

Биоорганическая химия, 2020, T. 46, № 3, стр. 332-336
Синтез и оптические свойства нового ацетиленового аналога хромофора белка Kaede
Э. Р. Зайцева 1, 2, А. Ю. Смирнов 1, А. С. Мишин 1, М. С. Баранов 1, 3
1 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 Российский химико-технологический университет имени Д.И. Менделеева
125047 Москва, Миусская площадь, 9, Россия
3 Российский национальный исследовательский медицинский университет имени Н.И. Пирогова
117997 Москва, ул. Островитянова, 1, Россия
Поступила в редакцию 22.11.2019
После доработки 29.11.2019
Принята к публикации 26.12.2019
Аннотация
В результате использования модифицированной реакции Соногаширы впервые синтезирован ацетиленовый аналог хромофора белка Kaede – (Z)-2-((4-(диэтиламино)фенил)этинил)-5-(4-гидроксибензилиден)-3-метил-3,5-дигидро-4H-имидазол-4-он. Изучение оптических свойств данного соединения установило, что максимумы абсорбции и эмиссии сдвинуты в длинноволновую область по сравнению с классическим хромофором GFP, а стоксов сдвиг достигает почти 150 нм.
Ключевыми из современных подходов к изучению процессов, протекающих в живых клетках, являются методы визуализации, связанные с испусканием света. Важнейшей составляющей данных методов является введение в изучаемый объект люминесцентной либо флуоресцентной метки. Среди последних стоит отдельно отметить многочисленные производные хромофора зеленых флуоресцентных белков (GFP), содержащие в своем составе бензилиденимидазолон [1]. Данный класс соединений обладает большой вариативностью оптических свойств, значительным стоксовым сдвигом, хорошей растворимостью в воде, высокой проницаемостью через цитоплазматическую мембрану, низкой токсичностью и синтетической доступностью [2–4].
Для биологических объектов характерно высокое поглощение коротковолнового излучения (которое зачастую нарушает протекающие биологические процессы) и низкое поглощение длинноволнового. Поэтому при проведении биомедицинских исследований предпочтительным является использование меток с максимумами поглощения и эмиссии флуоресценции в длинноволновой части видимого спектра. Одной из основных синтетических стратегий достижения батохромного сдвига является увеличение размера сопряженной π-системы молекулы хромофора. Примером такого подхода в случае бензимидазолонов являются аналоги хромофора белка Kaede, содержащие в своем составе стирольный заместитель во втором положении имидазолонового цикла [5]. Данные соединения уже используются как флуоресцентные [6, 7] или флуорогенные (вещества, увеличивающие квантовый выход флуоресценции при связывании с целевым объектом) метки [8, 9] в биомедицинских исследованиях. Однако, за все время изучения аналогов хромофора белка Kaede не были получены их ацетиленовые производные. В настоящей работе мы впервые разработали подход к синтезу данного класса соединений и на одном примере рассмотрели их доступность и оптические свойства.
В качестве целевого соединения нами было выбрано вещество (V) (рис. 1). Одним из самых распространенных методов синтеза ацетиленов является реакция Соногаширы, ее применение предполагает вовлечение в реакцию терминального алкина и арил- либо гетарилгалогенида. В литературных источниках не описаны 2-галогенимидазолоны, однако существуют примеры проведения реакции Соногаширы с участием алкилтиопроизводных, в которых происходит катализируемое комплексами палладия замещение серосодержащей группы на ацетилен [10, 11]. Также существует несколько примеров модифицирования тиогидантоинов [12] и 2-алкилтиоимидазолонов [13] в результате реакции Либскайнда-Срогля. Поэтому для синтеза целевого соединения мы выбрали модифицированную методику получения ацетиленов реакцией Соногаширы с десульфированием соответствующего тиогидантоинового производного.

Соединение (IV) было синтезировано с использованием достаточно редкой модификации реакции Соногаширы с использованием производного, содержащего группу SCH3 [14]. В качестве катализатора были использованы йодид меди (I) и Pd(dppf)Cl2 · CH2Cl2. В начале в результате конденсации 4-гидроксибензальдегида с 2-тиооксоимидазолин-4-оном было получено соединение (I). Так как алкилирование тиооксоимидазолина могло протекать и с участием гидрокси-группы бензилиденового фрагмента, следующей стадией стала ее защита триизопропилсилильной группой. Полученное в результате соединение (II) метилировали метилйодидом с образовaнием продукта (III). После этого проводилась реакция Соногаширы, в результате чего было выделено соединение (IV) с ацетиленовым фрагментом в структуре. Для удаления триизопропилсилильной защиты соединение (IV) обрабатывали фторидом тетрабутиламмония. В результате нами было получено новое соединение (V) с хорошим выходом.
Далее были определены оптические свойства нового соединения (V) в ацетонитриле (рис. 1). Было обнаружено, что эмиссия флуоресценции соединения (V) невелика, что согласуется с полученными ранее данными для других аналогов хромофора белка Kaede [6]. Максимумы абсорбции (457 нм) и эмиссии (примерно 605 нм) соединения (V) сдвинуты в значительно более длинноволновую область по сравнению с классическим хромофором GFP (Z)-5-(4-гидроксибензилиден)-2,3-диметил-3,5-дигидро-4H-имидазол-4-оном (максимумы абсорбции и эмиссии 368 и 438 нм соответственно) [8]. Также обращает на себя внимание тот факт, что стоксов сдвиг нового ацетиленового производного составляет почти 150 нм, что делает полученный краситель перспективным в задачах многоцветного мечения биологических образцов.
Таким образом, нами был впервые разработан подход к синтезу ацетиленовых аналогов хромофора белка Kaede на примере соединения (V). Максимумы абсорбции и эмиссии данного соединения лежат в более длинноволновой области, чем у аналогичного хромофора GFP из-за наличия сопряженной ацетиленовой системы. Этот факт, а также низкий уровень флуоресценции данного соединения позволяют предполагать, что новое производное (V) или его аналоги могут оказаться перспективными флуорогенными красителями.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры ЯМР (δ м.д., J Гц) зарегистрированы на приборе Bruker Avance III (700 МГц, США) в DMSO-d6 (внутренний стандарт – Me4Si). Спектры поглощения в УФ и видимом диапазоне регистрировали на спектрофотометре Varian Cary 100 Bio (США). Спектры флуоресценции регистрировались на спектрофлуориметре Varian Cary Eclipse (США). Температуры плавления определены на приборе SMP 30 (Великобритания) и не исправлены. Масс-спектры высокого разрешения записаны на приборе Bruker micrOTOF II, ионизация электрораспылением.
(Z)-5-(4-Гидроксибензилиден)-2-тиооксоимидазолин-4-он (I). 4-Гидроксибензальдегид (793 мг, 6.5 ммоль), 2-тиооксоимидазолин-4-он (580 мг, 5.0 ммоль) и ацетат натрия (1.76 г, 21.5 ммоль) растворяли в 15 мл ледяной уксусной кислоты, полученную смесь кипятили в течение 3 ч и остужали до комнатной температуры. При интенсивном перемешивании добавляли 50 мл воды, выпавший осадок отфильтровывали и промывали водой (20 мл) и диэтиловым эфиром (10 мл), после чего сушили в вакууме. Желтый порошок (935 мг, 85%), т. пл. 280°C с разложением; 1H-ЯМР: 6.43 (с, 1 H), 6.81 (д, J2 8.6, 2 H), 7.63 (д, J2 8.6, 2 H), 10.02 (ушир. с., 1 H), 11.95 (ушир. с., 1 H), 12.22 (ушир. с., 1 H).
(Z)-2-Тиоксо-5-(4-((триизопропилсилил)окси)бензилиден)имидазолидин-4-он (II). (Z)-5-(4-Гидроксибензилиден)-2-тиооксоимидазолин-4-он (I, 1.10 г, 5.0 ммоль), триизопропилсилилхлорид (1.06 г, 5.5 ммоль), DIPEA (0.78 г, 6.0 ммоль) и имидазол (20 мг) растворяли в ТГФ (50 мл), после чего перемешивали в течение ночи. Полученный раствор упаривали, остаток растворяли в этилацетате (200 мл), промывали насыщенным раствором хлорида натрия (3 × 50 мл), сушили над безводным Na2SO4 и упаривали. Продукт очищали флеш-хроматографией (элюент – хлороформ). Желтый порошок (1.48 г, 79%), т. пл. 275°C с разложением; 1H-ЯМР: 1.08 (д, J2 7.4, 18 H), 1.25–1.31 (септ., J2 7.4, 3 H), 6.45 (с, 1 H), 6.89 (д, J2 8.6, 2 H), 7.68 (д, J2 8.6, 2 H), 12.03 (ушир. с., 1 H), 12.28 (ушир. с., 1 H); 13C-ЯМР: 11.9, 17.5, 111.6, 119.8, 125.4, 126.1, 132.0, 156.5, 165.6, 178.6; HRMS (ESI), m/z: найдено М, 377.1711; рассчитано для C19H29N2O2SSi+, [M + H]+ 377.1714.
(Z)-3-Метил-2-(метилтио)-5-(4-((триизопропилсилил)окси)бензилиден)-3,5-дигидро-4H-имидазол-4-он (III). (Z)-2-Тиооксо-5-(4-((триизопропилсилил)окси)бензилиден)имидазолидин-4-он (II, 1.13 г, 3.0 ммоль), йодметан (1.70 г, 12.0 ммоль) и поташ (1.66 г, 12.0 ммоль) смешивали в ацетонитриле (30 мл) и кипятили в течение 8 часов. Реакционную смесь остужали, фильтровали, раствор упаривали на три четверти и добавляли этилацетат (150 мл). Полученный раствор промывали насыщенным раствором хлорида натрия (3 × 30 мл), сушили над безводным Na2SO4 и упаривали. Продукт очищали флеш-хроматографией (элюент – хлороформ). Желтый порошок (820 мг, 69%), т. пл. 160–163°C; 1H-ЯМР: 1.07 (д, J2 7.3, 18 H), 1.27 (септ., J2 7.3, 3 H), 2.72 (с, 3 H), 3.08 (с, 3 H), 6.83 (с, 1 H), 6.94 (д, J2 8.7, 2 H), 8.16 (д, J = 8.8, 2 H); 13C-ЯМР: 12.1, 12.5, 17.7, 26.3, 120.0, 122.1, 126.5, 133.8, 136.7, 157.1, 161.7, 170.9; HRMS (ESI), m/z: найдено М, 405.2022; рассчитано для C21H33N2O2SSi+, [M + H]+ 405.2027.
(Z)-2-((4-(Диэтиламино)фенил)этинил)-3-метил-5-(4-((триизопропилсилил)окси)бензилиден)-3,5-дигидро-4H-имидазол-4-он (IV). В колбу Шленка насыпали (Z)-3-метил-2-(метилтио)-5-(4-((триизопропилсилил)окси)бензилиден)-3,5-дигидро-4H-имидазол-4-он (III, 406 мг, 1 ммоль), йодид меди(I) (190 мг, 1 ммоль) и комплекс (1,1'‑бис(дифенилфосфино)ферроцена хлорида палладия(II) с дихлорметаном (41 мг, 0.05 ммоль). Колбу вакуумировали, заполнили аргоном, в токе аргона добавляли DIPEA (2.58 г, 20 ммоль) и N,N-диэтил-4-этиниланилин (340 мг, 2 ммоль). Реакционную смесь выдерживали при температуре 100°С в течение 12 ч, затем добавляли этилацетат (100 мл) и промывали насыщенным раствором хлорида натрия (3 × 30 мл). Органическую фазу сушили над безводным Na2SO4 и упаривали. Продукт очищали флеш-хроматографией (элюент – смесь этилацетата и гексана, 1 : 5). Темный порошок (227 мг, 43%), т. пл. 166–168°C. Продукт частично (до 20%) загрязнен соединением (V), использован в следующей стадии без проведения полной очистки. 1H-ЯМР: 1.08 (д, J2 7.4, 18 H), 1.13 (септ., J2 7.1, 6 H), 1.30 (т, J2 7.6, 1 H), 3.22 (с, 3 H), 3.42 (кв., J2 7.1, 4 H), 6.75 (д, J2 9.0, 2 H), 6.97 (д, J2 8.8, 2 H), 7.02 (с, 1 H), 7.52 (д, J2 9.0, 2 H), 8.16 (д, J2 8.6, 2 H); HRMS (ESI), m/z: найдено М, 530.3198; рассчитано для C32H44N3O2Si+, [M + H]+ 530.3197.
(Z)-2-((4-(Диэтиламино)фенил)этинил)-5-(4-гидроксибензилиден)-3-метил-3,5-дигидро-4H-имидазол-4-он (V). (Z)-2-((4-(Диэтиламино)фенил)этинил)-3-метил-5-(4-((триизопропилсилил)окси)бензилиден)-3,5-дигидро-4H-имидазол-4-он (IV, 159 мг, 0.3 ммоль) растворяли в ТГФ (4 мл), добавляли фторид тетрабутиламмония тригидрата (474 мг, 1.5 ммоль), после чего полученный раствор перемешивали 1 ч. Добавляли этилацетат (100 мл) и промывали насыщенным раствором хлорида натрия (10 × 15 мл). Органическую фазу сушили над безводным Na2SO4 и упаривали. Продукт очищали флеш-хроматографией (элюент – смесь хлороформа и этанола, 50 : 1). Темно-красный порошок (64 мг, 57%), т. пл. 188–190°C; 1H-ЯМР: 1.12 (т, J2 7.0, 6 H, NCH2CH3), 3.21 (с, 3 H, NCH3), 3.41 (кв., J2 6.9, 4 H, NCH2CH3), 6.73 (д, J2 9.0, 2 H, H5', H7'), 6.87 (д, J2 8.8, 2 H, H3'', H5''), 6.99 (с, 1 H, H7''), 7.51 (д, J2 8.8, 2 H, H4', H8'), 8.10 (д, J2 8.6, 2 H, H2'', H6''), 10.24 (ушир. с., 1 H, OH); 13C-ЯМР: 12.3 (NCH2CH3), 27.0 (NCH3), 43.8 (NCH2CH3), 78.4 (C1'), 102.6 (C2'), 103.6 (C3'), 111.3 (C5', C7'), 115.9 (C3'', C5''), 125.4 (C7''), 127.4 (C1''), 134.2 (C2'', C6''), 134.6 (C4', C8'), 136.4 (C2), 146.2 (C5), 149.1 (C6'), 160.1 (C4''), 168.5 (C4); HRMS (ESI), m/z: найдено М, 374.1864; рассчитано для ${{{\text{C}}}_{{{\text{23}}}}}{{{\text{H}}}_{{{\text{24}}}}}{{{\text{N}}}_{{\text{3}}}}{\text{O}}_{2}^{ + },$ [M + H]+ 374.1863.
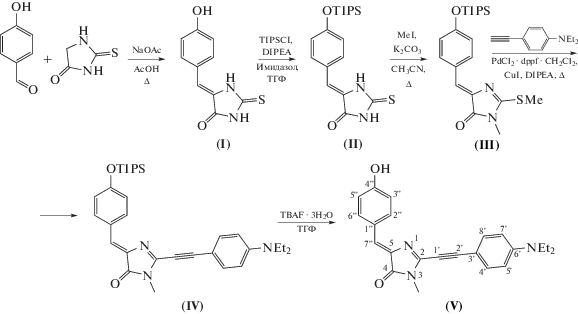
Схема 1. Схема синтеза соединения (V).
Список литературы
Walker C.L., Lukyanov K.A., Yampolsky I. V, Mishin A.S., Bommarius A.S., Duraj-Thatte A.M., Azizi B., Tolbert L.M., Solntsev K.M. // Curr. Opin. Chem. Biol. 2015. V. 27. P. 64–74.
Ge S., Deng H., Su Y., Zhu X. // RSC Adv. 2017. V. 7. P. 17 980–17 987.
Povarova N.V., Bozhanova N.G., Sarkisyan K.S., Gritcenko R., Baranov M.S., Yampolsky I.V., Lukyanov K.A., Mishin A.S. // J. Mater. Chem. C. 2016. V. 4. P. 3036–3040.
Feng G., Luo C., Yi H., Yuan L., Lin B., Luo X., Hu X., Wang H., Lei C., Nie Z., Yao S. // Nucl. Acids Res. 2017. V. 45. P. 10380–10392.
Yampolsky I.V., Kislukhin A.A., Amatov T.T., Shcherbo D., Potapov V.K., Lukyanov S., Lukyanov K.A. // Russ. J. Bioorg. Chem. 2008. V. 36. P. 96–104.
Baleeva N.S., Myannik K.A., Yampolsky I.V., Baranov M.S. // Eur. J. Org. Chem. V. 2015. P. 5716–5721.
Ermakova Y.G., Sen T., Bogdanova Y.A., Smirnov A.Y., Baleeva N.S., Krylov A.I., Baranov M.S. // J. Phys. Chem. Lett. 2018. V. 9. P. 1958–1963.
Povarova N.V., Zaitseva S.O., Baleeva N.S., Smirnov A.Y., Myasnyanko I.N., Zagudaylova M.B., Bozhanova N.G., Gorbachev D.A., Malyshevskaya K.K., Gavrikov A.S., Mishin A.S., Baranov M.S. // Chem. – A Eur. J. 2019. V. 25. P. 9592–9596.
Jung K.H., Fares M., Grainger L.S., Wolstenholme C.H., Hou A., Liu Y., Zhang X.A. // Org. Biomol. Chem. 2019. V. 17. P. 1906–1915.
Mehta V.P., Sharma A., Van der Eycken E. // Org. Lett. 2008. V. 10. P. 1147–1150.
Iwasaki M., Topolovčan N., Hu H., Nishimura Y., Gagnot G., Nanakorn R., Yuvacharaskul R., Nakajima K., Nishihara Y. // Org. Lett. 2016. V. 18. P. 1642–1645.
Gosling, S., Rollin, P., Tatibouët, A. // Synthesis. 2011. P. 3649–3660.
Oumouch S., Bourotte M., Schmitt M., Bourguignon J.-J. // Synthesis. 2005. P. 25–27.
Shook B.C., Chakravarty D., Jackson P.F. // Tetrahedron Lett. 2009. V. 50. P. 1013–1015.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия