

Журнал аналитической химии, 2023, T. 78, № 8, стр. 695-702
Адаптивная градуировка в электротермической атомно-абсорбционной спектрометрии
Ю. М. Садагов a, *, А. Ю. Садагов a
a ООО “Кортэк”
119602 Москва, Никулинская ул., 27, к. 2, Россия
* E-mail: sadagov@yandex.ru
Поступила в редакцию 20.02.2023
После доработки 23.03.2023
Принята к публикации 23.03.2023
- EDN: SHQAHS
- DOI: 10.31857/S0044450223080157
Аннотация
Разработан метод определения концентраций в зеемановской электротермической атомно-абсорбционной спектрометрии, основанный на трансформации функции преобразования атомно-абсорбционного спектрометра в градуировочную зависимость, адаптированную к матрице анализируемого образца. Функция преобразования выражена однопараметрической функцией насыщения, которая является интегральной оптической характеристикой спектрометра, независимой от состава анализируемой пробы. Алгоритм метода включает измерения аналитических сигналов анализируемого образца и этого же образца с известной добавкой элемента и расчет по этим сигналам матричного коэффициента и искомой концентрации элемента в анализируемом образце. Метод адаптивной градуировки опробован для As, Au, Cd, Cu, Mo, Pd, Pb, Mo, Pd, Pb, Ti, V. Относительная систематическая погрешность определения концентрации элемента в диапазоне измерения оптической плотности атомного пара не превышает 10%. Диапазоны определяемых концентраций элементов превышают три порядка.
Градуировку аналитического прибора считают ключевой процедурой методики анализа. Подразумевается, что градуировка и последующие измерения аналитического сигнала выполняются в идентичных условиях, задаваемых методикой [1, 2]. Реально ли обеспечить идентичность условий градуировки и измерений? Согласно определению [3], градуировочный образец – это объект аналитического контроля с известным содержанием аналита. Это означает, что для градуировки необходим набор стандартных образцов состава (СОС) анализируемой пробы с различными содержаниями аналита. Как правило, это требование невыполнимо из-за отсутствия соответствующих СОС. В электротермической атомно-абсорбционной спектрометрии (ЭТААС) используются “простые” градуировочные образцы неадекватного состава, которые готовятся из стандартных образцов (ГСО или МСО) элементов. Однако степень атомизации элемента для градуировочного и анализируемого образцов могут отличаться, что является источником систематической погрешности результатов измерений.
Аналитическим сигналом в атомно-абсорбционной (АА) спектрометрии является атомная абсорбционность (оптическая плотность атомного пара):
где ${{I}_{0}}$ и $I$ – интенсивности излучения аналитической резонансной линии на входе и выходе аналитической ячейки атомизатора соответственно.Для строго монохроматичной линии излучения по закону Бугера–Ламберта–Бера $I = {{I}_{0}} \times {{10}^{{ - kc}}},$ где с – концентрация определяемого элемента в анализируемом образце, пропорциональная концентрации атомного пара в аналитической ячейке, $k$ – многофакторный коэффициент, зависящий, в том числе, от степени атомизации анализируемого образца.
Наличие в источнике рассеянного излучения [4], не поглощаемого атомным паром, приводит к трансформации линейной функции A = kc в функцию насыщения:
где ${{I}_{S}}$ – интенсивность рассеянного излучения.Ранее нами показано [5], что функция преобразования входного сигнала концентрации с в выходной сигнал $A$ атомной абсорбционности ЭТАА-спектрометра с обратным эффектом Зеемана в переменном магнитном поле аппроксимируется двухпараметрической функцией насыщения:
где $~s$ – параметр, который определяется долей рассеянного излучения.Для установления функции преобразования (3) использовали два градуировочных образца с концентрациями с1 и с2 = 2с1 определяемого элемента. Очевидно, что при этом выполняется полноценная градуировка.
При определении концентрации элемента в анализируемом образце значение параметра k уточняли ведением добавки аналита в анализируемый образец. Установление функции преобразования или первичная градуировка уже не является процедурой методики анализа, а первичная градуировка после уточнения параметра $k$ может быть использована в разных методиках. Описанный способ опробован при определении (0.0n–10n) мкг/г As, Cd Pb, Sb, Se и Te в растворах после разложения СОС илистых почв методом ЭТААС [6].
Целью данной работы является исследование функций преобразования в зеемановской ЭТААС c целью установления функции преобразования АА-спектрометра без градуировки, разработка и апробация альтернативной процедуры определения концентрации элементов в анализируемых образцах сложного состава.
ТЕОРИЯ
Функция (3) является функцией насыщения, которая стремится к значению ${{A}_{{{\text{НАС}}}}} = \lg \left( {1 + {{s}^{{ - 1}}}} \right)$ при увеличении концентрации с. Из данного выражения для уровня ${{A}_{{{\text{НАС}}}}}$ насыщения следует: для того чтобы установить значение параметра $s$ достаточно измерить уровень насыщения:
Уровень насыщения, как и параметр $s,$ в основном определяется характеристиками излучающего слоя [4]. Следовательно, при данном токе источника излучения и фиксированной температуре поглощающего слоя параметр $k$ будет зависеть только от степени атомизации анализируемого образца. Таим образом, $s$ и $k$ являются взаимно независимыми параметрами.
Введя новую переменную $a = kc,$ получим функцию преобразования $A = f\left( a \right){\text{:}}$
По сути, аргумент $a$ является входным сигналом идеальной атомной абсорбционности, а функция (5) отображает преобразование идеального сигнала $a$ в реальный сигнал атомной абсорбционности $A.$ Функция (5) является первичной характеристикой АА-спектрометра, независимой от параметров процесса атомизации и свойств анализируемого образца.
Для определения концентрации сX элемента в анализируемом образце используется обратная функция преобразования $a = {{\varphi }}\left( A \right){\text{:}}$
(6)
$a = - \lg \left[ {\left( {1 + s} \right){{{10}}^{{ - A}}} - s} \right] = \lg \frac{{1 - {{{10}}^{{ - {{A}_{{{\text{НАС}}}}}}}}}}{{{{{10}}^{{ - A}}} - {{{10}}^{{ - {{A}_{{{\text{НАС}}}}}}}}}}.$Алгоритм включает измерение двух аналитических сигналов: ${{A}_{{1~}}}$ – для анализируемого образца с искомой концентрацией сX и ${{A}_{{2~}}}$ – для анализируемого образца с добавленной концентрацией (сX + сД). Этим сигналам соответствуют значения ${{a}_{1}} = {{\varphi }}\left( {{{A}_{1}}} \right)$ и ${{a}_{2}} = {{\varphi }}\left( {{{A}_{2}}} \right)$ обратной функции преобразования. Так как a = kc, то решив систему двух уравнений относительно неизвестных $k$ и сX:
(7)
$\left\{ \begin{gathered} k{{c}_{{\text{Х}}}} = \varphi \left( {{{A}_{1}}} \right), \hfill \\ k\left( {{{c}_{{\text{Х}}}} + {{c}_{{\text{Д}}}}} \right) = \varphi \left( {{{A}_{2}}} \right), \hfill \\ \end{gathered} \right.$получим:
(8)
$k = \frac{{{{\varphi }}\left( {{{A}_{2}}} \right) - {{\varphi }}\left( {{{A}_{1}}} \right)}}{{{{c}_{{\text{Д}}}}}},$(9)
${{c}_{{\text{Х}}}} = \frac{{{{c}_{{\text{Д}}}}}}{{\frac{{{{\varphi }}\left( {{{A}_{2}}} \right)}}{{{{\varphi }}\left( {{{A}_{1}}} \right)}} - 1}}.$Установленное значение (8) параметра $k$ трансформирует функцию преобразования (5) в градуировочную зависимость, адаптированную к анализируемому образцу. Следовательно, параметр $k$ можно назвать матричным коэффициентом.
По сути, алгоритм измерений, основанный на решении системы уравнений (7), является вариантом метода стандартных добавок, а точнее метода одной стандартной добавки [7]. Однако в методе стандартных добавок используется градуировочная зависимость $A = f\left( c \right),$ нелинейность которой является источником систематической погрешности определения концентрации, что является существенным ограничением метода добавок. В методе адаптивной градуировки используется линейная зависимость $a = kc$ идеальной атомной абсорбционности от концентрации, что позволяет корректно использовать метод одной стандартной добавки во всем концентрационном диапазоне.
Предложенный алгоритм измерений будет работать, если для данной матрицы коэффициент $k\left( c \right) = {\text{const,}}$ т.е. не зависит от концентрации определяемого элемента. Такое предположение представляется логичным, поскольку концентрация матричных компонентов существенно больше концентрации аналита.
Уровень насыщения – это аналитический сигнал для концентрации элемента, которая гарантирует выполнение условия ${{10}^{{--kc}}} \approx 0.$ Однако атомизация очень большой массы элемента связана с риском загрязнения графитовой печи. Кроме того, импульсные сигналы атомной абсорбционности для больших концентраций могут быть искажены эффектом roll-over [8]. Установлено, что для элементов, определяемых на ЭТАА-спектрометрах типа КВАНТ.Z, которые использовались в наших экспериментах, значения параметра $s$ лежат в диапазоне от 0.05 до 0.5 [5]. Это позволяет оценить минимальную концентрацию ${{c}_{{{\text{НАС}}}}}$ как обеспечивающую выполнение условия ${{10}^{{--k{{c}_{{{\text{НАС}}}}}}}} \approx 0.005,$ т.е. $k{{c}_{{{\text{НАС}}}}} \approx 2.3.$ Для того чтобы установить значение концентрация ${{c}_{{{\text{НАС}}}}},$ достаточно оценить характеристическую концентрацию ${{c}_{{{\text{ХАР}}}}}$ элемента. Из определения характеристической концентрации следует, что $k{{c}_{{{\text{ХАР}}}}} = 0.0044,$ следовательно, минимальную концентрацию насыщения можно вычислить по формуле ${{c}_{{{\text{НАС}}}}} \approx 500{{c}_{{{\text{ХАР}}}}}.$
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование и реактивы. Эксперименты проводился на серийных экземплярах ЭТАА-спектрометра типа КВАНТ.Z, производимом ООО “КОРТЭК” (Москва). Для коррекции фонового поглощения в спектрометре используется обратный эффект Зеемана – продольно нагреваемая графитовая печь располагается в продольном переменном магнитном поле сетевой частоты. Легкая графитовая печь нагревается со скоростью около 10°С/мс, что обеспечивает высокую чувствительность АА-измерений при использовании амплитуды импульса атомной абсорбционности в качестве аналитического сигнала (амплитудная регистрация).
Спектрометр оборудован автоматической шестиламповой турелью и автоматическим дозатором. Источники резонансного излучения – одноэлементные лампы с полым катодом (ЛПК) типа ЛТ-6М, производимые ООО “КОРТЭК”. В спектрометре используются графитовые печи с пиролитическим покрытием (П) и без покрытия (НП), производимые фирмами ООО “ВИРИАЛ” (Санкт-Петербург) и “SCHUNK” (Германия). АА-измерения проводили для элементов (резонансная линия, нм): As (193.7), Au (242.8), Cd (228.8), Cu (324.7), Mo (313.3), Pb (283.3), Pd (340.5), Ti (364.3), V (318.5).
Анализируемые и промежуточные растворы готовили в пластмассовых пробирках для центрифугирования объемом 1.5 или 2.0 мл, которые используются в автоматическом дозаторе прибора. Для приготовления растворов использовали пипеточные одноканальные дозаторы переменного объема (Ленпипет).
В экспериментах использовали дистиллированную воду, которую получали на лабораторном аквадистилляторе типа ДЭ-4М (ЭМО). В качестве стандартного образца состава использовали СОС сухого молока GBW10117 (Китай). Промежуточные и анализируемые растворы элементов готовили из соответствующих ГСО (МСО). Для приготовления растворов использовали следующие реактивы: конц. HNO3 (ос. ч. 27-5 ГОСТ 11125-84), конц. HCl (ос. ч. ГОСТ 14261-77), нитрат палладия концентрации 10 г/л (Palladium matrix modifier, Merck, Германия), нитрат магния концентрации 10 г/л (Magnesium matrix modifier, Merck, Германия), поверхностно активное вещество Triton X-100 (Thermo Scientific, США).
В качестве защитного инертного газа использовали аргон высшего сорта (чистота 99.993%).
Условия и процедуры. Pb, Cd, As и Au определяли в графитовых печах без пиролитического покрытия, Cu, Mo, Pd, Ti и V – в графитовых печах с пиролитическим покрытием. Во всех экспериментах дозируемая аликвота составляла 5 мкл. Для всех элементов испарение проводили в две стадии: время каждой стадий 6 с, температуры 90 и 110°С. Внутренний поток аргона закрывали только на стадии атомизации, на остальных стадиях испарения, пиролиза и очистки этот поток открывали.
При проверке модели анализируемые образцы готовили разбавлением промежуточного раствора элемента соответствующим фоновым раствором: дистиллированной водой для Cu, Cd, Mo, Pb, Pd, Ti, V, 1%-ной HCl в дистиллированной воде для Au, раствором палладиевого модификатора с концентрацией 50 мг/л палладия в дистиллированной воде для As. При проверке модели для всех элементов использовали первую стадию пиролиза со следующими параметрами: время – 6 с, температура − 200°С. На стадиях испарения и пиролиза внутренний поток аргона был открыт. Температуры стадии атомизации: 1800°С для Cd и Pb, 2100°С для Au, 2200°С для Cu, 2400°С для As и Pd, 2750°С для Mo, Ti, V. Для всех элементов длительность стадии атомизации была равна 500 мс, а внутренний поток аргона на стадии атомизации был закрыт.
Для приготовления анализируемых образцов из СОС сухого молока GBW10117 (КНР) использовали прямой метод подготовки, без минерализации. Навеску пробы СОС сухого молока массой около 1 г разбавляли в 50 мл дистиллированной воды в кварцевой колбе при нагревании до 50–60°С. Затем объем 980 мкл раствора пробы молока переносили в пробирку автодозатора и добавляли в этот раствор по 5 мкл палладиевого и магниевого модификаторов и 10 мкл 0.1%-ного раствора Triton X-100 в дистиллированной воде. В полученном анализируемом образе определяли As, Cd и Pb. При приготовлении холостого образца вместо пробы молока в пробирку вводили 980 мкл дистиллированной воды, в которую добавляли указанные выше модификаторы. Для определения Cu анализируемый и холостой образцы разбавляли в 10 раз дистиллированной водой.
Палладиевый и магниевый модификаторы позволяют увеличить температуру графитовой печи на стадии пиролиза и предотвратить существенные потери аналита. Добавка ПАВ Triton X-100 уменьшает поверхностное натяжение раствора, что способствует локализации дозируемой капли в центральной части графитовой печи и приводит к улучшению сходимости результатов измерений.
Для минимизации фонового поглощения, возникающего при атомизации анализируемых образцов молока, на последней (перед атомизацией) стадии пиролиза графитовую печь нагревали до 700°С для Cd, 900°С для Pb, 1100°С для Cu и 1400°С для As. Время выдержки установленной температуры на этой стадии пиролиза составляло 5 с для всех элементов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В первую очередь необходимо было апробировать предложенный способ измерения уровня насыщения и доказать, что при данном токе ЛПК уровень насыщения не зависит от типа графитовой печи и состава используемого раствора элемента. Ниже приведены результаты таких экспериментов для золота. Установили, что характеристическая масса золота в графитовой печи Вириал НП ${{c}_{{{\text{ХАР}}}}} \approx 0.5$ мкг/л, следовательно, минимальная концентрация насыщения ${{c}_{{{\text{НАС}}}}} \approx 250$ мкг/л.
Из рис. 1 следует, что концентрации 250 мкг/л достаточна для установления уровня насыщения золота, поскольку при дальнейшем увеличении концентрации значение ${{A}_{{{\text{НАС}}}}}$ практически остается постоянным. Установленное значение ${{A}_{{{\text{НАС}}}}}$ является средним арифметическим трех параллельных измерений. Относительное среднеквадратичное отклонение (ОСКО) результатов параллельных измерений не превышало 1%.
Рис. 1.
Сигналы атомной абсорбционности для 125 (1; Амакс = 1.034), 250 (2; Амакс = 1.318) и 500 (3; Амакс = 1.320) мкг/л золота (фон – 1%-ная HCl) в графитовой печи Вириал НП.
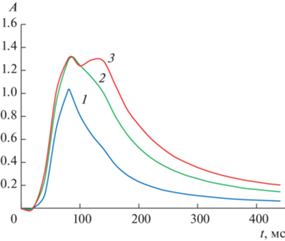
Уровни насыщения золота для различных матриц, приведенные в табл. 1, отличаются на 3% от среднего значения $\overline {{{A}_{{{\text{НАС}}}}}} = 1.31~{\text{Б}},$ которое можно принять постоянным для данного эксперимента. Аналогично установили значения уровней насыщения для других элементов (см. табл. 2).
Таблица 1.
Уровни насыщения (${{A}_{{{\text{НАС}}}}}$) функции преобразования золота в различных графитовых печах и матрицах
| Тип графитовой печи | Матрица | ${{A}_{{{\text{НАС}}}}},$ Б |
|---|---|---|
| Вириал НП | 1% HCl | 1.317 |
| 1% HCl + 50 мг/л Pd | 1.332 | |
| 1% HCl + 5% HNO3 + по 50 мг/л Pd и Mg | 1.298 | |
| Вириал П | 1% HCl | 1.32 |
| 1% HCl + 50 мг/л Pd | 1.325 | |
| Диоктисульфид в толуоле (1 : 20) | 1.308 | |
| Шунк НП | 1% HCl | 1.243 |
| 1% HCl + 50 мг/л Pd | 1.319 | |
| 1% HCl + 5% HNO3 + 50 мг/л Pd | 1.254 | |
| Шунк П | 1% HCl | 1.394 |
| 1% HCl + 50 мг/л Pd | 1.324 | |
| Среднее арифметическое значение ± СКО | 1.31 ± 0.04 | |
Таблица 2.
Значения температуры стадии атомизации $\left( {{{T}_{А}}} \right),$ уровня насыщения $\left( {{{A}_{{{\text{НАС}}}}}} \right),$ параметра $~\left( s \right)$ и коэффициента детерминации $\left( {{{R}^{2}}} \right)$ зависимости $a$ от cВХ элементов
| Элемент | ${{T}_{А}}$, °С | ${{A}_{{{\text{НАС}}}}},$ Б | $s$ | ${{R}^{2}}$ |
|---|---|---|---|---|
| Ванадий | 2750 | 0.823 | 0.177 | 0.9975 |
| Золото | 2100 | 1.320 | 0.0503 | 0.9986 |
| Кадмий | 1800 | 0.682 | 0.263 | 0.9968 |
| Медь | 2200 | 1.307 | 0.0519 | 0.9992 |
| Молибден | 2750 | 1.012 | 0.108 | 0.9973 |
| Мышьяк | 2400 | 0.565 | 0.374 | 0.9982 |
| Палладий | 2200 | 1.126 | 0.0810 | 0.9981 |
| Свинец | 1800 | 0.680 | 0.264 | 0.9979 |
| Титан | 2750 | 0.870 | 0.156 | 0.9983 |
Для аналитического прибора, каким после градуировки становится АА-спектрометр, входной величиной является определяемая концентрация ${{c}_{{{\text{ВХ}}}}},$ а выходной величиной – найденная концентрация ${{c}_{{{\text{ВЫХ}}}}}.$ Для получения искомой информации сначала проводят прямое преобразование $A = f\left( {{{c}_{{{\text{ВХ}}}}}} \right)$ определяемой концентрации в аналитический сигнал, а затем обратное преобразование ${{c}_{{{\text{ВЫХ}}}}} = {{\varphi }}\left( A \right)$ аналитического сигнала в найденную концентрацию. Так как ${{\varphi }}\left( {f\left( x \right)} \right) = x,$ то идеальная функция преобразования аналитического прибора имеет вид ${{c}_{{{\text{ВЫХ}}}}} = {{c}_{{{\text{ВХ}}}}}.$ Однако любая реальная модель, являясь приближением, искажает эту функцию. Мы попытались оценить степень искажения идеального преобразования ${{c}_{{{\text{ВЫХ}}}}} = {{c}_{{{\text{ВХ}}}}}$ для предлагаемой модели на примере палладия (табл. 3). Каждое значение ${{A}_{i}}$ является средним арифметическим четырех параллельных измерений. ОСКО результатов параллельных измерений не превышало 3%.
Таблица 3.
Определяемые (cВХ) и найденные (cВЫХ – cФ) концентрации (мкг/л) палладия, относительные погрешности $\left( {{{\Delta }_{{{\text{ОТН}}}}}} \right),$ измеренные сигналы атомной абсорбционности $\left( A \right)$ и соответствующие сигналы идеальной атомной абсорбционности $\left( a \right)$
| cВХ | А, Б | $a$, Б | $\bar {k}~$ = 0.01902 | $k\left( {400} \right)~$ = 0.02017 | ||
|---|---|---|---|---|---|---|
| cВЫХ – cФ | ${{\Delta }_{{{\text{ОТН}}}}},$ % | cВЫХ – cФ | ${{\Delta }_{{{\text{ОТН}}}}},$ % | |||
| 0 | 0.00136 | 0.0015 | 0 | – | 0 | – |
| 2 | 0.00483 | 0.0051 | 1.893 | –5.36 | 1.861 | –6.98 |
| 10 | 0.02064 | 0.0223 | 10.930 | 9.36 | 10.351 | 3.51 |
| 50 | 0.09694 | 0.1058 | 54.837 | 9.67 | 51.731 | 3.46 |
| 100 | 0.19225 | 0.2123 | 110.831 | 10.83 | 104.356 | 4.36 |
| 200 | 0.3563 | 0.4034 | 211.304 | 5.65 | 199.444 | -0.28 |
| 400 | 0.66017 | 0.8083 | 424.185 | 6.05 | 400.721 | 0.18 |
| 600 | 0.8633 | 1.1728 | 615.825 | 2.64 | 580.323 | 3.28 |
| 800 | 0.97236 | 1.4649 | 769.401 | –3.83 | 725.710 | 9.29 |
| 1000 | 1.06384 | 1.9061 | 1001.472 | 0.15 | 944.789 | –5.52 |
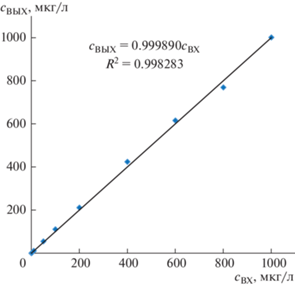
Полученная зависимость идеальной атомной абсорбционности ${{a}_{i}}$ от определяемой ${{c}_{{{\text{ВХ}}i}}}$ является линейной (a = 0.001902сВХ), т. к. коэффициент детерминации ${{R}^{2}}~$ = 0.998101. Коэффициент наклона линии регрессии $a = 0.001902{{c}_{{{\text{ВХ}}}}}$ является усредненным матричным коэффициентом $\bar {k},$ который использовали для вычисления ${{c}_{{{\text{ВЫХ}}}}} = {a \mathord{\left/ {\vphantom {a {\bar {k}}}} \right. \kern-0em} {\bar {k}}}$ и установления зависимости ${{c}_{{{\text{ВЫХ}}}}}$ от ${{c}_{{{\text{ВХ}}}}}$ (см. табл. 3). Так как в идеале ${{c}_{{{\text{ВЫХ}}}}} = {{c}_{{{\text{ВХ}}}}},$ то отличие от единицы коэффициента наклона линии регрессии зависимости ${{c}_{{{\text{ВЫХ}}}}}$ от ${{c}_{{{\text{ВХ}}}}}$ (см. рис. 2) является критерием правильности модели. Для палладия это отличие равно −0.011%, а для других элементов значения этого параметра лежат в диапазоне от −0.03 до 0.03%, что подтверждает правильность модели.
Рис. 2.
График зависимости и линия регрессии найденной концентрации ${{c}_{{{\text{ВЫХ\;}}}}}$ от определяемой концентрации ${{c}_{{{\text{ВХ}}}}}.$
Значение относительной погрешности определения концентрации элемента в анализируемом образце вычисляли по формуле:
(10)
${{\Delta }_{{{\text{ОТН}}}}} = \frac{{\left( {{{c}_{{{\text{ВЫХ}}}}}--{{c}_{{\text{Ф}}}}} \right)--{{c}_{{{\text{ВХ}}}}}}}{{{{c}_{{{\text{ВХ}}}}}}} \times 100\% ,$Установление среднего значения матричного коэффициента необходимо для верификации модели. Для реализации алгоритма адаптивной градуировки, основанной на предположении о независимости матричного коэффициента от концентрации элемента, достаточно установления значения матричного коэффициента для одной концентрации. Например, в табл. 3 приведены значения найденной концентрации, рассчитанные по матричному коэффициенту $k\left( {400} \right) = \left( {0.8083--0.0015} \right)/400 = 0.002017$. Градуировочная зависимость получается из функции преобразования путем измерения сигналов для фонового образца и анализируемого образца с концентрацией 400 мкг/л. Средняя относительная погрешность, которую вычисляли по формуле $\bar {\Delta } = {{\sum \left| {{{\Delta }_{i}}} \right|} \mathord{\left/ {\vphantom {{\sum \left| {{{\Delta }_{i}}} \right|} 9}} \right. \kern-0em} 9},$ в этом случае составила 4%, что в 1.5 раза меньше, чем для градуировки по усредненному матричному коэффициенту.
Из табл. 3 следует, что для палладия диапазон измерения аналитического сигнала простирается от 0.0044 до 1.0638 Б, что близко к уровню насыщения, равному 1.13 Б. Предел обнаружения палладия (по 3СКО-критерию) для нашего эксперимента составил 0.8 мкг/л. Таким образом, диапазон определяемых концентраций палладия составляет 1–1000 мкг/л, т.е. три порядка величины.
Кроме палладия, модель адаптивной градуировки проверили для элементов различной летучести. В табл. 2 приведены температуры атомизации, параметры функции преобразования (5) и коэффициенты детерминации зависимостей $~a$ от ${{c}_{{{\text{ВХ}}}}}$ этих элементов.
На рис. 3 показаны графики функций преобразования некоторых элементов. Для легколетучих элементов (Cd и Pb) в быстро нагреваемой графитовой печи скорость атомизации гораздо больше скорости диссипации атомного пара из аналитической ячейки и амплитуда ${{A}_{{{\text{МАКС}}}}}$ импульса $A\left( t \right)$ атомной абсорбционности равна общему числу ${{N}_{0}}$ атомов элемента в анализируемом образце [9, 10]. С уменьшением летучести элемента скорость атомизации становится сравнимой со скоростью диссипации для среднелетучих элементов (As, Au, Cu, Pd). И, наконец, для труднолетучих элементов (Mo, Ti, V), к тому же образующих термостойкие карбиды, амплитуда ${{A}_{{{\text{МАКС}}}}}$ почти целиком определяется скоростью атомизации.
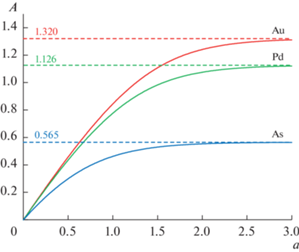
Мы полагаем, что близость коэффициента детерминации к единице для элементов различной летучести (см. табл. 3) является надежным доказательством адекватности способа адаптивной градуировки при применении амплитудного способа регистрации. Адаптивная градуировка использует весь диапазон измерения атомной абсорбционности элемента, что позволяет расширить диапазон определяемых концентраций до трех порядков. При определении концентрации элемента в анализируемом образце с помощью одной добавки систематическая погрешность в основном обусловлена погрешностью приготовления концентрации добавки и практически не превышает 10% во всем концентрационном диапазоне.
Все изложенное выше демонстрирует работу метода адаптивной градуировки для образцов с простой матрицей. Прямые доказательства эффективности предлагаемого метода для сложных матриц представлены ранее [6].
С целью подтверждения эффективности метода адаптивной градуировки и сравнения влияния методов адаптивной и внешней градуировки на правильность реультатов анализа определили концентрации элементов в СОС сухого молока GBW10117 (табл. 4). Если для адаптивной градуировки результаты измерений для всех элементов совпадают с аттестованными значениями, то для внешней градуировки результаты измерений оказались заниженными. При этом для As, Cd и Pb результаты измерений не совпадают с аттестованными значениями даже с учетом погрешностей. Это может быть обусловлено и депрессирующим влиянием матрицы, и разными потерями аналита при пиролизе “чистых” градуировочных и анализируемых образцов. Способ адаптивной градуировки компенсирует и первое, и второе.
Таблица 4.
Концентрации (мкг/г) элементов в стандартном образце состава сухого молока GBW10117
| Элемент | Аттестовано | Найдено по адаптивной градуировке (n = 3, P = 0.95) | Найдено по внешней градуировке (n = 3, P = 0.95) |
|---|---|---|---|
| Свинец | 0.416 ± 0.010 | 0.43 ± 0.03 | 0.32 ± 0.02 |
| Кадмий | 0.111 ± 0.002 | 0.118 ± 0.008 | 0.082 ± 0.006 |
| Мышьяк | 0.20 ± 0.01 | 0.23 ± 0.02 | 0.13 ± 0.02 |
| Медь | 5.96 ± 0.32 | 6.05 ± 0.42 | 5.84 ± 0.38 |
* * *
Способ адаптивной градуировки используется в аттестованных методиках количественного химического анализа, разработанных ООО “Кортэк”: “Методика измерений массовой концентрации свинца, кадмия, мышьяка, цинка, меди, железа, олова в продовольственном сырье и пищевых продуктах методом электротермической атомно-абсорбционной спектрометрии” (ФР.1.37.2020.38609) и “Методика измерений массовой концентрации свинца, кадмия, мышьяка, цинка, меди, железа в пищевых продуктах без минерализации методом электротермической атомно-абсорбционной спектрометрии” (ФР.1.31.2021.39780).
Таким образом, функция преобразования, которая является интегральной оптической характеристикой ЭТАА-спектрометра, позволяет определить концентрацию элемента в анализируемом образце непосредственно в процессе адаптивной градуировки. Два измерения аналитического сигнала для анализируемого образца и того же образца с добавкой аналита достаточны и для установления реальной концентрационной шкалы во всем диапазоне измерения атомной абсорбционности и для установления значения искомой концентрации элемента в анализируемом образце. Адаптивная градуировка объединяет процедуры градуировки и измерения и позволяет скорректировать не только матричные влияния, но и изменения эффективности атомизации, обусловленные другими причинами, например старением графитовой печи. Кроме того, способ адаптивной градуировки позволяет определять концентрацию аналита во всем диапазоне измерения аналитического сигнала атомной абсорбционности, не ограничиваясь его узкой линейной частью.
Список литературы
Основы аналитической химии. В 2-х тт. Т. 1 / Под ред. Золотова Ю.А. М.: Издательский центр “Академия”, 2012. 384 с.
Дворкин В.И. Метрология и обеспечение качества химического анализа. М.: Техносфера, 2020. 317 с.
ГОСТ Р 52361-2018. Контроль объекта аналитический. Термины и определения. М.: Стандартинформ, 2018. 11 с.
De Loos-Vollebregt M.T.C., de Galan L. Stray light in Zeeman and pulsed hollow cathode lamp atomic absorption spectrometry // Spectrochim. Acta B. 1986. V. 41. № 6. P. 597.
Садагов Ю.М., Левин А.Д., Бирюкова И.В. Функции преобразования в электротермической атомно-абсорбционной спектрометрии // Измерительная техника. 2021. № 4. С. 63. https://doi.org/10.32446/0368-1025it.2021-4-63-67
Садагов Ю.М., Тютюник О.А., Кубракова И.В., Садагов А.Ю. Учет матричных эффектов при спектрометрическом определении следов элементов с использованием метода одной стандартной добавки // Журн. аналит. химии. 2022. Т. 77 № 6. С. 563.
Frenich A.G., Vidal J.L.M., Moreno J.L.F., Romero-Gonzalez R. Compensation for matrix effects in gas chromatography–tandem mass spectrometry using a single point standard addition // J. Chromatogr. A. 2009. V. 1216. № 2. P. 4798.
Yasuda K., Koizumi H., Ohishi K, Noda T. Zeeman effect atomic absorption // Prog. Anal. Atom. Spectrosc. 1980. V. 3. № 4. P. 299.
Sadagoff Yu.M. A longitudinally heated graphite furnace for a longitudinal magnetic field. Formation of absorbance signals // Spectrochim. Acta B. 1997. V. 52. № 9–10. P. 1395.
Садагов Ю.М., Лаптев С.А. Формирование аналитических сигналов в графитовых печах // Журн. аналит. химии. 1998. Т. 53. № 10. С. 1051.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии