

Биоорганическая химия, 2019, T. 45, № 5, стр. 451-471
Никующие эндонуклеазы как уникальные инструменты в биотехнологии и генетической инженерии
Л. А. Абросимова 1, *, О. В. Кисиль 2, Е. А. Романова 1, Т. С. Орецкая 1, Е. А. Кубарева 1
1 Московский государственный университет имени М.В. Ломоносова, химический факультет и НИИ физико-химической биологии имени А.Н. Белозерского
119991 Москва, Ленинские горы, 1, Россия
2 Научно-исследовательский институт по изысканию новых антибиотиков имени Г.Ф. Гаузе
119021 Москва, ул. Большая Пироговская, 11, Россия
* E-mail: abrludmila@gmail.com
Поступила в редакцию 14.04.2019
После доработки 22.04.2019
Принята к публикации 08.05.2019
Аннотация
Уникальным семейством эндонуклеаз рестрикции являются никующие эндонуклеазы (nicking endonucleases, НЭ). Эти необычные ферменты катализируют гидролиз только одной цепи ДНК в определенном положении относительно узнаваемой ими двутяжевой последовательности. В обзоре суммированы способы получения “искусственных” НЭ, включающие инактивацию каталитического центра эндонуклеаз рестрикции, нарушение их димеризационного интерфейса или случайный мутагенез генов этих ферментов. Рассмотрены основные методы использования НЭ в биотехнологии и генетической инженерии, описано применение НЭ для усиления аналитического сигнала в системах детекции нуклеиновых кислот, белков и малых молекул.
ВВЕДЕНИЕ
Наименее изученным семейством эндонуклеаз рестрикции (ЭР, англ., restriction endonucleases, R) являются ферменты, которые узнают в двутяжевой ДНК специфическую последовательность и вносят разрыв (“ник”, англ., nick) только в одну ее цепь, – никующие эндонуклеазы (НЭ), или никазы (англ., nicking endonucleases, N). НЭ, гидролизующие “верхнюю” цепь дуплекса (5'→3'/3'→5'), обозначают Nt. (англ., top), а гидролизующие “нижнюю” цепь – Nb. (англ., bottom) [1].
Первая НЭ BstSEI была обнаружена в штамме Bacillus stearothermophilus SE-589 в 1996 г. [2]. На сегодняшний день, согласно базе данных REBASE (http://rebase.neb.com/cgi-bin/azlist?nick), известно 36 НЭ. Для более чем 7000 эндонуклеаз предсказывают никующую активность. Коммерчески доступными препаратами являются всего 15 НЭ. Известна также группа НЭ, которые ассоциированы в клетке с C5-цитозиновыми ДНК-метилтрансферазами и вносят разрыв рядом с неканонической парой G/T [1], например, V.HpaII (символ “V” происходит от названия наиболее изученной НЭ этого типа Vsr (very short patch repair)). Встречающиеся в природе НЭ довольно разнообразны и узнают различные по протяженности последовательности. НЭ могут представлять собой одну из субъединиц гетеродимерных ЭР, например, R.BtsI, R.BsrDI, R.BstNBI [3]. Релаксазы, присутствующие в конъюгативных плазмидах и принимающие участие в инициации репликации и переноса цепи ДНК, также могут выступать в качестве НЭ [4]. В вирусах хлореллы найдены частощепящие НЭ: Nt.CviPII [5], Nt.CviQII [6]. В бактериофаге f1 закодирована НЭ, представляющая собой белок, необходимый для репликации ДНК вируса [7]. Существуют и редкощепящие никующие “хоуминг”-эндонуклеазы (эндонуклеазы генной конверсии) с участком узнавания более 20 пар нуклеотидов (п.н.), например, I-HmuI и I-BasI [8–10].
За два десятилетия были разработаны и внедрены в практику изотермические методы амплификации нуклеиновых кислот с участием НЭ [11]. НЭ используют также для картирования генома [12–16], для экономичного клонирования ДНК, не требующего ферментативного лигирования [17, 18], на основе НЭ могут быть сконструированы химерные белки с заданной специфичностью [19, 20]. Были разработаны стратегии амплификации сигналов, опосредованные НЭ, для простого, быстрого и высокочувствительного обнаружения разнообразных мишеней, начиная от ДНК и РНК до клеток, белков, малых молекул и ионов. НЭ успешно используются для введения меток и модификаций во внутренние участки ДНК. С помощью НЭ были созданы генетические конструкции для исследования различных биологических процессов, например, репарации неканонических пар нуклеотидов ДНК [21, 22]. На сегодняшний день известно лишь несколько литературных источников, касающихся свойств и структуры НЭ, а также областей их применения [10, 23, 24]. В настоящем обзоре суммированы известные способы получения “искусственных” НЭ из ЭР и “хоуминг”-эндонуклеаз, а также описаны современные методы биотехнологии, использующие в качестве инструмента НЭ.
КОНСТРУИРОВАНИЕ “ИСКУССТВЕННЫХ” НЭ
Ограниченная доступность НЭ привела к созданию “искусственных” НЭ (не обнаруженных в природе) с новой специфичностью действия на основе ЭР. Способы их получения суммированы в табл. 1.
Таблица 1.
Примеры природных НЭ и искусственно созданных НЭ, полученных из ЭР
| Исходная ЭР (подтип) | Участок узнавания (5'-3'/3'-5', ↓ и ↑ ‒ места гидролиза) |
Способ получения | НЭ, ссылка |
|---|---|---|---|
| Гетеродимерные ЭР | |||
| BstNBI/ BspD6I (IIS) |
GAGTCNNNN↓NN CTCAGNNNN-NN↑ |
Природная НЭ: экспрессия гена только большой субъединицы | Nt.BstNBI/ Nt.BspD6I [4] |
| BtsI (IIS) | GCAGTG-NN↓ CGTCAC↑NN |
Природная НЭ: экспрессия гена только большой субъединицы “Искусственная” НЭ: смесь инактивированной большой субъединицы и активной малой субъединицы |
Nb.BtsI, Nt.BtsI [28] |
| BsrDI (IIS) | GCAATG-NN↓ CGTTAC↑NN |
Nb.BsrDI, Nt.BsrDI [28] | |
| Bpu10I (IIA) | CC↓TNA-GC GG-ANT↑CG |
Инактивация каталитического центра одной из субъединиц | Nt.Bpu10I, Nb.Bpu10I [29] |
| BbvCI (IIA) | CC↓TCA-GC GG-AGT↑CG |
Nt.BbvCI, Nb.BbvCI [30] | |
| Гомодимерные ЭР | |||
| MlyI (IIS) | GAGTCNNNNN↓ CTCAGNNNNN↑ |
Нарушение димеризационного интерфейса | Nt.MlyI [25] |
| AlwI (IIS) | GGATCNNNN↓N CCTAGNNNN-N↑ |
Замена фрагмента N-концевой области R.AlwI на гомологичный фрагмент из Nt.BstNBI | Nt.AlwI [27] |
| FokI (IIS) | GGATGNNNNNNNNN↓NNNN CCTACNNNNNNNNN-NNNN↑ |
Комбинирование субъединиц с введенными заменами АО | Nt.FokI, Nb.FokI [38] |
| Мономерные ЭР с непалиндромным участком узнавания | |||
| BsmI/Mva1269I (IIS) | GAATG-CN↓ CTTAC↑GN |
Инактивация одного из двух каталитических центров ЭР | Nb.Mva1269I [32] Nb.BsmI [10] |
| BtsCI (IIS) | GGATG-NN↓ CCTAC↑NN |
Nt.BtsCI, Nb.BtsCI [33] | |
| BspQI (IIS) | GCTCTTCN↓NNN CGAGAAGN-NNN↑ |
Получение мутантных форм с тремя заменами АО в ходе “аланинового сканирования” | Nt.BspQI, Nb.BspQI [13] |
| BsmBI (IIS) | CGTCTCN↓NNNN GCAGAGN-NNNN↑ |
Замена АО по аналогии с заменами АО в R.BsaI, приведшими к получению НЭ | Nt.BsmBI, Nb.BsmBI [37] |
| BsmAI (IIS) | GTCTCN↓NNNN CAGAGN-NNNN↑ |
Nt.BsmaI [37] | |
| BsaI (IIS) | GGTCTCN↓NNNN CCAGAGN-NNNN↑ |
Случайная замена АО | Nt.BsaI, Nb.BsaI [37] |
| SapI (IIS) | GCTCTTCN↓NNN CGAGAAGN-NNN↑ |
Nb.SapI, Nt.SapI [39] | |
| Мономерные ЭР с палиндромным участком узнавания | |||
| R.BcnI (IIP) | CC↓S-GG GG-S↑CC |
Направленная замена АО | Nt.BcnI, Nb.BcnI [34] |
Нарушение димеризационного интерфейса ЭР
Первая “искусственная” НЭ была получена путем нарушения димеризационного интерфейса R.MlyI [25]. R.MlyI и Nt.BstNBI имеют высокую степень идентичности первичной структуры и узнают одну и ту же нуклеотидную последовательность [26]. R.MlyI содержит единственный каталитический центр и гидролизует двутяжевую ДНК сначала в одной цепи и только затем расщепляет вторую цепь, причем эффективность гидролиза первой цепи намного выше, чем второй. Nt.BstNBI и R.MlyI существуют в растворе в виде мономеров, но при связывании с ДНК R.MlyI димеризуется. Конструирование мутантной формы R.MlyI(Y491A/K494A) или Nt.MlyI, которая утратила способность димеризоваться, позволило добиться эффективного расщепления ею только одной цепи ДНК [25].
С помощью реконструкции участка, ответственного за димеризацию, из R.AlwI была получена Nt.AlwI [27]. Близким гомологом R.AlwI является Nt.BstNBI. Участок, ответственный за димеризацию в R.AlwI, был заменен на гомологичный ему участок Nt.BstNBI. Nt.AlwI взаимодействовала с участком узнавания R.AlwI, но гидролизовала только одну цепь ДНК на расстоянии четырех нуклеотидов с 3'-конца от узнаваемой последовательности, как и Nt.BstNBI [27]. Полученные результаты показали, что с помощью методов белковой инженерии можно кардинально менять свойства ЭР, расщепляющих ДНК на некотором расстоянии от участка узнавания.
Инактивация одного из каталитических центров ЭР
В гетеродимерных ЭР BspD6I, BstNBI, BtsI, BsrDI большая субъединица в отсутствие малой проявляет никующую активность. Экспрессируя, например, ген большой субъединицы R.BtsI отдельно от гена ее малой субъединицы, можно получить Nb.BtsI, которая будет гидролизовать только “нижнюю” цепь ДНК-субстрата (табл. 1). Из R.BtsI была сконструирована также Nt.BtsI, гидролизующая “верхнюю” цепь ДНК. Для этого заменяли остатки каталитического центра большой субъединицы R.BtsI. Комплекс малой субъединицы и инактивированной большой субъединицы ЭР гидролизовал “верхнюю” цепь ДНК-субстрата [28].
Помимо указанных существуют другие гетеродимерные ЭР, такие как Bpu10I, BbvCI, каждая из субъединиц которых отдельно не способна гидролизовать ДНК. Смесь двух субъединиц гидролизует обе цепи ДНК [29]. Замена отдельных аминокислотных остатков (АО) в каталитическом центре одной из субъединиц приводит к его инактивации [30]. В работе [31] получены мутантные формы R.BbvCI − R1(E167G) + R2 и R2(E177G) + R1 (R1, R2 – субъединицы R.BbvCI, каждая из которых содержит один каталитический центр и кодируется своим геном). Эти ферменты гидролизовали ДНК с образованием в большей степени продукта с одноцепочечным (ОЦ) разрывом, чем с двуцепочечным.
Аналогично, путем “выключения” одного из двух каталитических центров, были получены НЭ из гомодимерных ЭР, таких как Mva1269I, BsmI, BtsCI [10, 32, 33]. Мутантная форма R.BtsCI(D121A/E128A) вносила ОЦ-разрыв в “верхнюю” цепь участка узнавания, тогда как мутантные формы R.BtsCI(D388A) и R.BtsCI(E403A/E405A) – в “нижнюю” цепь (табл. 1). Эти результаты позволили предположить, что каталитический центр в N-концевой области белка отвечает за гидролиз “нижней” цепи двуцепочечной ДНК, а каталитический центр в C-концевой области – за гидролиз “верхней” цепи [33]. Данное предположение может быть распространено и на ЭР, подобные R.BtsCI (R.Mva1269I, R.BsrI, R.BsmI). Существует небольшая группа ферментов, которые, в отличие от большинства классических представителей ЭР типа II, функционируют в виде мономеров и имеют один каталитический центр: R.MspI, R.HinP1I, R.MvaI и R.BcnI [34]. Для этих ЭР был предложен механизм последовательного гидролиза каждой из цепей. R.BcnI узнает псевдопалиндромную последовательность 5'-CC↓SGG-3' (S = C или G) и “подстраивается” под различные нуклеотиды, находящиеся в центре участка узнавания. Методом рентгеноструктурного анализа (РСА) определены аминокислотные остатки (АО) R.BcnI, непосредственно взаимодействующие с центральной парой нуклеотидов участка узнавания – His77, His219 и Gly217 (PDB-коды: 2ODI и 3IMB). Посредством переноса протона между двумя остатками His реализуются два разных набора водородных связей, осуществляемых ферментом для распознавания альтернативных пар G/C и C/G в центральной позиции участка узнавания [34]. Были получены мутантные формы R.BcnI, в которых His77 или His219 (или оба остатка) были заменены на Ala, Asn или Gln. Анализ продуктов гидролиза ДНК полученными белками показал, что R.BcnI(H77A) и R.BcnI(H219Q) являются сайт-специфическими НЭ, причем R.BcnI(H77A) проявляет большую специфичность к “C-цепи”, а R.BcnI(H219Q) – к “G-цепи” ДНК [34]. Предполагается, что такой метод дизайна ферментов, вносящих ОЦ-разрыв в ДНК, может быть применен и к другим похожим ЭР.
Инактивация одного из каталитических центров “хоуминг”-эндонуклеаз
“Хоуминг”-эндонуклеазы являются высокоспецифичными нуклеазами и кодируются открытыми рамками считывания, которые располагаются в составе интронов других генов либо в составе интеинов. Эти ферменты узнают протяженные участки ДНК длиной 14-40 п.н. и гидролизуют обе цепи двутяжевой ДНК или одну из них [35].
На основе “хоуминг”-эндонуклеазы I-SceI были получены две НЭ. I-SceI кодируется интроном, входящим в состав гена 21S митохондриальной РНК в Saccharomyces cerevisiae. I-SceI представляет собой мономерную “хоуминг”-эндонуклеазу, которая узнает в ДНК асимметричную последовательность из 18 п.н. и вносит в нее двуцепочечный разрыв. В одной молекуле I-SceI содержатся два симметрично расположенных каталитических центра, а гидролиз двух цепей ДНК является двухстадийным процессом, причем в первую очередь предпочтительно происходит гидролиз “нижней” (3'→5') цепи [36]. Для получения НЭ остатки Lys122 или Lys223, симметрично расположенные на периферии каталитических центров и участвующие в координации молекул воды, были заменены на остатки Ile. Это привело к значительному снижению эффективности гидролиза “верхней” или “нижней” цепи участка узнавания, соответственно.
Случайный мутагенез генов ЭР
Часто при конструировании НЭ из ЭР заряженные АО заменяют на нейтральные остатки Ala (“аланиновое сканирование”). Этот метод был применен к R.BspQI [13], для которой предсказаны два возможных каталитических центра; каждый из которых гидролизует “свою” цепь ДНК. В результате “аланинового сканирования” были получены мутантные формы R.BspQI, проявляющие никующую активность. Так, мутант R.BspQI(E172A/E248A/E255K) с тремя заменами АО гидролизовал только “верхнюю” цепь ДНК-дуплекса ‒ Nt.BspQI. Другой мутант R.BspQI(N235A/K331A/R428A) также оказался НЭ, но по отношению к “нижней” цепи ДНК ‒ Nb.BspQI (табл. 1).
Конструирование искусственных НЭ возможно на основе ЭР, для которых отсутствует информация об активных центрах. Примером является создание НЭ на основе R.BsaI [37]. В ходе ПЦР был осуществлен случайный мутагенез всего гена R.BsaI, в результате была получена библиотека генов неактивной R.BsaI. На следующем этапе исходную и мутированные последовательности гена R.BsaI гидролизовали определенными ЭР, а полученные фрагменты соединяли в различных комбинациях. Затем проводили трансформацию клеток E. coli сконструированными плазмидами, выращивали клеточную биомассу и проводили скрининг на присутствие НЭ в клеточных лизатах. Анализ клонов с никующей активностью показал, что замена в R.BsaI Arg236 на Gly или Asp приводит к образованию Nt.BsaI. Мутантная форма R.BsaI(N441D/R442G), получившая название Nb.BsaI, также является НЭ, которая, однако, гидролизует “нижнюю” цепь ДНК (табл. 1) [37].
НЭ были также получены при комбинировании субъединиц R.FokI, в которых заменены отдельные АО [38], а также в результате случайной замены АО R.SapI [39]. Таким образом, “искусственные” НЭ конструируют в ходе случайного и сайт-направленного мутагенеза генов известных ЭР и “хоуминг”-эндонуклеаз.
ПРИМЕНЕНИЕ НИКУЮЩИХ ЭНДОНУКЛЕАЗ
Изотермическая амплификация ДНК с использованием НЭ
Изотермическая амплификация ДНК является альтернативой ПЦР из-за упрощенного экспериментального протокола – весь цикл амплификации можно быстро и эффективно провести в одних температурных условиях. Одной из самых востребованных и популярных разновидностей изотермической амплификации ДНК является разработанный в 1992 г. метод, получивший название амплификация с вытеснением цепи (strand displacement amplification, SDA) [40]. Большинство последующих вариантов метода амплификации ДНК в той или иной степени опирались на SDA.
В классическом методе SDA использовалась R.HincII, катализирующая гидролиз участка узнавания ДНК в две стадии. Фактически еще за несколько лет до открытия НЭ Уолкер и соавт. [40] “заставили” ЭР вносить разрыв только в одну цепь ДНК путем замены расщепляемой фосфодиэфирной связи на тиофосфатную в процессе полимеразной реакции. Наиболее широкое распространение получил метод SDA, в котором используются четыре праймера (рис. 1). Два из них, S1 и S2, содержат ОЦ-последовательность участка узнавания R.HincII и инициируют синтез ДНК большим фрагментом ДНК-полимеразы I (фрагмент Кленова) в присутствии dGTP, dCTP, dTTP и 2'-дезоксиаденозин-5'-(α-тио)трифосфата (dATPαS). Синтез ДНК с двух других праймеров В1 и В2 (их называют праймеры смещения, bumper primer) приводит к вытеснению оцДНК-фрагментов, полученных в процессе “удлинения” праймеров S1 и S2, из комплекса с матричной ДНК. В ходе следующего раунда амплификации с участием праймеров S1, S2, В1 и В2 на матрицах S1-ext и S2-ext происходит накопление ДНК-продуктов с модифицированным участком узнавания R.HincII, причем гидролизу подвергается только одна цепь ДНК. В результате образуется ДНК-матрица с ОЦ-последовательностью участка узнавания R.HincII. С ней гибридизуется праймер S1 или S2, что инициирует следующий цикл синтеза ДНК с вытеснением цепи. Накопление ДНК происходит экспоненциально. Удается синтезировать 107 копий ДНК за 2 ч при 37°С. Однако синтезированная ДНК содержит тиофосфатные межнуклеотидные связи, что может негативно повлиять на ее использование в исследованиях. Кроме того, лишь небольшое число известных ЭР катализируют гидролиз ДНК по двустадийному механизму. Замена ЭР на НЭ в методе SDA полностью решает обе проблемы [41]. Использование в SDA только двух праймеров (S1 и В1 или S2 и В2) приводит к линейному накоплению продуктов.
Рис. 1.
Метод изотермической амплификации ДНК с экспоненциальным накоплением продукта реакции (strand displacement amplification, SDA). Праймеры S1 и S2 содержат ОЦ-фрагмент 5'-GTTGAC-3' участка узнавания R.HincII и добавляются в реакционную смесь в большом избытке по отношению к исходной ДНК. Модифицированный участок узнавания R.HincII представлен на вставке, стрелки указывают на позиции гидролиза ДНК данным ферментом. Предложено согласно [41].

Высокий уровень синтеза неспецифических побочных продуктов амплификации ограничивает применение метода SDA [42]. Действительно, для наработки целевого продукта необходимо, чтобы НЭ в каждом цикле вносила только один разрыв, необходимый для инициации реакции изотермической амплификации ДНК. При амплификации достаточно протяженного участка ДНК возрастает вероятность того, что на этом участке встретится второй, третий и т.д. участки узнавания фермента, что приведет к появлению побочных продуктов. Для предотвращения данного процесса необходимо иметь определенные “рычаги” воздействия извне на ферментативную активность НЭ.
Нами предложен подход [43, 44], позволяющий “выключать” и “включать” Nt.BspD6I в заданный момент времени с помощью внешнего сигнала (изменение температуры) за счет использования термочувствительных аналогов субстрата. Были сконструированы 13−30-звенные синтетические ДНК-дуплексы – как немодифицированные, так и модифицированные, которые образовывали непродуктивный комплекс с ферментом и блокировали его активность при 20°С. Повышение температуры вызывало диссоциацию дуплексов-ингибиторов и приводило к инициации ферментативного гидролиза протяженной ДНК при ~40–50°С. На наш взгляд прием временного блокирования активности НЭ может являться основой для усовершенствования метода SDA.
Метод экспоненциальной изотермической амплификации коротких 8‒16-звенных целевых олигодезоксирибонуклеотидов, предусматривающий использование НЭ – EXPAR (англ., exponential amplification reaction), был разработан Ван Нессом и соавт. (рис. 2) [45]. После гибридизации ДНК-матрицы и праймера происходит “удлинение” праймера, что приводит к формированию участка узнавания НЭ. После расщепления одной цепи дуплекса НЭ (Nt.BstNBI в [45]) целевой олигонуклеотид, не содержащий ОЦ-последовательность участка узнавания НЭ, “выходит” из состава дуплекса, так как температура “плавления” образованного им дуплекса ниже температуры протекания реакции (55°С). Целевой олигонуклеотид также может являться праймером на первой стадии данного метода, что обеспечивает экспоненциальный характер амплификации. Присутствующая в реакционной смеси ДНК-полимераза, используя образовавшийся свободный 3'-концевой гидроксил оставшегося на матрице фрагмента ДНК, вновь начинает синтез целевого олигонуклеотида. Как только ДНК-полимераза достроит недостающий фрагмент, НЭ снова вносит разрыв в синтезированную цепь. Цикличность процесса обеспечивает синтез около 106 копий олигонуклеотида нужной длины.
Рис. 2.
Метод экспоненциальной изотермической амплификации олигодезоксирибонуклеотидов (exponential amplification reaction, EXPAR). Черным цветом показана ДНК-матрица, голубым – целевой олигонуклеотид. Здесь и далее прямоугольники розового и серого цветов обозначают ОЦ-последовательности участка узнавания НЭ, стрелка красного цвета указывает на позицию гидролиза НЭ. Предложено согласно [45].
К недостаткам метода относят альтернативные пути протекания реакции вследствие синтеза ДНК ab initio и неспецифического взаимодействия некоторых олигонуклеотидов с ДНК-полимеразой [46, 47]. Кроме того, специфичность EXPAR зависит от первичной структуры амплифицируемого олигонуклеотида. На основе анализа 384 нуклеотидных последовательностей был предложен метод прогнозирования производительности EXPAR и, как следствие, целесообразности его применения [47].
Метод амплификации протяженных фрагментов ДНК с линейным накоплением продукта реакции, основанный на применении НЭ, был предложен в работе [48] и получил название NEMDA (англ., nicking-endonuclease-mediated DNA amplification). Для создания ОЦ-разрывов использовали природную НЭ Nt.CviPII, входящую в систему рестрикции-модификации вируса NYs-1 хлореллы. Гидролиз одной из цепей ДНК с помощью НЭ приводил к возникновению праймера для ДНК-полимераз, обладающих способностью вытеснять имеющуюся цепь ДНК в процессе синтеза новой последовательности нуклеотидов: ДНК-полимеразы Bst, ДНК-полимеразы U29 и секвеназы версии 2.0 (модифицированной Т7-ДНК-полимеразы). Выбор ДНК-полимераз с цепь-вытесняющей активностью обусловлен тем, что при амплификации протяженной ДНК при температуре проведения реакции (55°С) диссоциации ДНК-дуплекса не происходит.
В работе [49] впервые были амплифицированы протяженные ДНК длиной 500 и 5000 п.н. Авторы протестировали эффективность использования всех коммерчески доступных к 2010 г. НЭ: Nt.AlwI, Nt.BbvCI, Nb.BsmI, Nt.BspQI, Nb.BsrDI, Nt.BstNBI; а также большой набор ДНК-полимераз. Показано, что эффективность амплификации с участием любой из полимераз главным образом определяется концентрацией НЭ, которая уникальна для каждого фермента и требует тщательного подбора. Слишком высокая концентрация НЭ приводит к ингибированию амплификации, в то время как низкая концентрация НЭ может быть недостаточной для поддержания скорости процесса. Известно, что некоторые НЭ образуют прочный комплекс с продуктом реакции, представляющим собой ДНК с ОЦ-разрывом [46, 50]. Диссоциация такого комплекса лимитирует скорость реакции гидролиза. При повышении концентрации НЭ агрегируют [51], что может понижать их активность. Однако точный механизм снижения скорости гидролиза ДНК при высоких концентрациях НЭ пока не установлен.
Отметим, что соотношение концентраций ДНК-полимеразы и НЭ не оказывает существенного влияния на эффективность амплификации. Известно, что добавление в реакционную смесь белков, взаимодействующих с оцДНК, в значительной степени способствует синтезу ДНК многими полимеразами [52]. Действительно, эффективность амплификации ДНК с использованием НЭ существенно повышалась в присутствии белка гена 32 фага Т4 и белка SSB (англ., single-stranded binding protein). Использование белка SSB также предотвращало неспецифическую амплификацию ДНК.
Если на начальной стадии амплификации ДНК проводить ее гибридизацию с циклическими ДНК-зондами, то можно осуществлять амплификацию по типу катящегося кольца (primer-generation rolling circle amplification, PG-RCA) (рис. 3) [53]. Преимущество методики состоит в отсутствии нескольких праймеров, которые часто, вследствие их димеризации, являются причиной неспецифичной амплификации. Метод PG-RCA предполагает использование целевого олигонуклеотида в качестве праймера для инициации процесса амплификации. Сначала циклический зонд, содержащий фрагмент участка узнавания НЭ, гибридизуется с целевым олигонуклеотидом. Затем происходят последовательные циклические реакции ДНК-полимеризации и гидролиза под действием НЭ. Амплификация ДНК в PG-RCA идет в экспоненциальном режиме благодаря постоянно возобновляющемуся количеству праймеров. В работе [53] использовали Vent(exo-)-ДНК-полимеразу и Nb.BsmI.
Рис. 3.
Амплификация ДНК по типу катящегося кольца (primer generation rolling circle amplification, PG-RCA). Целевой олигонуклеотид представлен как линия, циклический зонд – как квадрат. Предложено согласно [53].

Все описанные в данном разделе методы амплификации ДНК с использованием НЭ демонстрируют высокую производительность и чувствительность обнаружения исходной ДНК, поэтому с момента их разработки и внедрения в научно-исследовательскую практику они нашли успешное применение для детекции нуклеиновых кислот.
Усиление аналитического сигнала с помощью НЭ при детекции различных соединений
При анализе биологических объектов исследователи стремятся к большей чувствительности и селективности определения в них целевых соединений. Главным требованием к образцу является обеспечение достаточного количества материала для детекции в амплификационном цикле.
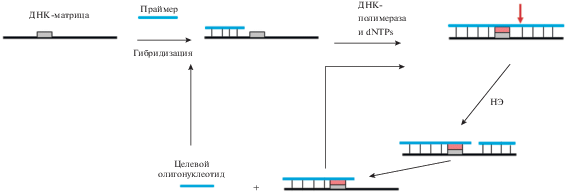
Детекция ДНК. В работе [54] был впервые предложен опосредованный НЭ метод детекции ДНК (nicking endonuclease signal amplification, NESA), получивший в дальнейшем широкое распространение (рис. 4а). Ключевой стадией NESA является гибридизация детектируемой ДНК с флуоресцентно меченным ДНК-зондом. При этом необходимо, чтобы ОЦ-фрагменты участка узнавания НЭ входили в нуклеотидные последовательности как ДНК-мишени, так и зонда. После формирования участка узнавания НЭ происходит гидролиз зонда, а флуоресцентно меченный продукт реакции детектируется методом капиллярного электрофореза. В NESA одна молекула ДНК-мишени способна поочередно гибридизоваться с несколькими флуоресцентно меченными зондами, т.к. НЭ вносит разрыв только в последовательность зонда. Благодаря этому происходит многократное усиление сигнала (увеличение интенсивности флуоресценции продукта гидролиза зонда) и, как следствие, повышается чувствительность обнаружения целевой ДНК.
Рис. 4.
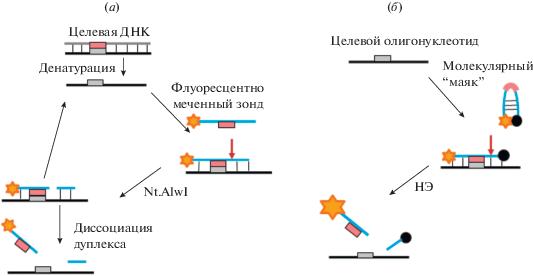
Детекция ДНК с использованием НЭ и ДНК-зондов. Детектируемая оцДНК показана черным цветом. Здесь и далее флуорофор в составе ДНК изображен в виде звездочки. (а) Метод NESA (nicking endonuclease signal amplification). Предложено согласно [54]. (б) Схема детекции ДНК с использованием молекулярного “маяка” (nickase-molecular beacon analysis, NMB-анализ). Здесь и далее “тушитель” флуоресценции показан кружком. Предложено согласно [55, 56].
Кислинг и соавт. [54] применили в методе NESA 6 НЭ: Nt.AlwI, Nt.BbvCI, Nb.BbvCI, Nb.BsmI, Nb.BsrDI, Nt.BstNBI.
Удачным дополнением методу NESA стало использование олигонуклеотидного зонда шпилечной структуры. Первые работы в этой области были опубликованы Л.А. Железной и соавт. [55, 56], предложившими метод детекции ДНК, который получил название nickase-molecular beacon analysis, NMB-анализ. Одноцепочечные олигодезоксирибонуклеотиды длиной около 25 нт содержали в своем составе фрагмент участка узнавания НЭ, их концевые последовательности были комплементарны, причем на одном конце находилась флуоресцентная метка, а на другом – тушитель флуоресценции (рис. 4б). Подобные структуры зонда называются молекулярными “маяками”.
При взаимодействии ОЦ-участков “маяка” и целевой ДНК формируется линейный двутяжевой фрагмент с участком узнавания НЭ. НЭ гидролизует “маяк”, который распадается на два фрагмента и “уходит” из комплекса с ДНК-мишенью. Происходит разобщение флуорофора и тушителя, и интенсивность флуоресценции резко возрастает. Целевая ДНК, освобожденная от фрагментов “маяка”, гибридизуется со следующим молекулярным зондом, последовательно участвуя в формировании нескольких субстратов для НЭ. Это вызывает увеличение сигнала флуоресценции, приводя к повышению чувствительности обнаружения ДНК почти на три порядка по сравнению со стандартным методом NESA [57–60].
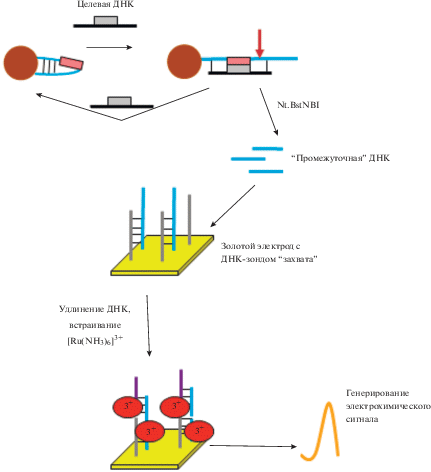
Следующим этапом развития методов высокочувствительной детекции ДНК стала разработка электрохимических подходов с участием НЭ. Одной из первых была разработана методика, описанная Чен и соавт. (рис. 5) [61]. На первом этапе шпилечная ДНК, закрепленная на магнитной частице, взаимодействует с целевой ДНК (показана черным цветом) и образует ДНК-дуплекс, который содержит участок узнавания Nt.BstNBI. Фермент вносит разрыв в иммобилизованную на частице цепь дуплекса, высвобождая “промежуточную” оцДНК. Целевая ДНК при этом остается интактной и способна взаимодействовать со следующей шпилечной ДНК, а “промежуточная” ДНК гибридизуется с комплементарной ей ДНК, иммобилизованной на золотом электроде. Процесс детекции включает в себя последовательное наращивание ДНК-дуплекса на электроде за счет гибридизации “липких” концов при участии “промежуточной” ДНК и “дополнительных” молекул ДНК, находящихся в растворе. В такой протяженный ДНК-дуплекс за счет электростатического взаимодействия встраиваются ионы гексаамминрутения [Ru(NH3)6]3+, благодаря которым и генерируется электрохимический сигнал. Минимальная концентрация детектируемой этим методом ДНК составляет 0.36 фМ. Описываемый метод [61] оказался чрезвычайно высокоспецифичным: при использовании в качестве детектируемой ДНК фрагмента, отличающегося всего одним нуклеотидом от целевой ДНК, регистрируемый электрохимический сигнал был в несколько раз слабее и по уровню интенсивности сопоставим с сигналом, наблюдаемым при использовании в качестве контроля полностью некомплементарной ДНК.
Рис. 5.
Схема электрохимического метода детекции ДНК. Черным цветом показана целевая ДНК; голубым цветом ‒ шпилечная ДНК, иммобилизованная на магнитной частице (коричневый круг). Дополнительная ДНК, необходимая для удлинения иммобилизованного на электроде ДНК-дуплекса, показана фиолетовым цветом. Ион гексаамминрутения [Ru(NH3)6]3+ обозначен как “3+” в красном круге. Предложено согласно [61].
Другим вариантом повышения чувствительности детекции ДНК является так называемое каскадное усиление, объединяющее два или более метода амплификации, что приводит к экспоненциальному усилению сигнала. Ключевое требование для реализации каскадного усиления состоит в том, чтобы ДНК-продукт первого этапа действовал в качестве инициирующего реагента (“триггера”, англ. trigger) второго этапа.
В методе CESA (cascade enzymatic signal amplification), предложенном в работе [62] (рис. 6), объединены техники инвазивной амплификации, в которой используется Flap-эндонуклеаза из археи Archaeoglobus fulgidus [63], и рассмотренного выше метода NESA. Flap-эндонуклеаза способна гидролизовать фосфодиэфирную связь на стыке одно- и двуцепочечной ДНК. При перекрывании ДНК-дуплекса, образованного целевой ДНК и зондом оранжевого цвета, с другим зондом (показан зелено-голубым цветом) на один нуклеотид, Flap-эндонуклеаза вносит ОЦ-разрыв в зелено-голубой зонд. Фрагмент этого зонда будет являться “триггерной” ДНК (показана голубым цветом) для следующего этапа, при этом одна детектируемая ДНК может приводить к образованию нескольких тысяч молекул-“триггеров”. Таким образом, детектируемая ДНК может не содержать участок узнавания НЭ, что является значительным преимуществом рассматриваемого метода.
Рис. 6.
Метод каскадного усиления сигнала при детекции ДНК (cascade enzymatic signal amplification, CESA). Черным цветом показана целевая ДНК, оранжевым и сине-зеленым цветом ‒ зонды для инвазивной амплификации. Место гидролиза Flap-эндонуклеазой из Archeoglobus fulgidus отмечено красной стрелкой. Молекулярный “маяк” представлен серым цветом, дополнительный зонд с фрагментом участка узнавания НЭ – фиолетовым цветом. Предложено согласно [62, 63].

На следующем этапе “триггерная” ДНК и дополнительная ДНК (показана фиолетовым цветом) гибридизуются со следующим олигонуклеотидным зондом – молекулярным “маяком”, и “раскрывают” его шпилечную структуру. Присутствующая в растворе Т4-ДНК-лигаза соединяет концы олигонуклеотидов, приводя к формированию участка узнавания НЭ, гидролиз которого ферментом приводит к разгоранию флуоресценции. Поскольку наработка детектируемой ДНК идет в CESA каскадом, предел обнаружения целевой ДНК снижается до нескольких фМ.
Из методов детекции ДНК с использованием НЭ, предложенных в последние годы, следует отметить SAMP (single primer-triggered isothermal amplification) [64]. В SAMP задействован только один праймер, который должен содержать фрагменты исходной ДНК в определенном порядке, что накладывает ограничения на применение данного метода. В 2018 г. на основе EXPAR был разработан метод детекции ДНК с использованием Nt.BstNBI, получивший название aligner-mediated cleavage-triggered exponential amplification, AMCEA [65]. По сравнению с классическим EXPAR в AMCEA ДНК-матрица имеет шпилечную структуру. Причем “ножка” шпильки содержит последовательность участка узнавания НЭ, благодаря чему образуется прочный комплекс НЭ с ДНК-матрицей еще до стадии гидролиза.
Детекция точечных мутаций в ДНК. НЭ нашли применение при определении однонуклеотидных замен в последовательностях генов. На основе SDA разработана техника анализа замены С→Т в гене цитохром-Р450-зависимой монооксигеназы 2D6*10 (CYP2D6*10) [66]. CYP2D6 отвечает за метаболизм большинства лекарственных препаратов. Точечная мутация в аллеле CYP2D6*10 приводит к образованию фермента со сниженной каталитической активностью. В методике используют два зонда, которые гибридизуются только с мутантной формой аллеля (рис. 7). Оба зонда имеют шпилечную структуру. Один из них содержит ОЦ-последовательность участка узнавания НЭ и остаток аденозина на 3'-конце, второй – фосфатную группу на 5'-конце. После лигирования зондов на ДНК-матрице следует стадия классической SDA-амплификации в присутствии Bst-ДНК-полимеразы (3'-конец зонда фиолетового цвета выступает в качестве праймера) и Nt.BstNBI. В ходе SDA синтезируется значительное количество фрагмента ДНК, который является аптамером гемина и выполняет функцию ДНКзима при образовании пероксидаза-подобного комплекса. Данный метод детекции высокочувствителен – минимальная концентрация аналита в пробе составляет 0.1 пМ; он также малозатратен и прост в применении. Предложенный метод был успешно использован для анализа точечных мутаций в гене k-ras [67], которые характерны для рака прямой кишки.
Рис. 7.
Метод детекции точечной мутации С→Т в гене цитохром-Р450-зависимой монооксигеназы 2D6*10. Детектируемая мутантная форма гена показана черным цветом. Удлиняющийся в процессе SDA 3'-конец ДНК отмечен галочкой. Предложено согласно [66].

Детекция РНК. МикроРНК представляют собой класс малых некодирующих молекул РНК (около 22 нт), принимающих участие в посттранскрипционной регуляции экспрессии генов. Аберрантная, отклоняющаяся от нормы экспрессия микроРНК тесно связана с различными заболеваниями человека, включая онкологические. Таким образом, микроРНК можно рассматривать как перспективные клинически значимые биомаркеры. Все основные, хорошо зарекомендовавшие себя методы амплификации ДНК, опосредованные НЭ – SDA, PG-RCA и EXPAR, адаптированы для определения микроРНК [68].
Джиа и соавт. показали [69], что для амплификации РНК подходит классический EXPAR (рис. 2). ДНК-матрица в данном случае содержит 2 участка, комплементарные целевой микроРНК, между которыми находятся ОЦ-последовательности участка узнавания и гидролиза НЭ. Специфичность может быть повышена за счет введения дополнительной стадии синтеза с целевой микроРНК “промежуточной” детектируемой ДНК. В работе [70] представлен быстрый, высокочувствительный и специфический анализ микроРНК, включающий два последовательных цикла EXPAR и предполагающий использование наносенсора на основе квантовой точки. Такой подход позволяет достигнуть предела обнаружения в 0.1 амоль и дискриминировать гомологичные последовательности с единичными нуклеотидными заменами.
Во избежание неспецифического усиления сигнала при обнаружении микроРНК были предложены также каскадные стратегии на основе SDA [71, 72]. В работе [72] использовали олигодезоксирибонуклеотидные зонды двух типов: имеющий форму шпильки и комплементарный ему молекулярный “маяк” (рис. 8). ДНК-шпилька содержит на 5'-конце ОЦ-участок, комплементарный детектируемой РНК, а также фрагмент, комплементарный “маяку” и содержащий ОЦ-последовательность участка узнавания Nb.BbvCI. На 5'- и 3'-концах “маяка” находятся флуорофор и тушитель флуоресценции. При гибридизации с РНК двутяжевой участок первого зонда диссоциирует и образует ДНК-дуплекс с “маяком”. Nb.BbvCI гидролизует сформировавшийся участок узнавания, что приводит к разобщению флуорофора и тушителя и появлению сигнала флуоресценции.

Теоретически все опосредованные НЭ методы усиления сигнала могут быть использованы не только для детекции микроРНК, но и для детекции протяженных фрагментов РНК. Однако на сегодняшний день предложена единственная, основанная на PG-RCA, каскадная схема детекции (рис. 3) 742-звенного фрагмента CD4 мРНК человека [73]. Предел обнаружения CD4 мРНК составил 143 змоль.
Детекция белков. В отличие от ДНК или РНК белки не могут выступать в качестве праймеров для инициации цикла амплификации. Поэтому методы детекции белков часто включают использование аптамеров – синтетических ДНК- или РНК-фрагментов длиной около 50–60 нт, которые способны специфически взаимодействовать с различными биомолекулами [74]. Так, для детекции тромбина использовали аптамер, который, взаимодействуя с белком, меняет свою шпилечную структуру (рис. 9а) [75]. 3'-Конец аптамера выступает в качестве праймера для ДНК-полимеразы (фрагмента Кленова). В ходе амплификации в варианте SDA формируется участок узнавания Nt.BbvCI. Образующийся при гидролизе ферментом короткий ДНК-фрагмент детектируется в результате его взаимодействия с молекулярным “маяком” (рис. 4б). В работе [76] объединили методы SDA и EXPAR для детекции тромбоцитарного фактора роста PDGF-BB, с чрезмерной экспрессией гена которого связаны злокачественные новообразования.
Рис. 9.
Методы детекции белков в пробе с использованием НЭ. (а) Аптамер в виде “шпильки” изменяет свою структуру при связывании с тромбином. Предложено согласно [75]. (б) Метод детекции субъединицы p50 транскрипционного фактора NF-κB. Двуцепочечная ДНК с участком узнавания субъединицы p50 NF-κB показана черным цветом, субъединица p50 обозначена серым овалом. Предложено согласно [77].

ДНК-связывающие белки, содержащие домены, взаимодействующие с последовательностями ДНК, например, факторы транскрипции, могут быть обнаружены с помощью методов амплификации ДНК, опосредованной НЭ [77, 78]. Однако все эти методики предполагают использование экзонуклеаз. В работе [77] представлен метод детекции субъединицы p50 транскрипционного фактора NF-κB (рис. 9б). На первой стадии детектируемый транскрипционный фактор взаимодействует с участком узнавания в ДНК. Затем полученный ДНК-белковый комплекс обрабатывают экзонуклеазой ExoIII, которая гидролизует с 3'-конца все фосфодиэфирные связи за исключением тех, которые “закрыты” транскрипционным фактором. Образующаяся оцДНК инициирует цикл ЕXPAR с использованием Nb.BbvCI, в результате которого синтезируется линкерная ДНК. Линкерная ДНК способна образовывать комплекс с наночастицами золота (AuNPs), в результате этого цвет раствора увеличенных частиц становится фиолетовым. В отсутствие субъединицы p50 NF-κB амплификации ДНК не происходит, и раствор с частицами AuNPs остается красного цвета.
В методах детекции белка НЭ были вовлечены в цикл изотермической амплификации ДНК наряду с ДНК-полимеразой (методы SDA и EXPAR). В работе [79] предложен метод, получивший название nicking endonuclease-assisted amplification, NEA, в котором отсутствует стадия синтеза детектируемой ДНК. НЭ непосредственно выступает как биокаталитический усилитель сигнала. На первой стадии происходит связывание олигонуклеотидного зонда, содержащего на конце остаток фолиевой кислоты, с рецепторами фолиевой кислоты, значительное количество которых экспрессируется в клетках злокачественных опухолей. Все не связавшиеся с рецепторами зонды удаляются из реакции в результате их гидролиза экзонуклеазой ExoI. Далее зонды с рецепторами гибридизуются с олигонуклеотидами, иммобилизованными на поверхности золотого электрода. Nt.BstNBI гидролизует присоединенную к подложке цепь, после чего зонд высвобождается и гибридизуется со следующим олигонуклеотидом на электроде, который в результате также “укорачивается”. После нескольких циклов количество исходных олигонуклеотидов, закрепленных на подложке, понижается и повышается доступность ионов [Fe(CN)6]3–/4– к электроду. В результате возрастает аналитический сигнал, получаемый методом дифференциальной импульсной полярографии. Предел обнаружения рецептора фолиевой кислоты составляет 0.19 нг/мл. Данный метод может быть использован также для детекции других белков.
В настоящее время базовые методы амплификации ДНК SDA и EXPAR применяются в основном в комбинации с методом детекции целевых биомолекул с образованием ДНКзима, мимикрирующего под пероксидазу [80].
Идентификация раковых клеток и патогенов. Идентификация раковых клеток и патогенов основывается главным образом на обнаружении специфических для них нуклеиновых кислот или белков. На базе аптамера, связывающего клетки, были разработаны методы обнаружения опухолевых клеток на основе усиления сигнала с помощью НЭ [81, 82].
В работе [82] предложен простой, экономичный и высокочувствительный колориметрический метод визуального обнаружения раковых клеток на основе циклического усиления сигнала с помощью НЭ. При этом используются аптамерные зонды шпилечной структуры к клеткам-мишеням и ДНК-линкеры, комплементарные ОЦ-участку аптамера, который формируется при взаимодействии аптамера с опухолевой клеткой. В отсутствие клеток-мишеней ДНК-линкер не связан с аптамером, но образует комплекс с наночастицами золота – раствор имеет фиолетовый цвет. При взаимодействии аптамера с опухолевой клеткой его структура претерпевает конформационные изменения, и ДНК-линкер, способный гибридизоваться с образовавшимся ОЦ-участком аптамера, инициирует циклическую реакцию с участием Nb.BbvCI. В результате гидролиза ДНК-линкера образующиеся короткие фрагменты теряют способность к ассоциации с AuNPs, и раствор меняет свой цвет на красный.
Разработано большое число эффективных методик изотермической амплификации с использованием аптамеров и НЭ для обнаружения патогенов пищевого происхождения. Для детекции Salmonella enteritidis и E. coli (штамм O157:H7) метод SDA, опосредованный НЭ, был объединен с биосенсором бокового потока [83, 84]. Предел обнаружения бактерий этим методом составляет всего 10 колониеобразующих единиц. На основе EXPAR был предложен коммерчески доступный набор ANSR™ для определения Listeria spp., Salmonella spp., Campylobacter spp. и E. coli O157:H7 [83–86]. Он включает ДНК-полимеразу и НЭ, которые действуют совместно при одной температуре; за результатами амплификации можно следить в режиме реального времени. Время обнаружения патогена составляет 10 мин при 56°С.
Определение активности ферментов. НЭ используют в методах детекции ДНК-метилтрансферазы [87, 88], теломеразы [89, 90], Т4-полинуклеотидкиназы [91], ДНК-лигазы [92] и урацил-ДНК-гликозилазы [93, 94]. Рассмотрим два из них.
Хьюэ и соавт. [88], взяв за основу EXPAR, предложили флуоресцентный метод анализа наличия в пробе ДНК-метилтрансферазы Dam, которая метилирует дезоксиаденозин в обеих цепях последовательности 5'-GATC-3'/3'-CTAG-5' в дуплексном участке ДНК-шпильки (рис. 10). R.DpnI гидролизует такие метилированные участки, а продукт гидролиза выступает в качестве праймеров в цикле EXPAR: он гибридизуется с матрицей, затем “удлиняется” ДНК-полимеразой и получившийся дуплекс разрезается Nt.BstNBI. Образующиеся олигонуклеотиды формируют G‑квадруплексы и, связываясь с цинк-протопорфирином IX (ZnPPIX), дают флуоресцентный сигнал. Предел обнаружения метилтрансферазы Dam в описанном методе составляет 8.6 × 10–5 ед./мл.
Рис. 10.
Флуоресцентный метод анализа наличия в пробе ДНК-метилтрансферазы Dam. ДНК с участком узнавания метилтрансферазы Dam представляет собой “шпильку”. ZnPPIX – комплекс протопорфирина IX и Zn2+ (zinc(II)-proto-porphyrin IX). Предложено согласно [88].

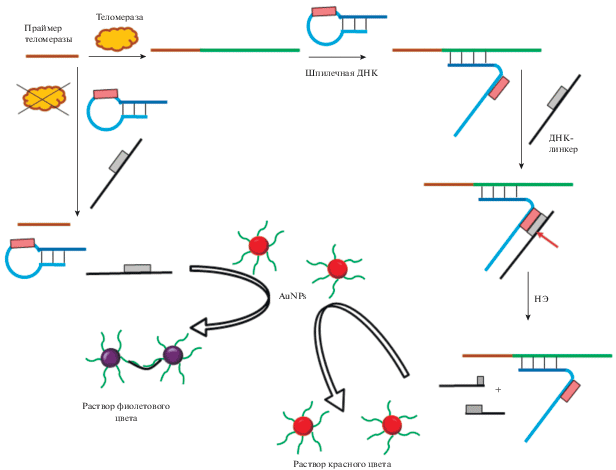
Теломераза представляет собой рибонуклеопротеин, присоединяющий определенные повторяющиеся последовательности ДНК к 3'-концу цепи ДНК в участках теломер, располагающихся на концах хромосом в эукариотических клетках. Повышенная активность теломеразы наблюдается примерно в 90% опухолевых клеток, что делает фермент ценным биомаркером для ранней диагностики. Разработан способ определения теломеразной активности, основанный на SDA и EXPAR [95]. В работе [90] для этой цели предложен колориметрический метод с использованием Nb.BbvCI (рис. 11). Для анализа активности фермента в образец добавляют праймер теломеразы, ДНК-шпильку, ДНК-линкер, НЭ и AuNPs. При наличии в образце теломеразы происходит удлинение праймера. Удлиненный праймер образует дуплекс с ДНК-шпилькой, “раскрывая” ее. В результате становится возможной гибридизация фрагмента шпильки с ДНК-линкером. В первичной структуре шпильки и ДНК-линкера содержатся ОЦ-последовательности участка узнавания Nb.BbvCI. Короткие продукты гидролиза линкера (в отличие от него самого) не могут ассоциировать с AuNPs, и, как следствие, раствор меняет свой цвет с фиолетового (в отсутствие теломеразы) на красный.
Рис. 11.
Колориметрический метод анализа наличия в пробе теломеразы. Праймер теломеразы показан коричневым цветом, удлиненный участок праймера – зеленым цветом. Предложено согласно [90].
Детекция малых молекул и ионов металлов. Малые молекулы, такие как кокаин [96, 97], охратоксин А [98], никотинамидадениндинуклеотид (NAD+) [99] и L-гистидин [100] могут быть обнаружены с помощью изотермических методов амплификации ДНК с применением НЭ и аптамеров. Так, для определения кокаина в пробе используют ДНК-зонд, образующий шпильку при связывании с детектируемой молекулой [96]. В результате 3'-конец зонда становится праймером для ДНК-полимеразы. Матрицей для ДНК-полимеразы является удлиненный 5'-конец зонда, содержащий ОЦ-последовательность участка узнавания Nt.BbvCI. Гидролиз “верхней” цепи приводит к высвобождению олигонуклеотида заданной последовательности, с которым гибридизуется молекулярный “маяк”. Данный метод сравним по чувствительности с иммуноферментным анализом: предел обнаружения кокаина составляет 5 мкМ.
Загрязнение ионами тяжелых металлов, такими как Pb2+ и Hg2+, является серьезной проблемой для здоровья человека. Методы изотермического усиления сигнала, опосредованные НЭ, успешно используются для повышения чувствительности биосенсоров при анализе наличия в пробе ионов Pb2+ [101, 102]; Hg2+ [103, 104] и K+ [105]. Недавно созданный электрохимический биосенсор [104] был успешно применен для определения солей ртути в питьевой воде и озерных водах, показав высокую селективность определения Hg2+ и крайне низкий уровень ложноположительных сигналов.
Детекция метилирования ДНК. Известно, что изменение “паттернов” метилирования остатков дезоксицитидина играет значительную роль в возникновении и развитии различных заболеваний, в том числе онкологических. Для определения степени метилирования были модифицированы базовые стратегии амплификации ДНК, опосредованной НЭ, − SDA, NASE и EXPAR. Так, был разработан метод определения метилированной ДНК, получивший название COEXPAR [106], в котором EXPAR инициируется химическим гидролизом ДНК в участках с 5-метилцитозином. В работе [107] предложен каскадный метод обнаружения метилированной ДНК (рис. 12). ДНК предварительно обрабатывают бисульфитом натрия, при этом остатки цитозина превращаются в урацил, в то время как 5-метилцитозин не модифицируется. Только метилированная ДНК комплементарна ДНК-матрице, задействованной в первом амплификационном цикле, который по сути является SDA. С помощью фрагмента Кленова и НЭ синтезируется “триггерная” ДНК, которая используется во втором цикле амплификации с участием молекулярного “маяка” (метод NASE). Каскадный метод позволяет детектировать до 0.1% метилированной ДНК в смеси с неметилированной ДНК и даже единичный локус метилирования.
Рис. 12.
Каскадный метод обнаружения метилированной ДНК в пробе. Исходная ДНК показана черным цветом. Предложено согласно [106].

Использование НЭ в картировании генома
В работе [12] предложен метод оптического картирования генома с использованием НЭ. На первом этапе ДНК подвергают гидролизу этим ферментом. С образовавшегося свободного 3'-конца ДНК-полимераза синтезирует новую цепь, включая флуоресцентно-меченные нуклеотиды. Меченые молекулы ДНК растягиваются в линейную форму на модифицированной поверхности стекла и визуализируются с помощью флуоресцентной микроскопии полного внутреннего отражения (англ., total internal reflection fluorescence, TIRF). Определяя положение флуоресцентных меток вдоль ДНК, устанавливают распределение участков узнавания НЭ. Большим преимуществом использования НЭ в этом методе по сравнению с ЭР в TIRF-микроскопии является возможность введения флуоресцентной метки непосредственно рядом с позицией гидролиза НЭ. Метод TIRF-микроскопии с использованием Nb.BbvCI был успешно применен в работе [12] для расшифровки геномов аденовируса человека и некоторых штаммов риновирусов, в том числе для получения оптической карты генома фага λ, а с использованием Nt.BspQI и Nb.BspQI в работе [13] − для картирования генома фага Т7.
В работе [15] после гидролиза исследуемой ДНК Nt.BsmAI в места ОЦ-разрывов с помощью терминальной трансферазы вводили биотин-16-дезоксиуридинтрифосфат, который эффективно связывался со стрептавидином, закрепленным на поверхности подложки. В молекуле ДНК в местах ОЦ-разрывов появлялся “довесок”, который детектировали методом атомно-силовой микроскопии. Авторам удалось определить индивидуальный образец ДНК в смеси 15 видов ДНК, представленных не менее 100 копиями. Для повышения точности картирования могут быть использованы несколько НЭ одновременно. Применение комбинации Nt.BsmAI, Nt.BstNBI и Nt.AlwI позволило достичь точности определения образца 97.1% ± ± 5.6%, в то время как при использовании одной НЭ точность составляла около 51% [16].
ЗАКЛЮЧЕНИЕ
Свойство НЭ гидролизовать только одну цепь ДНК в определенном положении относительно участка узнавания успешно используется для разработки различных методов детекции, в которых усиление сигнала опосредовано циклическим НЭ-катализируемым расщеплением ДНК. В научно-исследовательскую практику вошли такие методы изотермической амплификации нуклеиновых кислот с участием НЭ, как SDA и EXPAR [12]. Многие их варианты интегрированы с портативными приборами диагностики, включая микрофлюидные чипы [108] и тест-полоски [109]. Предложены разнообразные аналитические устройства для обнаружения нуклеиновых кислот, основанные на SDA с AuNPs в качестве индикатора цвета [110, 111]. Биопробы, полученные в результате опосредованной НЭ изотермической амплификации нуклеиновых кислот, являются неотъемлемой частью коммерчески доступных наборов для диагностики патогенов [112]. Востребованность НЭ в современной генетической инженерии подводит нас к необходимости дальнейшего изучения этого перспективного класса ферментов.
Список литературы
Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S.Kh., Dryden D.T., Dybvig K. et al. // Nucl. Acids Res. 2003. V. 31. P. 1805–1812.
Абдурашитов М.А., Беличенко О.А., Шевченко А.В., Дегтярев С.Х. // Молекул. биология. 1996. Т. 30. С. 754–758.
Железная Л.А., Перевязова Т.А., Альжанова Д.В., Матвиенко Н.И. // Биохимия. 2001. Т. 66. С. 1215–1220.
Francia M.V., Clewell D.B., de la Cruz F., Moncalian G. // Proc. Natl. Acad. Sci. U.S.A. 2013. V. 110. P. 13606–13611.
Chan S.H., Zhu Z., Van Etten J.L., Xu S.Y. // Nucl. Acids Res. 2004. V. 32. P. 6187–6199.
Chan S.H., Zhu Z., Dunigan D.D., Van Etten J.L., Xu S.Y. // Protein Expr. Purif. 2006. V. 49. P. 138–150.
Geider K., Meyer T.F., Baumel I., Reimann A. // Adv. Exp. Med. Biol. 1984. V. 179. P. 45–54.
Landthaler M., Shub D.A. // Nucl. Acids Res. 2003. V. 31. P. 3071–3077.
Landthaler M., Shen B.W., Stoddard B.L., Shub D.A. // J. Mol. Biol. 2006. V. 358. P. 1137–1151.
Chan S.H., Stoddard B.L., Xu S. // Nucl. Acids Res. 2011. V. 39. P. 1–18.
Zhao Y., Chen F., Li Q., Wang L., Fan C. // Chem. Rev. 2015. V. 115. P. 12491–12545.
Xiao M., Phong A., Ha C., Chan T.F., Cai D., Leung L., Wan E., Kistler A.L., De Risi J.L., Selvin P.R., Kwok P.Y. // Nucl. Acids Res. 2007. V. 35. e16.
Zhang P.H., Too P.H., Samuelson J.C., Chan S.H., Vincze T., Doucette S., Backstrom S., Potamousis K.D., Schramm T.M., Forrest D. et al. // Protein Expr. Purif. 2010. V. 69. P. 226–234.
Mak A.C., Lai Y.Y., Lam E.T., Kwok T.P., Leung A.K., Poon A., Mostovoy Y., Hastie A.R., Stedman W., Anantharaman T. et al. // Genetics. 2016. V. 202. P. 351–362.
Reed J., Hsueh C., Lam M.L., Kjolby R., Sundstrom A., Mishra B., Gimzewski J.K. // J. R. Soc. Interface. 2012. V. 9. P. 2341–2350.
Lam M.L., Chen B., Chen T.H. // J. Lab. Autom. 2015. V. 20. P. 411–417.
Yang J., Zhang Z., Zhang X.A., Luo Q. // Biotechniques. 2010. V. 49. P. 817–821.
Wang R.Y., Shi Z.Y., Guo Y.Y., Chen J.C., Chen G.Q. // PLoS One. 2013. V. 8. e57943.
Gabsalilow L., Schierling B., Friedhoff P., Pingoud A., Wende W. // Nucl. Acids Res. 2013. V. 41. e83.
Gutjahr A., Xu S. // Nucl. Acids Res. 2014. V. 42. e77.
Kim Y., de la Torre A., Leal A.A., Finkelstein I.J. // Sci. Rep. 2017. V. 7. P. 2071.
Перевозчикова С.А. Характеристика начальных этапов функционирования системы репарации ДНК-“мисматчей” Escherichia coli с использованием модифицированных ДНК. М.: МГУ им. М.В. Ломоносова, 2013.
Железная Л.А., Качалова Г.С., Артюх Р.И., Юнусова А.К., Перевязова Т.А., Матвиенко Н.И. // Биохимия. 2009. Т. 74. С. 107–128.
Xu S.Y. // Biomol. Concepts. 2015. V. 6. P. 253–267.
Besnier C.E., Kong H. // EMBO Rep. 2001. V. 2. P. 782–786.
Higgins L.S., Besnier C., Kong H. // Nucl. Acids Res. 2001. V. 29. P. 2492–2501.
Xu Y., Lunnen K.D., Kong H. // Proc. Natl. Acad. Sci. U.S.A. 2001. V. 98. P. 12990–12995.
Xu S.Y., Zhu Z., Zhang P., Chan S.H., Samuelson J.C., Xiao J., Ingalls D., Wilson G.G. // Nucl. Acids Res. 2007. V. 35. P. 4608–4618.
Stankevicius K., Lubys A., Timinskas A., Vaitkevicius D., Janulaitis A. // Nucl. Acids Res. 1998. V. 26. P. 1084–1091.
Heiter D.F., Lunnen K.D., Wilson G.G. // J. Mol. Biol. 2005. V. 348. P. 631–640.
Bellamy S.R.W., Milsom S.E., Scott D.J., Daniels L.E., Wilson G.G., Halford S.E. // J. Mol. Biol. 2005. V. 348. P. 641–653.
Armalyte E., Bujnicki J.M., Giedriene J., Gasiunas G., Kosiński J., Lubys A. // J. Biol. Chem. 2005. V. 280. P. 41584–41594.
Too P.H., Zhu Z., Chan S.H., Xu S.Y. // Nucl. Acids Res. 2010. V. 38. P. 1294–1303.
Kostiuk G., Sasnauskas G., Tamulaitiene G., Siksnys V. // Nucl. Acids Res. 2011. V. 39. P. 3744–3753.
Belfort M., Roberts R.J. // Nucl. Acids Res. 1997. V. 25. P. 3379–3388.
Niu Y., Tenney K., Li H., Gimble F.S. // J. Mol. Biol. 2008. V. 382. P. 188–202.
Zhu Z., Samuelson J.C., Zhou J., Dore A., Xu S. // J. Mol. Biol. 2004. V. 337. P. 573–583.
Bitinaite J., Wah D.A., Aggarwal A.K., Schildkraut I. // Proc. Natl. Acad. Sci. U.S.A. 1998. V. 95. P. 10570–10575.
Samuelson J.C., Zhu Z., Xu S.Y. // Nucl. Acids Res. 2004. V. 32. P. 3661–3671.
Walker G.T., Fraiser M.S., Schram J.L., Little M.C., Nadeau J.G., Malinowski D.P. // Nucl. Acids Res. 1992. V. 20. P. 1691–1696.
Shi C., Liu Q., Ma C., Zhong W. // Anal. Chem. 2014. V. 86. P. 336–339.
He Y., Jiang T. // Adv. Biosci. Biotechnol. 2013. V. 4. P. 539–542.
Абросимова Л.А., Мигур А.Ю., Кубарева Е.А., Венде В., Железная Л.А., Орецкая Т.С. // Известия ВУЗов. Прикладная химия и биотехнология. 2015. Т. 2. С. 48‒59.
Abrosimova L.A., Migur A.Y., Kubareva E.A., Zatsepin T.S., Gavshina A.V., Yunusova A.K., Perevyazova T.A., Pingoud A., Oretskaya T.S. // PLoS One. 2018. V. 13. e0207302.
Van Ness J., Van Ness L.K., Galas D.J. // Proc. Natl. Acad. Sci. U.S.A. 2003. V. 100. P. 4504–4509.
Tan E., Erwin B., Dames S., Ferguson T., Buechel M., Irvine B., Voelkerding K., Niemz A. // Biochemistry. 2008. V. 47. P. 9987–9999.
Qian J., Ferguson T.M., Shinde D.N., Ramírez-Borrero A.J., Hintze A., Adami C., Niemz A. // Nucl. Acids Res. 2012. V. 40. e87.
Chan S., Zhu Z., Van Etten J.L., Xu S. // Nucl. Acids Res. 2004. V. 32. P. 6187–6199.
Joneja A., Huang X. // Anal. Biochem. 2011. V. 414. P. 58–69.
Abrosimova L.A., Kubareva E.A., Migur A.Y., Gavshina A.V., Ryazanova A.Y., Norkin M.V., Perevyazova T.A., Wende W., Hianik T., Zheleznaya L.A. et al. // Biochim. Biophys. Acta. 2016. V. 1864. P. 1072−1082.
Секерина С.А., Гришин А.В., Рязанова А.Ю., Артюх Р.И., Рогулин Е.А., Юнусова А.К., Орецкая Т.С., Железная Л.А., Кубарева Е.А. // Биоорг. химия. 2012. Т. 38. С. 431–438.
Rapley R. // Mol. Biotechnol. 1994. V. 2. P. 295–298.
Murakami T., Sumaoka J., Komiyama M. // Nucl. Acids Res. 2009. V. 37. e19.
Kiesling T., Cox K., Davidson E.A., Dretchen K., Grater G., Hibbard S., Lasken R.S., Leshin J., Skowronski E., Danielsen M. // Nucl. Acids Res. 2007. V. 35. e117.
Железная Л.А., Перевязова Т.А., Железнякова E.H., Матвиенко Н.И. // Биохимия. 2002. Т. 67. С. 595–600.
Zheleznaya L.A., Kopein D.S., Rogulin E.A., Gubanov S.I., Matvienko N.I. // Anal. Biochem. 2006. V. 348. P. 123–126.
Li J.J., Chu Y., Lee B.Y., Xie X.S. // Nucl. Acids Res. 2008. V. 36. e36.
Yao G., Liang R., Yu X., Huang C., Zhang L., Qiu J. // Anal. Chem. 2015. V. 87. P. 929–936.
Yang X., Lv J., Yang. Z., Yuan R., Chai Y. // Anal. Chem. 2017. V. 89. P. 11636–11640.
Cui X., Li R., Liu X., Wang J., Leng X., Song X., Pei Q., Wang Y., Liu S., Huang J. // Anal. Chim. Acta. 2018. V. 997. P. 1–8.
Chen Y., Wang Q., Xu J., Xiang Y., Yuan R., Chai Y. // Chem. Commun. (Camb.). 2013. V. 49. P. 2052–2054.
Zou B., Ma Y., Wu H., Zhou G. // Angew. Chem. Int. Ed. Engl. 2011. V. 50. P. 7395−7398.
Hall J.G., Eis P.S., Law S.M., Reynaldo L.P., Prudent J.R., Marshall D.J., Allawi H.T., Mast A.L., Dahlberg J.E., Kwiatkowski R.W. et al. // Proc. Natl. Acad. Sci. U.S.A. 2000. V. 97. P. 8272−8277.
Ma C., Han D., Deng V., Wang J., Shi C. // Chem. commun. (Camb). 2015. V. 51. P. 553–556.
Wu W., Fan H., Lian X., Zhou J., Zhang T. // Talanta. 2018. V. 185. P. 141–145.
Wang H.Q., Wu Z., Zhang Y., Tang L.J., Yu R.Q., Jiang J.H. // Anal. Chim. Acta. 2012. V. 710. P. 111–117.
Wu D., Xu H., Shi H., Li W., Sun M., Wu Z.S. // Anal. Chim. Acta. 2017. V. 957. P. 55–62.
Mahdiannasser M., Karami Z. // Biosens. Bioelectron. 2018. V. 107. P. 123−144.
Jia H., Li Z., Liu C., Cheng Y. // Angew. Chem. Int. Ed. Engl. 2010. V. 49. P. 5498−5501.
Zhang Y., Zhang C.Y. // Anal. Chem. 2012. V. 84. P. 224−231.
Duan R., Zuo X., Wang S., Quan X., Chen D., Chen Z., Jiang L., Fan C., Xia F. // J. Am. Chem. Soc. 2013. V. 135. № 12. P. 4604−4607.
Hosoda K., Matsuura T., Kita H., Ichihashi N., Tsukada K., Urabe I., Yomo T. // RNA. 2008. V. 14. P. 584–592.
Murakami T., Sumaoka J., Komiyama M. // Nucl. Acids Res. 2012. V. 40. e22.
Kadioglu O., Malczyk A.H., Greten H.J., Efferth T. // Invest. New Drugs. 2015. V. 33. P. 513–520.
Ma C., Zhao C., Ge Y., Shi C. // Clin. Chem. 2012. V. 58. P. 384−390.
Zhang Z.Z., Zhang C.Y. //Anal. Chem. 2012. V. 84. P. 1623−1629.
Zhang Y., Hu J., Zhang C.Y. // Anal. Chem. 2012. V. 84. P. 9544−9549.
Ma F., Yang Y., Zhang C.Y. // Anal. Chem. 2014. V. 86. P. 6006−6011.
Cao Y., Zhu S., Yu J., Zhu X., Yin Y., Li. G. // Anal. Chem. 2012. V. 84. P. 4314–4320.
Peng H., Newbigging A.M., Wang Z., Tao J., Deng W., Le X.C., Zhang H. // Anal. Chem. 2018. V. 90. P. 190−207.
Bi S., Zhang J., Zhang S. // Chem. Commun. 2010. V. 46. P. 5509−5511.
Zhang X., Xiao K., Cheng L., Chen H., Liu B., Zhang S., Kong J. // Anal. Chem. 2014. V. 86. P. 5567−5572.
Mozola M., Norton P., Alles S., Gray R.L., Tolan J., Caballero O., Pinkava L., Hosking E., Luplow K., Rice J. // J. AOAC Int. 2013. V. 96. P. 842–853.
Viator R., Alles S., Le Q.N., Hosking E., Meister E., Pinkava L., Tovar E., Mozola M., Rice J. // J. AOAC Int. 2016. V. 99. P. 705–716.
Wendorf M., Feldpausch E., Pinkava L., Luplow K., Hosking E., Norton P., Biswas P., Mozola M., Rice J. // J. AOAC Int. 2013. V. 96. P. 1414–1424.
Chen Y., Odumeru J., Ziemer W. // J. AOAC Int. 2016. V. 99. P. 1555–1564.
Zhao Y., Chen F., Lin M., Fan C. // Biosens. Bioelectron. 2014. V. 54. P. 565−570.
Xue Q., Lv Y., Xu S., Zhang Y., Wang L., Li R., Yue Q., Li H., Gu X., Zhang S. et al. // Biosens. Bioelectron. 2015. V. 66. P. 547–553.
Wang W., Huang S., Li J., Rui K., Zhang J.R., Zhu J.J. // Sci. Rep. 2016. V. 6. P. 23504.
Yu T., Zhao W., Xu J.J., Chen H.Y. // Talanta. 2018. V. 178. P. 594–599.
Chen F., Zhao Y., Qi L., Fan C. // Biosens. Bioelectron. 2013. V. 47. P. 218−224.
He K., Li W., Nie Z., Huang Y., Liu Z., Nie L., Ya S. // Chemistry. 2012. V. 18. P. 3992−3999.
Liu X., Chen M., Hou T., Wang X., Liu S., Li F. // Biosens. Bioelectron. 2014. V. 54. P. 598–602.
Ahn J.K., Lee C.Y., Park K.S., Park H.G. // Biotechnol. J. 2018. V. 13. 1700603.
Wang L.J., Zhang Y., Zhang C.Y. // Anal. Chem. 2013. V. 85. P. 11509−11517.
Shlyahovsky B., Li D., Weizmann Y., Nowarski R., Kotler M., Willner I. // J. Am. Chem. Soc. 2007. V. 129. P. 3814−3815.
He J.L., Wu Z.S., Zhou H., Wang H.Q., Jiang J.H., Shen G. L., Yu R.Q. // Anal. Chem. 2010. V. 82. P. 1358−1364.
Hun X., Liu F., Mei Z., Ma L., Wang Z., Luo X. // Biosens. Bioelectron. 2013. V. 39. P. 145−151.
Jiang C., Kan Y.Y., Jiang J.H., Yu R.Q. // Anal. Chim. Acta. 2014. V. 844. P. 70−74.
Kong R.M., Zhang X.B., Chen Z., Meng H.M., Song Z.L., Tan W., Shen G.L., Yu R.Q. // Anal. Chem. 2011. V. 83. P. 7603−7607.
Li W., Yang Y., Chen J., Zhang Q., Wang Y., Wang F., Yu C. // Biosens. Bioelectron. 2014. V. 53. P. 245−249.
Zhao Y., Chen F., Zhang Q., Zhao Y., Zuo X., Fan C. // NPG Asia Materials. 2014. V. 6. e131.
Li D., Wieckowska A., Willner I. // Angew. Chem. Int. Ed. Engl. 2008. V. 47. 3927−3931.
Hong M., Wang M., Wang J., Xu X., Lin Z. // Biosens. Bioelectron. 2017. V. 94. P. 19–23.
Zhu X., Zhao J., Wu Y., Shen Z., Li G. // Anal. Chem. 2011. V. 83. P. 4085−4089.
Xu Y., C. Niu Y., Xiao X., Zhu W., Dai Z., Zou X. // Anal. Chem. 2015. V. 87. P. 2945−2951.
Zhu G., Yang K., Zhang C.Y. // Biosens. Bioelectron. 2013. V. 49. P. 170−175.
Faustino V, Catarino S.O., Lima R., Minas G. // J. Biomech. 2016. V. 49. P. 2280–2292.
Sedighi A., Krull U.J. // Talanta. 2018. V. 186. P. 568−575.
Wu W., Zhao S., Mao Y., Fang Z., Lu X., Zeng L. // Anal. Chim. Acta. 2015. V. 861. P. 62−68.
Wu W., Mao Y., Zhao S., Lu X., Liang X., Zeng L. // Anal. Chim. Acta. 2015. V. 881. P. 124−130.
Lie P., Liu J., Fang Z., Dun B., Zeng L. // Chem. Commun. (Camb.). 2012. V. 48. P. 236−238.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия