, в присутствии 0.5 М HCl при 80°C время гидролиза-дериватизации можно снизить до 20 мин.
Рис. 2.
Влияние концентрации соляной кислоты на выход продуктов дериватизации метаболитов нитрофуранов АМОЗ (а), АОЗ (б), АГД (в) и СЕМ (г) с 5-нитро-2-фуральдегидом в зависимости от времени проведения гидролиза-дериватизации в термостатированной ультразвуковой бане при 80°C. сHCl, М: 0.1 (1), 0.2 (2), 0.3 (3).
Сравнение результатов определения метаболитов нитрофуранов в курином мясе и субпродуктах. В выбранных условиях (0.5 М HCl; 80°C; термостатированная ультразвуковая баня, 37 кГц; 30 мин) определяли содержание метаболитов нитрофуранов в четырех образцах куриного мяса и субпродуктов (печень, желудки, сердце). Полученные результаты приведены в табл. 3. Там же представлены результаты анализа этих образцов с использованием официальной методики пробоподготовки (0.1 М HCl, 40°C, водяная баня, 16 ч) и двух дериватизирующих агентов – 2-НБА и 5-НФА. Установили, что результаты, полученные с использованием официальной методики пробоподготовки, не различаются между собой для двух указанных выше дериватизирующих агентов, тогда как при использовании ускоренного метода гидролиза-дериватизации для всех типов анализируемых образцов получены более высокие значения. Таким образом, при повышении температуры до 80°C и концентрации соляной кислоты до 0.5 М удается не только значительно сократить время гидролиза-дериватизации с 16 ч до 30 мин, но и повысить выход продуктов дериватизации на 11–49%.
Таблица 3.
Результаты (мкг/кг) определения метаболитов нитрофуранов в курином мясе и субпродуктах с использованием официальной методики пробоподготовки и 2-нитробензальдегида (1); официальной методики пробоподготовки и 5-нитро-2-фуральдегида (2); ускоренной методики пробоподготовки и 5-нитро-2-фуральдегида (3) (n = 3, P = 0.95)
| Дериватизирующий агент, условия проведения гидролиза и дериватизации | AМОЗ | AОЗ | АГД | СЕМ |
|---|---|---|---|---|
| Мясо | ||||
| (1) 2-НБА, 0.1 М HCl, 40°С, водяная баня, 16 ч (ГОСТ 32014-2012) | 40 ± 6 (0.06)* | 31 ± 4 (0.05) | 88 ± 11 (0.05) | 110 ± 10 (0.04) |
| (2) 5-НФА, 0.1 М HCl, 40°С, водяная баня, 16 ч | 41 ± 5 (0.05) | 30 ± 3 (0.04) | 91 ± 9 (0.04) | 105 ± 13 (0.05) |
| (3) 5-НФА, 0.5 М HCl, 80°С, УЗ-баня, 30 мин | 50 ± 6 (0.05) | 43 ± 5 (0.05) | 104 ± 13 (0.05) | 122 ± 21 (0.05) |
| Печень | ||||
| (1) 2-НБА, 0.1 М HCl, 40°С, водяная баня, 16 ч (ГОСТ 32014-2012) | 149 ± 30 (0.08) | 62 ± 10 (0.07) | 120 ± 24 (0.08) | 128 ± 20 (0.06) |
| (2) 5-НФА, 0.1 М HCl, 40°С, водяная баня, 16 ч | 143 ± 21 (0.06) | 64 ± 9 (0.06) | 125 ± 22 (0.07) | 122 ± 21 (0.07) |
| (3) 5-НФА, 0.5 М HCl, 80°С, УЗ-баня, 30 мин | 216 ± 43 (0.08) | 92 ± 16 (0.07) | 153 ± 30 (0.08) | 147 ± 21 (0.08) |
| Желудки | ||||
| (1) 2-НБА, 0.1 М HCl, 40°С, водяная баня, 16 ч (ГОСТ 32014-2012) | 96 ± 17 (0.07) | 67 ± 10 (0.06) | 96 ± 17 (0.06) | 115 ± 20 (0.07) |
| (2) 5-НФА, 0.1 М HCl, 40°С, водяная баня, 16 ч, | 92 ± 18 (0.08) | 64 ± 11 (0.07) | 100 ± 18 (0.07) | 123 ± 18 (0.06) |
| (3) 5-НФА, 0.5 М HCl, 80°С, УЗ-баня, 30 мин | 120 ± 21 (0.07) | 98 ± 15 (0.06) | 126 ± 16 (0.06) | 178 ± 31 (0.07) |
| Сердце | ||||
| (1) 2-НБА, 0.1 М HCl, 40°С, водяная баня, 16 ч (ГОСТ 32014-2012) | 104 ± 21 (0.08) | 53 ± 9 (0.07) | 125 ± 19 (0.06) | 97 ± 17 (0.07) |
| (2) 5-НФА, 0.1 М HCl, 40°С, водяная баня, 16 ч, | 109 ± 19 (0.07) | 52 ± 10 (0.08) | 134 ± 27 (0.08) | 103 ± 20 (0.08) |
| (3) 5-НФА, 0.5 М HCl, 80°С, УЗ-баня, 30 мин | 126 ± 22 (0.07) | 75 ± 15 (0.08) | 152 ± 26 (0.07) | 143 ± 25 (0.07) |
Авторы выражают благодарность Министерству науки и высшего образования Российской Федерации и Совету по грантам Президента Российской Федерации для государственной поддержки молодых российских ученых и по государственной поддержке ведущих научных школ Российской Федерации за финансовую поддержку исследований (проект МД-1448.2021.1.3).
Список литературы
Hird S.J., Lau, B.P.-Y., Schuhmacher R., Krska R. Liquid chromatography-mass spectrometry for the determination chemical contaminants in food // Trends Anal. Chem. 2014. V. 59. P. 59.
Wang B., Xie K., Lee K. Veterinary drug residues in animal-derived foods: Sample preparation and analytical methods // Foods. 2021. V. 10. № 3. P. 555.
Sun R., Chen J., Pan C., Sun Y., Mai B., Li Q.X. Antibiotics and food safety in aquaculture // J. Agric. Food Chem. 2020. V. 68. P. 11908.
Ture M., Fentie T., Regassa B. Veterinary drug residue: the risk, public health significance and its management college of veterinary medicine and animal science // J. Vet. Sci. 2019. V. 13. № 2. Article 555856.
Guardone L., Tinacci L., Armani A., Trevisani M. Residues of veterinary drugs in fish and fish products: An analysis of RASFF data over the last 20 years // Food Control. 2022. V. 135. Article 108780.
Vass M., Hruska K., Fránek M. Nitrofuran antibiotics: A review on the application, prohibition and residual analysis // Vet. Med. 2008. V. 53. № 9. P. 469.
EFSA Panel on Contaminants in the Food Chain. Scientific Opinion on nitrofurans and their metabolites in food // EFSA J. 2015. V. 13. № 6. Article 4140.
McCalla D.R. Mutagenicity of nitrofuran derivatives: Review // Environ. Mutagen. 1983. V. 5. P. 5745.
Commission Regulation (EC) 1995/1442/EC (1995) // Off. J. Eur. Commun. 1995. V. 38. № 143. P. 26.
Единые санитарно-эпидемиологические и гигиенические требования к продукции (товарам), подлежащей санитарно-эпидемиологическому надзору (контролю). 2010. С. 391.
Закревский В.В., Лелеко С.Н. Скрининг нитрофурановых соединений в импортном мясном сырье // Гигиена и санитария. 2014. Т. 5. С. 121.
Points J., Burns D.T., Walker M.J. Forensic issues in the analysis of trace nitrofuran veterinary residues in food of animal origin // Food Control. 2015. V. 50. P. 92.
Sniegocki T., Giergiel M., Sell B., Posyniak A. New method of analysis of nitrofurans and nitrofuran metabolites in different biological matrices using UHPLC-MS/MS // J. Vet. Res. 2018. V. 62. № 2. P. 161.
Cooper K.M., Mulder P.P.J., van Rhijn J.A., Kovacsics L., McCracken R.J., Young P.B., Kennedy D.G. Depletion of four nitrofuran antibiotics and their tissue-bound metabolites in porcine tissues and determination using LC-MS/MS and HPLC-UV // Food Addit. Contam. 2005. V. 22. № 5. P. 406.
Куликовский А.В., Горлов И.Ф., Сложенкина М.И., Вострикова Н.Л., Иванкин А.Н., Кузнецова О.А. Определение нитрофуранов в мышечной ткани методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием // Журн. аналит. химии. 2019. Т. 75. № 8. С. 703. (Kulikovskii A.V., Gorlov I.F., Slozhenkina M.I., Vostrikova N.L., Ivankin A.N., Kuznetsova O.A. Determination of nitrofuran metabolites in muscular tissue by high-performance liquid chromatography with mass spectrometric detection // J. Anal. Chem. 2019. V. 74. № 9. P. 906.)
Alkan F., Kotan A., Özdemir N. Development and validation of confirmatory method for analysis of nitrofuran metabolites in milk, honey, poultry meat and fish by liquid chromatography–mass spectrometry // Maced. Vet. Rev. 2016. V. 39. № 1. P. 15.
Øye B.E., Couillard F.D., Valdersnes S. Complete validation according to current international criteria of a confirmatory quantitative method for the determination of nitrofuran metabolites in seafood by liquid chromatography isotope dilution tandem mass spectrometry // Food Chem. 2019. V. 300. Article 125175.
Ryu E., Park J.S., Giri S.S., Park S.C. Development and validation of a LC-MS/MS method for the determination of nitrofuran metabolites in soft-shell turtle powder health food supplement // Int. J. Anal. Chem. 2021. Article 8822448.
Guichard P., Laurentie M., Hurtaud-Pessel D., Verdon E. Confirmation of five nitrofuran metabolites including nifursol metabolite in meat and aquaculture products by liquid chromatography-tandem mass spectrometry: validation according to european union decision 2002/657/ec // Food Chem. 2021. V. 342. Article 128389.
Verdon E., Couedor P., Sanders P. Multi-residue monitoring for the simultaneous determination of five nitrofurans (furazolidone, furaltadone, nitrofurazone, nitrofurantoine, nifursol) in poultry muscle tissue through the detection of their five major metabolites (AOZ, AMOZ, SEM, AHD, DNSAH) by liquid chromatography coupled to electrospray tandem mass spectrometry — In-house validation in line with Commission Decision 657/2002/EC // Anal. Chim. Acta. 2007. V. 586. № 1. P. 336.
Chen D., Delmas J.-M., Hurtaud-Pessel D., Verdon E. Development of a multi-class method to determine nitroimidazoles, nitrofurans, pharmacologically active dyes and chloramphenicol in aquaculture products by liquid chromatography-tandem mass spectrometry // Food Chem. 2020. V. 311. Article 125924.
Johnston L., Croft M., Murby J. Fortified versus incurred residues: Extraction of furazolidinone metabolite from prawn // Anal. Bioanal. Chem. 2015. V. 407. P. 4535.
Wu S., Yang B., Yu H. A rapid derivatization method for analyzing nitrofuran metabolites in fish using ultra-performance liquid chromatography-tandem mass spectrometry // Food Chem. 2020. V. 310. Article 125814.
Wang K., Kou Y., Wang M., Ma X., Wang J. Determination of nitrofuran metabolites in fish by ultraperformance liquid chromatography-photodiode array detection with thermostatic ultrasound-assisted derivatization // ACS Omega. 2020. V. 5. № 30. P. 18887.
Wang K., Wang M., Kou Y., Guo C., Ma X., Tian B., Qi Y., Wang W., Wang J. Determination of nitrofuran metabolites in meat products by UHPLC-fluorescence with ultrasonic-assisted derivatization // J. Food Compos. Anal. 2022. V. 107. Article 104375.
Regan G., Moloney M., Di Rocco M. Development and validation of a rapid LC–MS/MS method for the confirmatory analysis of the bound residues of eight nitrofuran drugs in meat using microwave reaction // Anal. Bioanal. Chem. 2022. V. 414. P. 1375.
Palaniyappan V., Nagalingam A.K., Ranganathan H.P., Kandhikuppam K.B., Kothandam H.P., Vasu S. Microwave-assisted derivatisation and LC–MS/MS determination of nitrofuran metabolites in farm-raised prawns (Penaeus monodon) // Food Addit. Contam. Part A. 2013. V. 30. № 10. P. 1739.
Veach B.T., Baker C.A., Kibbey J.H., Fong A., Broadaway B.J., Drake C.P. Quantitation of chloramphenicol and nitrofuran metabolites in aquaculture products using microwave-assisted derivatization, automated SPE, and LC-MS/MS // J. AOAC Int. 2015. V. 98. № 3. P. 588.
Luo X., Yu Y., Kong X., Wang X., Ji Z., Sun Z., You J. Rapid microwave assisted derivatization of nitrofuran metabolites for analysis in shrimp by high performance liquid chromatography–fluorescence detector // Microchem. J. 2019. V. 150. Article 104189.
Luo X., Sun Z., Wang X., Yu Y., Ji Z., Zhang S., Li G., You J. Determination of nitrofuran metabolites in marine products by high performance liquid chromatography–fluorescence detection with microwave-assisted derivatization // New J. Chem. 2019. V. 43. P. 2649.
Мелехин А.О., Толмачева В.В., Шубина Е.Г., Дмитриенко С.Г., Апяри В.В., Грудев А.И., Золотов Ю.А. Новый дериватизирующий агент для определения метаболитов нитрофуранов в яйцах методом ВЭ-ЖХ-МС/МС // Журн. аналит. химии. 2021. Т. 76. № 11. С. 1012. (Melekhin A.O., Tolmacheva V.V., Shubina E.G., Dmitrienko S.G., Apyari V.V., Grudev A.I., Zolotov Yu.A. A new derivatizing agent for determining nitrofuran metabolites in chicken eggs by high-performance liquid chromatography–tandem mass spectrometry // J. Anal. Chem. 2021. V. 76. № 11. P. 1312.)
Melekhin A.O., Tolmacheva V.V., Shubina E.G., Dmitrienko S.G., Apyari V.V., Grudev A.I. Determination of nitrofuran metabolites in honey using a new derivatization reagent, magnetic solid-phase extraction and LC–MS/MS // Talanta. 2021. V. 230. Article 122310.
ГОСТ 32014–2012. Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания метаболитов нитрофуранов с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором (с Изменением № 1, с Поправками). М.: Стандартинформ, 2013.
McCracken R.J., Kennedy D.G. Detection, accumulation and distribution of nitrofuran residues in egg yolk, albumen and shell // Food Addit. Contam. 2007. V. 24. № 1. P. 26.
McCracken R.J., van Rhijn J.A., Kennedy D.G. The occurrence of nitrofuran metabolites in the tissues of chickens exposed to very low dietary concentrations of the nitrofurans // Food Addit. Contam. 2005. V. 22. № 6. P. 567.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии