

Биоорганическая химия, 2020, T. 46, № 1, стр. 65-76
Структура О-специфического полисахарида и липида А типового штамма бактерий Azospirillum rugosum DSM-19657
Е. Н. Сигида *, М. С. Кокоулин **, П. С. Дмитренок **, В. С. Гринёв *, ***, Ю. П. Федоненко *, ***, С. А. Коннова *, ***
* Институт биохимии и физиологии растений и микроорганизмов РАН, Россия, 410049, Саратов, просп. Энтузиастов, 13
** Тихоокеанский институт биоорганической химии имени Г.Б. Елякова ДВО РАН, Россия, 690022, Владивосток, Проспект 100 лет Владивостоку, 159
*** Саратовский национальный исследовательский государственный университет имени Н.Г. Чернышевского, Россия, 410012, Саратов, ул. Астраханская, 83
Поступила в редакцию 08.07.2019
После доработки 28.07.2019
Принята к публикации 16.08.2019
Аннотация
Проведены структурные исследования липополисахарида типового штамма азотфиксирующих почвенных бактерий Azospirillum rugosum, выделенного из загрязненных нефтью почв. На основании данных химического анализа, 1D- и 2D- 1H- и 13С-ЯМР-спектроскопии установлено, что О-специфический полисахарид состоит из повторяющихся звеньев двух типов, идентичных обнаруженным ранее в О-специфическом полисахариде штамма Azospirillum brasilense Jm125A2. Структурный анализ липида А c применением ГЖХ и МАЛДИ-масс-спектрометрии выявил смесь пента-, тетра- и триацильных типов молекул. Первичными жирными кислотами в липиде А являются N-связанная 16:0(3-OH)- и О-связанная 14:0(3-OH). Микрогетерогенность в пределах каждого типа обусловлена природой вторичных жирных кислот (16:0, 18:1 или 19:0), ацилирующих 16:0(3-OH)-кислоту дистального кольца GlcN.
ВВЕДЕНИЕ
Грамотрицательные альфа-протеобактерии рода Azospirillum из семейства Rhodospirillaceae являются типичными представителями ризосферы диких и культивируемых злаков [1]. На сегодняшний день род Azospirillum включает 21 вид, большинство из которых способны увеличивать урожайность растений, оказывая множественное воздействие на молекулярном, клеточном и тканевом уровнях [2]. Рост-стимулирующий потенциал азоспирилл реализуется посредством продукции фитогормонов, улучшения минерального питания, фиксации атмосферного азота, снижения биотических и абиотических стрессов, биоконтроля патогенов и других механизмов [3, 4].
В начальные этапы формирования растительно-микробных ассоциаций с участием азоспирилл вовлечены гликополимеры бактериальной поверхности – капсульные полисахариды и липополисахариды (ЛПС) [5]. Стимулирующий эффект, оказываемый азоспириллами на растения, является весомой причиной для изучения молекулярного механизма ассоциативного взаимодействия и требует детального изучения структуры ЛПС.
ЛПС состоит из трех химически, биологически и биосинтетически различающихся компонентов, ковалентно связанных друг с другом: липида А, корового олигосахарида и О-специфического полисахарида (ОПС). ОПС является вариабельным доменом ЛПС; многообразие его возможных структур обусловлено природой, количеством, типом связей и конфигурацией моносахаридных остатков, образующих повторяющиеся звенья, а также наличием заместителей неуглеводной природы [6]. Липиду А, напротив, присуща общая консервативность строения в пределах бактериального рода при наличии микрогетерогенности, обусловленной разнообразием остатков – вторичных – жирных кислот (ЖК), ацилирующих ЖК, связанные с сахаром, степенью ацилирования и фосфорилирования. Гетерогенность строения липида А может быть связана со многими факторами, в том числе адаптацией бактерий к изменяющимся условиям среды, степенью завершенности биосинтеза и частичной модификацией препарата во время выделения [7].
Считается, что многие фундаментальные свойства бактерий определяются условиями экологической ниши, в которой они обитают. Известно, что азоспириллы хорошо адаптируются к условиям существования, обладают высокой конкурентоспособностью и не имеют строгой специфичности к растению-хозяину [8]. Кроме того, в последнее время описаны “неризосферные” виды, способные заселять нехарактерные для азоспирилл ниши. Из отработанного дорожного покрытия выделен представитель нового вида A. picis [9], из сульфидного источника – A. thiophilum [10], из пресной воды – A. griseum [11], из ферментированных молочных продуктов – A. ramasamyi [12], из микробной топливной ячейки – A. humicireducens [13]. Культура A. rugosum была выделена из загрязненных нефтью почв [14]. Активность в утилизации нефти в качестве источника углерода и энергии, возрастающая в присутствии малата, одного из компонентов корневых экссудатов растений, была отмечена и для ряда ризосферных штаммов A. brasilense и A. lipoferum [15], что делает их перспективными агентами для инокуляции при фиторемедиации загрязненных нефтью почв.
В последние годы установлены структуры OПС для 50 ризосферных штаммов A. brasilense, A. lipoferum [16], A. halopraeferens [17], A. fermentarium [18] и A. dobereinerae [19]. Данные о строении липида А азоспирилл в основном ограничены составом жирных кислот, а полная структура была опубликована для единственного штамма A. lipoferum SpBr17 [20].
В настоящей работе приводятся сведения о строении О-специфического полисахарида и липида А типового штамма ранее не изученной в этом отношении бактерии A. rugosum DSM-19657.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Из сухой биомассы бактерий A. rugosum DSM-19657 водно-фенольной экстракцией был выделен ЛПС, в результате деградации которого в мягких кислых условиях были получены ОПС и липид А. Моносахаридный анализ ОПС в виде ацетатов полиолов методом ГЖХ выявил наличие Rha, Glc, GlcN и Fuc3N в соотношении ~4.9 : 2.4 : 1.4 : 1 (отклик детектора). Анализ продуктов полного гидролиза ОПС методом ГЖХ в виде ацетилированных (S)-2-октигликозидов позволил установить D-конфигурацию Glc и GlcN и L-конфигурацию Rha.
13C-ЯМР-спектр ОПС (рис. 1) содержал сигналы различной интенсивности, свидетельствующие о нерегулярности строения полимера. В спектре присутствовали сигналы нескольких аномерных углеродов при δ 98.2–105.6 м.д., двух HOCH2-групп остатков Glc и GlcNAc при δ 61.9 и 62.2 м.д., двух углеродов, связанных с азотом (C2 GlcNAc и C3 Fuc3NAc) при δ 52.3 и 56.8 м.д., нескольких CH3–C-групп (отвечающих C6-атомам остатков Fuc3NAc и Rha) при δ 16.6 и 17.4–18.0 м.д., других углеродов моносахаридных колец при δ 67.6–83.1 м.д., двух N-ацетильных групп (CH3 при δ 23.3 и 23.5 м.д., CO при δ 175.1 и 175.6 м.д.). Отсутствие сигналов в диапазоне δ 83–88 м.д., характеристичных для фуранозидов, свидетельствовало о том, что все моносахариды в составе ОПС являются пиранозидами. Соответственно, в 1H-ЯМР-спектре ОПС присутствовали сигналы нескольких аномерных протонов при δ 4.76–5.37 м.д., ряда CH3–C-групп (H6 Rha и Fuc3NAc) при δ 1.23–1.32 м.д., другие протоны моносахаридных колец при δ 3.43–4.50 м.д., двух N-ацетильных групп при δ 2.06 м.д. Учитывая схожесть моносахаридного состава ОПС A. rugosum с составом ОПС A. brasilense Jm125A2, установленным нами ранее [21], мы провели сравнение 1D- и 2D-ЯМР-спектров ОПС этих штаммов. В результате сравнения была выявлена практически полная идентичность мажорных серий сигналов в спектрах, что свидетельствовало об идентичности повторяющихся звеньев ОПС. На основании этих данных сигналы в спектрах 1H- и 13С-ЯМР были отнесены с использованием 1H,1H COSY, TOCSY, ROESY, и 1H,13С HSQC-экспериментов (табл. 1).
Рис. 1.
13С-ЯМР-спектры ОПС A. rugosum DSM-19657 (вверху) и A. brasilense Jm125A2 (внизу). Арабские цифры относятся к атомам углерода в моносахаридных остатках, обозначенных в табл. 1 и в тексте.
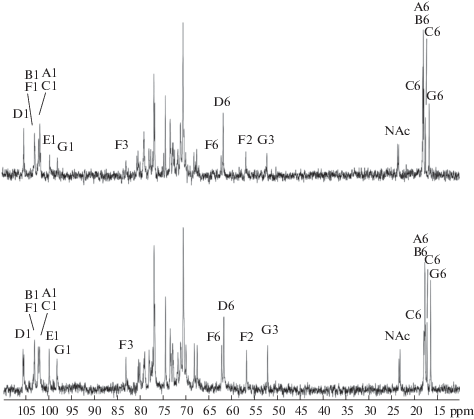
Таблица 1.
Данные 1H- и 13C-ЯМР-спектров ОПС A. rugosum (δ, м.д.)
| Моносахаридный остаток | H1 | H2 | H3 | H4 | H5 | H6 (a; b) | NAc |
|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | ||
| Повторяющееся звено 1 | |||||||
| →2,3)-α-L-Rhap-(1→ (A) | 5.37 102.4 |
4.20 80.5 |
4.02 78.0 |
3.66 73.6 |
3.88 70.6 |
1.31 17.8 |
|
| →3)- α-L-Rhap-(1→ (B) | 4.97 103.3 |
4.17 71.3 |
3.86 79.3 |
3.57 72.8 |
3.75 70.7 |
1.27 17.8 |
|
| →2)-α-L-Rhap-(1→ (C) | 5.22 102.1 |
4.08 79.3 |
3.91 71.5 |
3.50 73.7 |
3.79 70.8 |
1.32 18.0 |
|
| β-D-Glcp-(1→ (D) | 4.58 105.6 |
3.35 74.5 |
3.50 76.9 |
3.43 70.7 |
3.43 77.1 |
3.74, 3.86 61.9 |
|
| Повторяющееся звено 2 | |||||||
| →2,3)-α-L-Rhap-(1→ (E) | 4.89 99.8 |
3.98 77.2 |
3.96 78.1 |
3.66 72.9 |
3.99 70.7 |
1.25 17.4 |
|
| →3)-β-D-GlcpNAc-(1→ (F) | 4.79 103.2 |
3.82 56.8 |
3.62 83.1 |
3.48 70.1 |
3.43 77.2 |
3.78, 3.94 62.2 |
2.06 23.3, 175.1 |
| α-D-Fucp3NAc-(1→ (G) | 4.76 98.2 |
3.77 67.7 |
4.15 52.3 |
3.74 71.8 |
4.50 68.3 |
1.23 16.6 |
2.06 23.5, 175.5 |
Таким образом, ОПС A. rugosum состоит из повторяющихся звеньев двух типов, идентичных по структуре A. brasilense Jm125A2:

В составе липида А методом ГЖХ метиловых эфиров жирных кислот (ЖК) были выявлены преобладающие 14:0 (3-OH)-, 16:0 (3-OH)- и 18:1-кислоты, на долю которых приходилось более 85% от суммы идентифицированных ЖК. Также были обнаружены 16:0- (11%), 16:1- (2%) и 19:0- (1%) кислоты. Природа О-связанных жирных кислот была установлена с применением мягкого щелочного гидролиза в 12.5% NH4OH, в условиях которого эфирные ацильные и ацилоксиацильные группы менее устойчивы, по сравнению с ациламидными и ацилоксиациламидными [22]. В результате О-дезацилирования наблюдалось снижение содержания ЖК 14:0 (3-OH) по отношению к другим кислотам, что свидетельствовало о эфирном типе связи этой первичной кислоты, а вторичные кислоты ацилируют амидосвязанную ЖК 16:0 (3-OH). Методом ГЖХ ацетилированных метилгликозидов в составе липида А были идентифицированы GlcN и GalA в соотношении ~2 : 1. Полученные данные указывали на сходство строения липида А A. rugosum и A. lipoferum SpBr17 [20], для последнего продемонстрировано наличие трисахаридного остова следующей структуры: GlcpN(1→6)GlcpN(1↔1)GalpA, в котором оба остатка GlcpN ацилированы кислотой 16:0 (3-OH) в положении 2 и кислотой 14:0 (3-OH) в положениях 3 и 3', а остатки вторичных кислот этерифицируют первичную ЖК 16:0 (3-OH) в положении 2'. Учитывая консервативность строения липида А в пределах бактериального рода, мы приняли, что сахарный остов липида А A. rugosum идентичен таковому A. lipoferum.
МАЛДИ-масс-спектр отрицательных ионов липида А, записанный в режиме рефлектрона, содержал несколько групп сигналов депротонированных молекул [M – H]– в диапазоне m/z 1023.7–1756.1, отличающихся природой и количеством остатков жирных кислот (рис. 2). В спектре присутствовали три обособленные группы пиков с наиболее интенсивными сигналами в каждой группе при m/z 1287.9, 1514.0 и 1740.1 (рис. 3), которые соответствовали три-, тетра- и пентаацильным типам молекул, содержащих остаток GalA и два остатка GlcN, ацилированных кислотами 16:0 (3-OH), 14:0 (3-OH) и 18:1 (табл. 2). Разница в массах 26 и 16 Да между сигналами в группах была обусловлена различной длиной вторичных жирных кислот и соответствовала наличию идентифицированных ГЖХ-анализом остатков насыщенных кислот 16:0 и 19:0 вместо ненасыщенной 18:1 (табл. 2). Разница между наиболее интенсивными сигналами пента-, тетра- и триацильных типов составляла 226 Да, что соответствовало одному остатку первичной кислоты 14:0 (3OH). Сигнал при m/z 1023.7, отличающийся на 264 Да от наиболее интенсивного сигнала триацильного липида А, соответствовал диацильному типу, несущему только остатки 16:0 (3-OH) и лишенному остатка вторичной кислоты 18:1. Отсутствие в спектре сигналов с разницей 80 Да свидетельствовало об отсутствии фосфорилирования липида А.
Рис. 2.
МАЛДИ-масс-спектры липида А A. rugosum, записанные в режиме детектирования отрицательных (вверху) и положительных (внизу) ионов.


Таблица 2.
Результаты масс-спектрометрии и предполагаемый состав жирных кислот липида А A. rugosum
| [M – H]– эксп. (Да) | [M – H]– расч. (Да) | [M + Na]+ эксп. (Да) | [M + Na]+ расч. (Да) | [M – H + 2Na]+ эксп. Да) | [M – H + 2Na]+ расч. (Да) | Состав ЖК |
|---|---|---|---|---|---|---|
| Триацильные типы | ||||||
| 1261.919 | 1261.8518 | 1285.816 | 1285.8483 | Н.о. | 1307.8303 | 2 × 16:0 (3OH) 1 × 16:0 |
| 1287.942 | 1287.8675 | 1311.794 | 1311.8640 | Н.о. | 1333.8459 | 2 × 16:0 (3OH) 1 × 18:1 |
| 1303.913 | 1303.88988 | 1327.850 | 1327.8953 | Н.о. | 1349.8772 | 2 × 16:0 (3OH) 1 × 19:0 |
| Тетраацильные типы | ||||||
| 1488.064 | 1488.0451 | 1511.993 | 1512.0416 | Н.о. | 1534.0235 | 2 × 16:0 (3OH) 1 × 14:0 (3OH) 1 × 16:0 |
| 1514.041 | 1514.0607 | 1538.007 | 1538.0572 | 1559.987 | 1560.0392 | 2 × 16:0 (3OH) 1 × 14:0 (3OH) 1 × 18:1 |
| 1530.037 | 1530.0920 | 1554.010 | 1554.0885 | 1576.006 | 1577.1080 | 2 × 16:0 (3OH) 1 × 14:0 (3OH) 1 × 19:0 |
| Пентаацильные типы | ||||||
| 1714.101 | 1714.2384 | 1738.201 | 1738.2349 | 1760.199 | 1761.2246 | 2 × 16:0 (3OH) 2 × 14:0 (3OH) 1 × 16:0 |
| 1740.108 | 1740.2540 | 1764.225 | 1764.2505 | 1786.207 | 1787.2403 | 2 × 16:0 (3OH) 2 × 14:0 (3OH) 1 × 18:1 |
| 1756.082 | 1756.2853 | 1780.209 | 1780.2818 | 1802.187 | 1803.2716 | 2 × 16:0 (3OH) 2 × 14:0 (3OH) 1 × 19:0 |
МАЛДИ-масс-спектр положительных ионов липида А, записанный в режиме рефлектрона, содержал соответствующие сигналы натриевых аддуктов [M + Na]+ и [M – H + 2Na] + для три-, тетра- и пентаацильных типов липида А в диапазоне m/z 1285.8–1802.2 (рис. 2, табл. 2).
Для подтверждения строения липида А и доказательства положения вторичных кислот были записаны МС/МС-спектры наиболее интенсивных сигналов в режиме детектирования положительных и отрицательных ионов. Спектры второго порядка [M – H]-иона с m/z 1514.0 (рис. 3) подтверждали предполагаемое строение липида А и содержали сигналы осколочных ионов, указывающие на последовательное элиминирование остатков 14:0(3-OH)- и 18:1-ЖК. Отщепление вторичной кислоты 18:1 происходило в виде нейтральной молекулы, так как в спектре присутствовали только сигналы с характеристичной разницей масс 282 Да, но не наблюдалось таковых, возникающих в ходе отщепления кетена и альдегида с разницами масс 264 и 222 Да, соответственно [23].
Элиминирование остатка ЖК 14:0(3-OH) происходило различными путями, о чем свидетельствовало присутствие в спектре сигналов с разницей 244, 226 и 184 Да. Максимальная разница масс 244 Да соответствовала отщеплению кислоты в виде нейтральной молекулы, отличие в 226 Да отвечало потере кетена, а 184 Да – альдегида, протекающему через перегруппировку МакЛафферти. Хотя последний тип фрагментации наиболее часто упоминается для N-связанных ЖК [24, 25], он отмечен и для ЖК-остатков, связанных сложноэфирной связью [26].
В средней области МС/МС-спектра [M – H]–-иона с m/z 1514.0 наблюдались сигналы при m/z 987.8, 1005.6 и 1047.7 (рис. 3), соответствующие отщеплению одного ацильного остатка 18:1 и одного остатка 14:0(3-OH) от родительского иона (–(282 + 244), –(282 + 226), –(282 + 184) Да, соответственно). Образование промежуточного продукта фрагментации с m/z 742.3 происходило в результате потери родительским ионом с m/z 1514.0 остатка 18:1 в виде нейтральной молекулы (–282 Да) с образованием осколка с m/z 1232.0 и его последующей диссоциации по пути 0.2A2 (здесь и далее используется номенклатура фрагментации углеводов Домон и Кастелло [27]). Диацильный фрагмент с m/z 742.3 (рис. 3) в ходе дальнейшего расщепления теряет остаток 14:0(3-OH) в виде нейтральной ЖК (–244 Да) с образованием иона при m/z 498.3.
Последовательное отщепление остатка N-связанной ЖК 16:0(3-OH) в виде кетена и воды от осколка с m/z 1005.6 приводило к образованию фрагмента с m/z 731.7. В результате дегидратации последнего происходило образование иона при m/z 713.7, а его фрагментация по пути 0,4A2 приводила к возникновению осколка с m/z 456.2, отщепление гликольальдегида (–60 Да) от которого, предположительно, приводило к образованию иона, детектируемого при m/z 396.2. В области низкомолекулярных продуктов МС/МС-спектра также присутствовали ионы [M – H]– депротонированных молекул 14:0(3-OH)- и 18:1-ЖК при m/z 243.3 и 281.2 соответственно.
Предполагаемые структуры и пути фрагментации [M – H]–-иона с m/z 1514.1 липида А A. rugosum в МС/МС-спектрах приведены на рис. 4. МС/МС-спектр иона [M – H]– при m/z 1287.9 содержал характеристичные осколки при m/z 1005.6, 731.7, 456.2, образование которых происходило по аналогии с приведенной схемой.
Рис. 4.
Предполагаемые структуры и пути фрагментации [M – H]-иона липида А с m/z 1514.0. Структуры изображены в виде нейтральных молекул.

Таким образом, были отнесены наиболее интенсивные сигналы в МС/МС-спектре [M – H]–-ионов. Следует отметить, что представленная на рисунке схема фрагментации [M – H]–-иона тетраацильного липида А при m/z 1514.0, в котором 14:0(3-OH) находится в положении 3 проксимального остатка GlcN будет также справедлива и для липида А, в котором ЖК 14:0(3-OH) замещает положение 3 дистального кольца GlcN. В то же время, о положении вторичной кислоты 18:1, ацилирующей 16:0(3-OH) дистального звена GlcN, однозначно свидетельствовало наличие иона [0.4A2-H]– при m/z 456.2, поскольку фрагментация альтернативного по структуре иона, в котором ЖК 18:1 связана с 16:0(3-OH)-кислотой проксимального GlcN, не приводила бы к образованию осколков, наблюдаемых в МС/МС-спектрах.
МС/МС-спектр иона [M + Na]+ при m/z 1738.2 (рис. 5), соответствующего пентаацильному типу липида А, несущего по два остатка 14:0(3-OH) и 16:0(3-OH), а также один остаток 16:0, содержал характеристичные сигналы осколков, образующихся в результате последовательного отщепления GalA по путям С2 и B2 [27] с разницей масс 176 и 194 Да соответственно, остатка 14:0(3-OH) в виде кислоты или кетена (–244 или –226 Да), остатка 16:0 (–256 Да) и фрагмента 16:0(3-OH) в виде альдегида (–212 Да) в диапазоне m/z 605.0–1562.1. Серия наиболее интенсивных сигналов осколочных ионов, получающихся в ходе данного пути фрагментации, присутствовали в рассматриваемом спектре в диапазоне m/z 1073.5–1562.1 и соответствовали потере одного, двух или трех вышеуказанных фрагментов. Менее интенсивные сигналы [M + Na]+ при m/z 817.4, 831.2 и 861.3 соответствовали отщеплению четырех фрагментов, а ион при m/z 605.0 отвечал элиминированию GalA, двух остатков 14:0(3-OH), а также по одному остатку 16:0(3-OH) и 16:0 от родительского иона.

В средней области МС/МС-спектра присутствовали интенсивные сигналы осколка [0,4A2 + Na]+ при m/z 962.4, дальнейшая фрагментация которого сопровождалась последовательным элиминированием остатков 16:0 и 14:0(3-OH) с образованием осколка с m/z 461.8. МС/МС-спектр также содержал менее интенсивный сигнал иона при m/z 902.5, соответствующего продукту B1 фрагментации по гликозидной связи. В результате отрыва от этого фрагмента остатков 14:0(3-OH) (–226 и –244 Да) и 16:0 (–256 Да) наблюдалось образование ионов [M + Na]+ при m/z 646.2, 658.2 и 676.2 соответственно (рис. 6).
Рис. 6.
Предполагаемые структуры и пути фрагментации [M + Na]+-иона при m/z 1738.2. Структуры изображены в виде натриевых аддуктов нейтральных молекул.

На основании комплексного анализа химического состава и МАЛДИ-масс-спектров была установлена структура липида А A. rugosum (рис. 7), который представляет собой смесь молекул пента-, тетра- и триацильных типов, отличающихся друг от друга количеством остатков первичной О-связанной 14:0(3-OH). Микрогетерогенность в пределах каждого из типов липида А с разной степенью ацилирования, обусловлена природой вторичных жирных кислот, ацилирующих остаток N-связанной 16:0(3-OH) кольца дистального GlcN. Анализ масс-спектров второго порядка при установлении структуры липида А A. rugosum продемонстрировал распределение вторичных жирных кислот, аналогичное таковому для A. lipoferum SpBr17, предложенному ранее на основе данных масс-спектрометрии первого порядка. Отличительной особенностью липида А A. rugosum является присутствие остатков вторичной ЖК 19:0, не идентифицированной в липиде А штамма SpBr17.

Таким образом, на основании данных химического анализа, спектроскопии ЯМР и МАЛДИ-масс-спектрометрии для типового штамма нового вида бактерий A. rugosum было установлено сходство строения ОПС и липида А с таковыми компонентами изученных ранее видов A. brasilense и A. lipoferum. Межвидовая распространенность близких по строению повторяющихся звеньев ОПС продемонстрирована ранее и для других представителей рода Azospirillum [15] и может свидетельствовать об отсутствии видовой приуроченности в отношении структуры этих традиционно вариабельных компонентов ЛПС, которая, скорее всего, определяется особенностями экологической ниши обитания. Сходство строения липида А A. rugosum и A. lipoferum позволяет предположить наличие близкородственных структур у других представителей данного рода.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Культивирование бактерий. Штамм Azospirillum rugosum DSM-19657 (IBPPM 629) предоставлен Коллекцией ризосферных микроорганизмов ИБФРМ РАН (г. Саратов). Культивирование бактерий проводили в жидкой малатно-солевой среде с витаминами [28] до окончания экспоненциальной фазы роста при температуре 30°С и перемешивании на вибростенде. Клетки осаждали центрифугированием, ресуспендировали в 0.15 М растворе NaCl и смывали с поверхности капсульный материал механическим перемешиванием в течение 5 сут с ежедневной сменой отмывающего раствора.
Выделение ЛПС, ОПС и липида А. ЛПС выделяли из высушенных ацетоном бескапсульных клеток горячим 45% водным раствором фенола без разделения слоев [29]. Примеси белков осаждали из раствора ЛПС добавлением 40% CCl3CO2H до конечного значения рН 2.7. Деградацию ЛПС проводили 2% CН3CO2H при 100°С в течение 4 ч. Осадок липида А отделяли центрифугированием, промывали водой и трижды обрабатывали смесью CHCl3 : CH3OH : H2O (10 : 10 : 9, v/v) [30]. Супернатант, содержащий ОПС, разделяли гель-хроматографией на колонке с Toyopearl TSK HW-50 (Toyosoda, Япония) в 1% AcOH, контролируя элюцию с помощью дифференциального проточного рефрактометра Knauer (Германия). Фракцию высокомолекулярного О-специфического полисахарида концентрировали и лиофилизовали.
О-Дезацилированние ЛПС проводили в 12.5% NH4OH при 37°С в течение 16 ч. Модифицированный ЛПС выделяли гель-фильтрацией на колонке с Toyopearl TSK HW-40 (Toyosoda, Япония) в 1% AcOH.
Моносахаридный анализ. Анализ моносахаридного состава и абсолютных конфигураций сахаров проводили методом ГЖХ ацетатов полиолов [31], ацетилированных метилгликозидов и ацетилированных 2-(S)-октилгликозидов [32] на хроматографе Hewlett-Packard 7820А с капиллярной колонкой HP-5 (Hewlett-Packard, США). Градиент температуры от 160°C (1 мин) до 290°C, скорость нагрева 7°C/мин.
Состав жирных кислот. Состав жирных кислот ЛПС в виде метиловых эфиров жирных кислот определяли с помощью ГЖХ на хроматографе GС-2010 (Shimadzu, Япония), снабженном колонкой DB-5 (Agilent, США). Метилирование выполняли методом, описанным в работе [33].
ЯМР-спектроскопия. Спектры ЯМР записывали на спектрометре DRX-700 (“Bruker”, Германия) в растворе 99.96% D2O при 30°С (внутренний стандарт – ацетон, δC 31.45, и 3-триметилсилилпропаноат-d4, δH 0.0. Образцы предварительно лиофилизовали дважды из 99.9% D2O. Двумерные спектры записывали с использованием стандартного математического обеспечения (компании “Bruker”, Германия); для сбора и обработки данных использовали программу TOPSPIN 2.1. В экспериментах TOCSY и NOESY время смешивания составляло 150 и 200 мс соответственно.
МАЛДИ-масс-спектрометрия. К 1 мкл раствора липида А (1 мг/мл) добавляли 3 мкл раствора 2,5-дигидроксиацетофенона (10 мг/мл, CH3CN : H2O, 1 : 1 по объему), 1 мкл полученной смеси точечно наносили на стандартные планшеты для МАЛДИ и высушивали на воздухе. Масс-спектрометрию осуществляли на времяпролетном МАЛДИ-масс-спектрометре Autoflex speed компании “Bruker Daltonics” (Германия), снабженном азотным лазером (337 нм), в режиме рефлектрона с использованием программного обеспечения FlexControl (версия 3.4; Bruker Daltonics). Спектры записывали в режиме детектирования положительных и отрицательных ионов. МС/МС-спектры записывали в режиме диссоциации, индуцированной соударениями (CID). Для внешней калибровки масс использовали смесь сульфатированных олигосахаридов [34]. Обработку полученных результатов проводили с использованием программного обеспечения Flexanalysis (версия 3.4; Bruker Daltonics). МС-спектры получали аккумуляцией 1500 выстрелов лазера, МС/МС-спектры – 6000–7000 выстрелов при минимальной энергии лазера, требующейся для ионизации образца.
Список литературы
Steenhoudt O., Vanderleyden J. // FEMS Microbiol. Rev. 2000. V. 24. P. 487–506.
Bashan Y., de-Bashan L.E. // Adv. Agron. 2010. V. 108. P. 77–136.
Fukami J., Cerezini P., Hungria M. // AMB Express. 2018. V. 8. P. 73.
Fibach-Paldi S., Burdman S., Okon Y. // FEMS Microbiol. Lett. 2012. V. 326. P. 99–108.
Skvortsov I.M., Ignatov V.V. // FEMS Microbiol. Lett. 1998. V. 165. P. 223–229.
Knirel Y.A. // Bacterial Lipopolysaccharides / Eds. Knirel Y.A., Valvano M.A. Wien: SpringerWienNewYork, 2011. P. 41–115.
Molinaro A., Holst O., Di Lorenzo F., Callaghan M., Nurisso A., D’Errico G., Zamyatina A., Peri F., Berisio R., Jerala R., Jiménez-Barbero J., Silipo A., Martín-Santamaría S. // Chemistry. 2015. V. 21. P. 500–519.
Pereg L., Luz E., Bashan Y. // Plant Soil. 2016. V. 399. P. 389–414.
Lin S.Y., Young C.C., Hupfer H., Siering C., Arun A.B., Chen W.M., Lai W.A., Shen F.T., Rekha P.D., Yassin A.F. // Int. J. Syst. Evol. Microbiol. 2009. V. 59. P. 761–765.
Lavrinenko K., Chernousova E., Gridneva E., Dubinina G., Akimov V., Kuever J., Lysenko A., Grabovich M. // Int. J. Syst. Evol. Microbiol. 2010. V. 60. P. 2832–2837.
Yang Y., Zhang R., Feng J., Wang C., Chen J. // Int. J. Syst. Evol. Microbiol. 2019.
Anandham R., Heo J., Krishnamoorthy R., SenthilKumar M., Gopal N.O., Kim S.J., Kwon S.W. // Int. J. Syst. Evol. Microb-iol. 2019. V. 69. P. 1369–1375.
Zhou S., Han L., Wang Y., Yang G., Zhuang L., Hu P. // Int. J. Syst. Evol. Microbiol. 2013. V. 63. P. 2618–2624.
Young C.C, Hupfer H., Siering C., Ho M.J., Arun A.B., Lai W.A., Rekha P.D., Shen F.T., Hung M.H., Chen W.M., Yassin A.F. // Int. J. Syst. Evol. Microbiol. 2008. V. 58. P. 959–963.
Муратова А.Ю., Турковская О.В., Антонюк Л.П., Макаров О.Е., Позднякова Л.И., Игнатов В.В. // Микробиология. 2005. Т. 74. № 2. С. 248–254.
Федоненко Ю.П., Сигида Е.Н., Коннова С.А., Игнатов В.В // Изв. АН. Сер. хим. 2015. Т. 64. № 5. С. 1024–1031.
Sigida E.N., Fedonenko Y.P., Shashkov A.S., Arbatsky N.P., Zdorovenko E.L., Konnova S.A., Ignatov V.V., Knirel Y.A // Beilstein J. Org. Chem. 2016. V. 12. P. 636–642.
Sigida E.N., Fedonenko Y.P., Shashkov A.S., Zdorovenko E.L., Konnova S.A., Knirel Y.A. // Carbohydr. Res. 2019. V. 478. P. 54–57.
Sigida E.N., Fedonenko Y.P., Shashkov A.S., Konnova S.A., Ignatov V.V. // Carbohydr. Res. 2018. V. 465. P. 40–43.
Choma A., Komaniecka I. // Carbohydr. Res. 2008. V. 343. P. 799–804.
Sigida E.N., Fedonenko Y.P., Shashkov A.S., Zdorovenko E.L., Konnova S.A., Ignatov V.V., Knirel Y.A. // Carbohydr. Res. 2015. V. 416. P. 37–40.
Silipo A., Lanzetta R., Amoresano A., Parrilli M., Molinaro A. // J. Lipid Res. 2002. V. 43. P. 2188–2195.
Murphy R.C., Axelsen P.H. // Mass Spectrom. Rev. 2011. V. 30. P. 579–599.
Phillips N.J., Schilling B., McLendon M.K., Apicella M.A., Gibson B.W. // Infect. Immun. 2004. V. 72. P. 5340–5348.
Schilling B., McLendon M.K., Phillips N.J., Apicella M. A., Gibson B.W. // Anal. Chem. 2007. V. 79. P. 1034–1042.
Shaffer S.A., Harvey M.D., Goodlett D.R., Ernst R.K. // J. Am. Soc. Mass Spectrom. 2007. V. 18. P. 1080–1092.
Domon B, Costello C. // Glycoconjugate J. 1988. V. 5. P. 397–409.
Konnova S.A., Makarov O.E., Skvortsov I.M., Ignatov V.V. // FEMS Microbiol. Lett. 1994. V. 118. P. 93–99.
Westphal O., Jann K. // Methods Carbohydr. Chem. 1965. V. 5. P. 83–91.
Que N.L.S., Ribeiro A.A., Raetz C.R.H. // J. Biol. Chem. 2000. V. 275. P. 28017–28027.
Sawardeker J.S., Sloneker J.H., Jeanes A. // Anal. Chem. 1965. V. 37. P. 1602–1603.
Leontein K., Lindberg B., Lönngren J. // Carbohydr. Res. 1978. V. 62. P. 359−362.
Mayer H., Merkofer T., Warth C., Weckesser J. // J. Endotox. Res. 1996. V. 3. P. 345−352.
Шевченко Н.М., Анастюк С.Д., Герасименко Н.И., Дмитренок П.С., Исаков В.В., Звягинцева Т.Н. // Биоорг. хим. 2007. Т. 33. № 1. С. 96−107.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия