

Биоорганическая химия, 2021, T. 47, № 4, стр. 526-536
Синтез изонитрильных производных диглицеридов и углеводов для многокомпонентной реакции Уги
А. И. Ничуговский 1, *, А. А. Хрулёв 1, К. А. Перевощикова 1, Д. А. Чешков 2, Н. Г. Морозова 1, М. А. Маслов 1
1 Институт тонких химических технологий им. М.В. Ломоносова (МИРЭА – Российский технологический университет)
119454 Москва, просп. Вернадского, 78, Россия
2 Государственный научно-исследовательский институт химии и технологии элементоорганических соединений
105118 Москва, ш. Энтузиастов, 38, Россия
* E-mail: ashpwnz77@gmail.com
Поступила в редакцию 15.09.2020
После доработки 28.09.2020
Принята к публикации 30.09.2020
Аннотация
В последнее время возрос интерес к многокомпонентным реакциям, главным образом из-за возможности собирать сложные органические молекулы всего за несколько шагов. Ключевой реагент многокомпонентной реакции Уги – изоцианид, проявляющий двойственные свойства – как электрофила, так и нуклеофила. Существует несколько способов формирования изонитрильных производных, но один из основных – внутримолекулярная дегидратация формамида. В данной работе мы осуществили синтез изонитрильных производных диалкилглицерина и D-глюкозы, которые при дальнейшем взаимодействии в условиях многокомпонентных реакций способны служить новыми полезными блоками для построения химических библиотек липофильных полиаминов, содержащих остаток углевода при терминальном атоме азота. Структуры всех синтезированных соединений были подтверждены физико-химическими методами анализа и могут быть использованы в качестве строительных блоков для многокомпонентной реакции Уги.
ВВЕДЕНИЕ
В последние годы возрос интерес к многокомпонентными реакциям, главным образом из-за возможности собирать сложные органические молекулы за несколько шагов [1]. Одна из них – четырехкомпонентная реакция Уги, в которой первичный амин, карбонильное соединение, карбоновая кислота и изоцианид взаимодействуют с образованием α‑ациламиноамидов. Карбонильное соединение и первичный амин требуются для формирования промежуточного имина.
Изоцианид – ключевой реагент четырехкомпонентной реакции Уги, обладающий уникальными реакционными свойствами, поскольку атом углерода изоцианидной группы способен действовать как нуклеофил и электрофил [2, 3]. Кроме того, электроноакцепторный характер изоцианидной группы приводит к повышенной кислотности α-протонов на атоме углерода, примыкающем к изоцианидной группе. Существует несколько способов создания изонитрильных групп: по реакции Хоффмана [4], дегидратацией соответствующего формамида (оксихлоридом фосфора [5], комбинацией трифенилфосфина с четырехбромистым углеродом [6] или иодом [7]), в реакции замещения галогенида на изоцианид [8, 9], с раскрытием оксирановых циклов [10], превращением алкенов в изонитрилы [11] и конверсией спиртов [12].
Ранее нами были синтезированы липофильные катионные полиамины, содержащие остаток диалкилглицерина (рис. 1), на основе подхода, включающего взаимодействие 2-нитробензолсульфонильных производных полиаминов с бромпроизводными диглицеридов [13, 14]. Мы показали, что эти соединения угнетают рост раковых клеток (аденокарциномы хронического миелогенного лейкоза человека K562, толстой кишки HCT116 и молочной железы MCF7) в микромолярных концентрациях [15]. Было установлено, что наличие алкильных заместителей при терминальной аминогруппе этих соединений снижает значение IC50 (концентрации, при которой происходит гибель 50% клеток в популяции) в среднем на 30%. Также было показано, что соединения (I–III) обладают низким индексом селективности относительно клеток фибробластов кожи человека в тех же условиях.

Известно, что нейтральные гликозилированные глицеролипиды (IV) и (V) также проявляют антипролиферативный эффект на линиях клеток K562, HCT116, меланомы человека B16 и острого промиелоцитарного лейкоза HL60 по апоптоз-независимому механизму [16]. Их высокая токсичность в отношении опухолевых клеток и отсутствие мутагенности указывают на возможность использования этих соединений как индивидуально, так и в комбинации с другими противоопухолевыми агентами. Введение в структуру гликоглицеролипидов положительно заряженных групп может увеличивать антипролиферативное действие этих соединений [17, 18]. Следует отметить, что углеводы широко используются для создания новых противоопухолевых агентов [19–22].
Согласно разработанной нами ретросинтетической схеме (схема 1 ), для синтеза липофильных полиаминов, содержащих при терминальном атоме азота углеводный фрагмент, используется 2-(гидроксиметил)бензойная кислота, которая во время финальной стадии реакции Уги (перегруппировки Мамма) подвергается элиминированию с образованием фталида [23]. Для реализации такой схемы необходимо получить изонитрильные производные диалкилглицерина и углевода. Следует отметить, что в литературе отсутствует метод синтеза изонитрильных производных диглицеридов.
Цель данной работы – разработка подходов к получению изонитрильных производных диглицерида и углеводов.

Схема 1 . Ретросинтетическая схема синтеза липофильных полиаминов с помощью реакции Уги.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Предварительно нами было опробовано несколько синтетических подходов (схема 2 ) к получению липофильных изонитрилов из модельного октадециламина (VI).
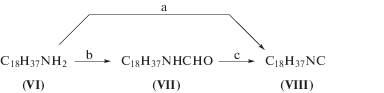
Схема 2 . Получение октадецилизонитрила. Реагенты и условия: a – NaOH, CHCl3, TBAB, DCM; b – HCOOCH2CN, DCM, DIPEA; c – 1) CBr4, Ph3P, TEA, DCM; 2) T3P®, TEA, EtOAc; 3) POCl3, TEA, DCM; 4) I2, Ph3P, TEA.
В первом подходе из исходного амина (VI) по реакции Хоффмана [4] получали изонитрил (VIII) в одну стадию с выходом 37% (схема 2 ). Данный метод – простой и удобный, однако выход недостаточен для получения целевых соединений. Второй подход основан на количественном получении 1-октадецилформамида (VII) действием (формилокси)ацетонитрила на амин (VI) и последующей трансформации формамида (VII) в изонитрил (VIII). Согласно первому способу, соединение (VII) вводили в реакцию с CBr4 и Ph3P в среде триэтиламина, получая изоцианид (VIII) с выходом 51% [24]. Во втором способе формамид (VII) обрабатывали ангидридом трипропилтриметафосфорной кислоты в безводном этилацетате, получая изоцианид с выходом 42% [25]. Третий способ включал взаимодействие соединения (VII) с хлорокисью фосфора в безводном дихлорметане в присутствии триэтиламина (выход 20%) [26], а четвертый – взаимодействие с молекулярным иодом и трифенилфосфином в присутствии триэтиламина [7]. Следует отметить, что только использование четвертого подхода позволило получить изоцианид (VIII) с выходом 72%, при этом сам метод характеризуется доступностью реагентов и легкостью масштабирования.
Структура октадецилизонитрила (VIII) была подтверждена данными спектроскопии 1H- и 13С-ЯМР. В спектре 1Н-ЯМР присутствуют сигналы протонов α-метиленового звена при изоцианидной группе (3.51 м.д.), а также сигналы протонов в сильном поле алкильного заместителя. В спектре 13С-ЯМР присутствуют характерные сигналы атомов углерода изоцианидной группы в виде триплета (δ = 155.9 м.д., J = 5.7 Гц), а также атомов углерода α-метиленового звена при изоцианидной группе (41.6 м.д.). Такой сигнал изоцианидного заместителя вызван симметричным распределением электронов вокруг атома азота, что приводит к спин-спиновому взаимодействию атомов 13С-14N.
Синтез изонитрильного производного 1-О-децил-2-О-этил-rac-глицерина осуществляли из rac-глицидола последовательным введением алкильных заместителей (схема 3 ). Исходный rac-глицидол (IX) обрабатывали 1.3-кратным избытком бензилбромида в присутствии гидрида натрия в DMF, получая после хроматографической очистки (система петролейный эфир–этилацетат, 85 : 15) соединение (X) с выходом 96%. Раскрытие оксиранового цикла соединения (X) проводили действием алкоголята децилового спирта, который предварительно получали при взаимодействии деканола с гидридом натрия. В ходе реакции также образовывался дидециловый эфир, имеющий близкую хроматографическую подвижность с соединением (XI), который не влиял на протекание следующей реакции. Этилирование вторичной гидроксильной группы соединения (XI) проводили этилбромидом в присутствии гидрида натрия. Соединение (XII) выделяли методом колоночной хроматографии на силикагеле, используя систему петролейный эфир – этилацетат (25 : 1), и получили его с выходом 30% (в расчете на соединение (X)). После удаления бензильной защиты гидрогенолизом в присутствии 5%Pd/C получали 1-О-децил-2-О-этил-rac-глицерин (XIII) с выходом 98%.

Схема 3 . Получение изонитрильного производного диглицерида. Реагенты и условия: a – BnBr, NaH, DMF; b – C10H21OH, NaH, THF; c – EtBr, NaH, THF; d – H2/Pd, EtOAc; e – CBr4, PPh3, СH2Cl2; f – NaN3, DMF; g – H2, 5% Pd/C, EtOAc; h – CH3COOCHO, DIPEA; i – 1) CBr4, Ph3P, TEA, DCM; 2) I2, Ph3P, TEA, DCM.
Последующую замену гидроксильной группы соединения (XIII) на атом брома проводили в условиях реакции Аппеля [27] действием CBr4 в присутствии трифенилфосфина и триэтиламина. Выход соединения (XIV) составил 99%. Поскольку хроматографическая подвижность бромида (XIV) сравнима с подвижностью азида (XV), протекание реакции введения азидной группы контролировали методом спектроскопии 13С-ЯМР, фиксируя исчезновение сигнала атома углерода CH2Br (δ = 32.7 м.д.) и появление сигнала атома углерода CH2N3-группы с химическим сдвигом 52.2 м.д. Восстановление азидной группы проводили в присутствии 5% Pd/C, получая амин (XVI) с выходом 95% на две стадии. Взаимодействие амина (XVI) с (формилокси)ацетонитрилом в присутствии DIPEA приводило к образованию формамида (XVII) с выходом 71%.
Синтез изонитрила (XVIII) проводили теми же способами, как описано для октадецилформамида (VII). Было замечено, что соединение (XVIII) образовывалось только при использовании в качестве реагентов смеси иода и трифенилфосфина в присутствии триэтиламина, во всех остальных случаях целевой продукт в реакционной смеси отсутствовал. На спектре 13С-ЯМР целевого изонитрила (XVIII) присутствуют сигналы атомов углерода алкильных заместителей в области сильного поля, глицеринового остова (66.2–76.0 м.д.), а также сигналы атомов углерода CH2N- (43.1 м.д.) и NC-групп (157.5 м.д.) в виде триплетных сигналов с J = 7.0 и 5.5 Гц соответственно, что подтверждает образование целевого изонитрила (XVIII).
Следующим этапом стала разработка синтеза 1-дезокси-1-изонитрил-2,3,4,6-О-тетраацетил-β-D-глюкопиранозы (XXIII) из ацетобромглюкозы (XIX) (схема 4 ), которая была получена по известной методике [28]. Взаимодействие бромида (XIX) с азидом натрия в DMF приводило к образованию соединения (XX), которое выделяли хроматографией на силикагеле в системе толуол–этилацетат (1 : 1) с выходом 90%. После восстановления азидной группы гидрогенолизом на катализаторе 5% Pd/C получали аминогликозид (XXI) с выходом 91%.

Схема 4 . Получение изонитрильного производного D-глюкозы. Реагенты и условия: a – NaN3, DMF; b – H2, 5%Pd/C, MeOH; c – CH3COOCHO, DIPEA; d – 1) CBr4, Ph3P, TEA, DCM; 2) I2, Ph3P, TEA, DCM.
В отличие от синтеза диалкилглицерина (XVII), формилирование 1-амино-1-дезокси-2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозы (XXI) действием (формилокси)ацетонитрила не приводило к образованию соединения (XXII). Литературный поиск показал, что возможна замена (формилокси)ацетонитрила на формилацетат [29]. Использование свежеприготовленного формилацетата, полученного при взаимодействии муравьиной кислоты с уксусным ангидридом, позволило получить амид (XXII) с выходом 82%.
Для синтеза изонитрила (XXIII) нами были опробованы два подхода. Согласно первому подходу, формамид (XXII) обрабатывали иодом в присутствии Ph3P и триэтиламина. После хроматографической очистки на силикагеле выход соединения (XXIII) составил 65%. Замена иода на CBr4 приводила к образованию изонитрила (XXIII) с выходом 60%.
Спектр 1Н-ЯМР изоцианида (XXIII) соответствует спиновой системе ABCDX, при этом сигнал аномерного протона представляет собой сложный мультиплет с сильно выраженными эффектами не первого порядка, которые также наблюдаются у сигналов протонов H-2, H-3 и H-4 из-за близости их резонансных частот, разность которых не превышает 6× КССВ между ними. Установление конфигурации аномерного протона основано на величинах КССВ, определение которых непосредственно из тонкой мультиплетной структуры сигналов не первого порядка невозможно и требует проведения анализа спектра по полной форме линии.
Поскольку анализ по полной форме линии спектров ЯМР основан на применении квантово-механического формализма для спиновых систем, сложность которого находится в экспоненциальной зависимости от числа спинов в спиновой системе, было решено применить селективную спиновую развязку для понижения числа спинов с 7 до 4. Был зарегистрирован спектр 1H{1H, sel)-ЯМР с развязкой от протона H-5 (Х) пиранозы и проанализирован в рамках спиновой системы ABCD по полной форме линии с помощью программы ANATOLIA [30] (рис. 2). В результате были определены уточненные значения химических сдвигов и КССВ для редуцированной спиновой системы. Исследование показало, что величина КССВ между аномерным протоном и протоном при атоме С-2 составляет 9.25 Гц, что указывает на β-конфигурацию изоцианида.

ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и оборудование. В работе использовали коммерчески доступные растворители российского (ООО “Химмед”, ООО “Компонент-Реактив”) и зарубежного производства (Sigma-Aldrich, США; Acros, США). Все реакции проводили под положительным давлением аргона с использованием растворителей класса HPLC (MeOH, DMF, CHCl3). Перед реакцией THF выдерживали над KOH, кипятили над натрием в присутствии бензофенона, DCM кипятили над CaH2, этилацетат кипятили над P2O5, перегоняли.
Ход реакции контролировали методом тонкослойной хроматографии (ТСХ) на пластинках Silica gel 60 F254 (Merck, Германия). Визуализацию пятен осуществляли в УФ-свете (254 нм) реактивом Драгендорфа, раствором фосфорномолибденовой кислоты – сульфат церия(IV) с последующим прогреванием. Колоночную хроматографию проводили на силикагеле Kieselgel 60 (0.040–0.063 мм или 0.063–0.200 мм; Merck, Германия).
Спектры 1Н- и 13С-ЯМР регистрировали на импульсных Фурье-спектрометрах Bruker DPX-300, Bruker Avance II 400, Bruker Avance II 600 (Bruker, Германия) в CDCl3. Химические сдвиги (δ) указаны в миллионных долях по отношению к пику остаточного протона растворителя, константы спин-спинового взаимодействия (J) – в Гц.
Масс-спектры регистрировали на масс-спектрометрах LCQ Deca XP Plus (Thermo Finnigan, США) и Advion Expression (Advion, США), оборудованных источниками ESI и детекторами ионной ловушки.
Октадецилформамид (VII). К 0.1 М раствору октадециламина (VI) (1.08 г, 4.0 ммоль) в безводном DCM добавляли (формилокси)ацетонитрил (0.43 мл, 6.0 ммоль) и перемешивали 2 ч при 50°С. Реакционную смесь охлаждали до 24°С, растворитель удаляли под вакуумом, остаток хроматографировали на силикагеле, элюируя системой петролейный эфир – этилацетат (9 : 1). Получили 287 мг (96%) соединения (VII) в виде желтого масла.
1H-ЯМР (300 МГц, основной ротамер): 0.87 (т, 3H, J 6.9, CH3), 1.25 (уш. с, 30H, (CH2)15), 1.52 (п, 2H, J 7.2, CH2CH2N), 3.28 (кв, 2Н, J 6.7, CH2N), 8.13 (д, 1H, J 1.9, NHCHO).
13C-ЯМР (75 МГц): 14.07, 22.64, 26.77, 29.18, 29.31, 29.33, 29.47, 29.50, 29.52, 29.57, 29.59, 29.61, 29.65, 31.87, 38.34, 48.61, 161.68.
HRMS-ESI, m/z: [M + H]+ рассчитано для C19H40NO 298.3104, найдено 298.3105.
Октадецилизоцианид (VIII). Способ А: к раствору 0.5 М октадециламина (6.2 г, 0.023 моль) в DCM добавляли 50%-ный водный раствор NaOH (28 г, 0.69 моль), CHCl3 (2.8 мл, 0.035 моль) и TBAB (8.5 г, 0.026 моль). Реакционную смесь перемешивали 12 ч при 24°С, затем экстрагировали DCM (3 × 50 мл). Органический слой сушили безводным Na2SO4, концентрировали в вакууме, остаток хроматографировали на силикагеле. Получили 2.38 г (37%) соединения (VIII).
Способ C1: к 0.1 М раствору октадецилформамида (VII) (298 мг, 1.0 ммоль) в DCM добавляли Ph3P (393 мг, 1.5 ммоль), CBr4 (332 мг, 1.0 ммоль) и TEA (0.14 мл, 1.0 ммоль). Реакционную смесь перемешивали 20 ч при 40°С. Охлаждали до 24°С, концентрировали, выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (95 : 5). Получили 143 мг (51%) соединения (VIII).
Способ C2: к 0.1 М раствору октадецилформамида (VII) (298 мг, 1.0 ммоль) в этилацетате добавляли 50%-ный раствор трипропилтриметафосфорной кислоты (1.3 мл, 2.2 ммоль) в этилацетате и TEA (0.28 мл, 2.0 ммоль). Реакционную смесь перемешивали в течение 20 ч при 75°С. Охлаждали до 24°С, концентрировали в вакууме, выделяли колоночной хроматографией на силикагеле. Получили 118 мг (42%) соединения (VIII).
Способ C3: к 0.1 М раствору октадецилформамида (VII) (298 мг, 1.0 ммоль) в DCM добавляли POCl3 (0.14 мл, 1.5 ммоль) и TEA (0.35 мл, 2.5 ммоль). Реакционную смесь перемешивали в течение 20 ч при 75°С. Охлаждали до 24°С, концентрировали, выделяли колоночной хроматографией на силикагеле. Получили 56 мг (20%) соединения (VIII).
Способ C4: к 0.1 М раствору октадецилформамида (VII) (1.0 ммоль) в DCM добавляли Ph3P (393 мг, 1.5 ммоль), I2 (380 мг, 1.5 ммоль) и TEA (0.42 мл, 3.0 ммоль). Реакционную смесь перемешивали в течение 48 ч при 24°С, концентрировали, выделяли колоночной хроматографией на силикагеле. Получили 202 мг (72%) соединения (VIII).
1H-ЯМР (400 МГц): 0.88 (т, 3H, J 6.8, CH3), 1.27 (уш. с, 28H, (CH2)14), 1.43 (п, 2H, J 6.9, CH2CH2CH2NC), 1.67 (м, 2H, CH2CH2CH2NC), 3.36 (тт, 2H, J 1.9, 6.7, CH2CH2CH2NC).
13C-ЯМР (101 МГц): 14.03, 22.63, 26.28, 28.66, 29.09, 29.31, 29.46, 29.55, 29.59, 29.61, 29.64, 31.88, 41.45 (т, J 6.4, CH2NC), 155.77 (т, J 5.8, NC).
rac-1-О-Бензилглицидиловый эфир (X). К раствору rac-глицидола (IX) (6.7 мл, 0.09 моль) в 75 мл безводного DMF добавляли бензилбромид (14 мл, 0.120 моль). Охлаждали до 0°С и вносили порционно NaH (2.39 г, 1.0 моль). Перемешивали 18 ч при 24°С. Реакционную смесь разбавляли водой (250 мл), экстрагировали диэтиловым эфиром (3 × 100 мл), органический слой промывали водой (3 × 70 мл), сушили Na2SO4, фильтровали, упаривали. Продукт выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (85 : 15). Получили 14.23 г (96%) соединения (X) в виде масла светло-желтого цвета.
1H-ЯМР (300 МГц): 2.56–2.68 (м, 1Н, CHaHbOCH) и 2.74–2.84 (м, 1Н, CHaHbOCH), 3.11–3.26 (м, 1H, CH2OCH), 3.44 (дд, 1H, CHaHb) и 3.77 (дд, 1H, J 3.0, 11.4, CHaHbOBn), 4.59 (с, 2H, OCH2Ph), 7.21–7.44 (м, 5H, Ph).
13C-ЯМР (75 МГц): 44.41, 50.99, 70.94, 73.44, 76.78, 77.20, 77.63, 127.89, 128.56, 138.07.
rac-3-Бензилокси-1-децилокси-пропан-2-ол (XI). К 0.25 М раствору н-деканола (6 г, 0.04 моль) в THF добавляли NaH (1.4 г, 0.06 моль) при 0°С и перемешивали 1 ч при 90°С. После охлаждения реакционной смеси до 25°С по каплям добавляли раствор соединения (X) (4.8 г, 0.03 ммоль) в 1 М растворе DMF и перемешивали 12 ч при 90°С. Органические растворители удаляли в вакууме, к остатку добавляли 150 мл DCM, промывали водой (5 × 70 мл). Водный слой дополнительно экстрагировали DCM (4 × 70 мл). Объединенный органический экстракт сушили Na2SO4, фильтровали, упаривали. Продукт выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (85 : 15). Получили 6.21 г соединения (XI).
rac-3-Бензилокси-1-децилокси-2-этоксипропан (XII). К раствору 6.21 г соединения (XI) в 150 мл безводном THF добавляли NaH (0.9 г, 0.04 ммоль) и перемешивали 2 ч при 90°С, охлаждали до 24°С, затем добавляли этилбромид (3 мл, 0.04 ммоль) и перемешивали 6 ч при 90°С. Реакционную смесь концентрировали в вакууме, добавляли DCM (10 мл) и воду (10 мл), экстрагировали DCM (4 × 50 мл), органический слой промывали водой (4 × 50 мл). Объединенный органический экстракт сушили Na2SO4, фильтровали, упаривали. Продукт выделяли колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир–этилацетат (50 : 1). Получили 3.06 г (32% по двум стадиям) соединения (XII) в виде бесцветного масла.
1H-ЯМР (300 МГц): 0.88 (т, 3H, J 6.8, (СН2)9СН3), 1.14 (т, 3H, J 7.0, OCH2CH3), 1.17–1.32 (уш. с., 14H, (CH2)7CH3), 1.38–1.58 (м, 2H, OCH2CH2), 3.32–3.65 (м, 9H, OCH2CH(OCH2CH3)CH2OCH2), 4.49 (с, 2H, CH2Ph), 7.16–7.31 (м, 5H, C6H5).
13C-ЯМР (75 МГц): 14.07. 15.62, 22.66, 26.10, 29.31, 29.46, 29.56, 29.60, 29.64, 31.89, 65.70, 70.36, 70.77, 71.67, 73.37, 77.72, 127.48, 127.57, 128.29, 138.46.
rac-3-Децилокси-2-этоксипропан-1-ол (XIII). К суспензии 5% Pd/C (0.2 г) в безводном THF (80 мл) добавляли соединение (XII) (3.06 г, 8.7 ммоль) и вакуумировали в течение 5 мин. Затем выдерживали при перемешивании в атмосфере водорода в течение 12 ч. Далее реакционную смесь фильтровали через Celite® 545 (Sigma, США), целевое вещество выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (25 : 1). Получили 2.27 г (98%) соединения (XIII) в виде бесцветного масла.
1H-ЯМР (300 МГц): 0.88 (т, 3H, J 6.7, (СН2)9СН3), 1.15 (т, 3H, J 7.0, OCH2CH3), 1.21–1.47 (уш. с., 14H, (CH2)7CH3), 1.47–1.62 (м, 2H, OCH2CH2), 2.13 (с, 1H, CH2OH), 3.34–3.82 (м, 9H, OCH2CH(OCH2CH3)CH2OCH2).
13C-ЯМР (75 МГц): 13.96, 15.46, 22.56, 25.98, 29.20, 29.34, 29.45, 29.48, 29.51, 31.79, 62.89, 65.44, 70.83, 71.73, 78.18.
MS-ESI, m/z: [M + NH4]+ рассчитано для C15H36NO3 278.27, найдено 277.85.
rac-1-Бромо-3-децилокси-2-этоксипропан (XIV). К раствору соединения (XIII) (2.27 г, 8.7 ммоль) в безводном DCM (20 мл) добавляли Ph3P (3.43 г, 13.1 ммоль) при 0°C. Через 15 мин добавляли CBr4 (4.34 г, 13.1 ммоль) и перемешивали 2 ч при 25°С. Добавляли 25 мл MeOH и перемешивали 15 мин. Полученный раствор упаривали, выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (30 : 1). Получили 2.80 г (99%) соединения (XIV) в виде бесцветного масла.
1H-ЯМР (300 МГц): δ 0.88 (т, 3H, J 6.7, O(СН2)9СН3), 1.15–1.49 (уш. с, 17H, (CH2)7CH3; OCH2CH3), 1.48–1.66 (м, 2H, OCH2CH2), 3.39–3.73 (м, 9H, BrCH2CH(OCH2CH3)CH2OCH2).
13C-ЯМР (75 МГц): δ 14.08, 15.45, 22.66, 26.07, 29.30, 29.43, 29.55, 29.59, 31.89, 32.66, 65.75, 70.75, 71.78, 77.71.
HRMS-ESI, m/z: [M + Na]+ рассчитано для C15H31NaBrO2 345.1405, найдено 345.1405.
rac-1-Азидо-3-децилокси-2-этоксипропан (XV). К раствору бромида (XIV) (2.76 г, 8.5 ммоль) в DMF (40 мл) добавляли NaN3 (0.8 г, 12.8 ммоль) и перемешивали 4 ч при 90°С. Реакционную смесь охлаждали, полученный раствор концентрировали в вакууме, добавляли воду (25 мл) и экстрагировали этилацетатом (3 × 50 мл). Органический слой промывали водой (5 × 50 мл), сушили над безводным Na2SO4, фильтровали и упаривали. Получили 2.38 г (98%) соединения (XV) в виде бесцветного масла.
1H-ЯМР (300 МГц): 0.88 (т, 3H, J 6.8, (CH2)7CH3), 1.15–1.40 (м, 17H, CH2CH3, (CH2)7CH3), 1.55 (п, 2H, J 6.9, OCH2CH2), 3.25–3.75 (м, 9H, 3CH2O, CHO, CH2N3).
13C-ЯМР (75 МГц): 14.07, 15.49, 22.66, 26.08, 29.30, 29.43, 29.55, 29.58, 31.88, 52.03, 65.86, 70.22, 71.79, 77.70.
HRMS-ESI, m/z: [M + Na]+ рассчитано для C15H31O2N3Na 308.2314, найдено 308.2307.
rac-1-Амино-3-децилокси-2-этоксипропан (XVI). К раствору азида (XV) (856 мг, 3.0 ммоль) в этилацетате (30 мл) добавляли каталитическое количество 10% Pd/C (0.03 ммоль). Реакционную смесь охлаждали до 5°С и перемешивали в атмосфере водорода в течение 6 ч. Реакционную смесь фильтровали через подложку с Celite® 545, промывали этилацетатом (15 мл), растворитель удаляли в вакууме. Остаток хроматографировали на силикагеле, элюируя системой DCM–MeOH (30 : 1). Получили 350 мг (45%) соединения (XVI) в виде бесцветного масла.
1H-ЯМР (300 МГц): 0.86 (т, 3H, J 6.6, (CH2)7CH3), 1.14–1.37 (м, 23H, (CH2)7CH3, CH2CH3), 1.54 (п, 3Н, J 6.6, OCH2CH2), 2.20 (уш. с, 2H, NH2), 2.64–2.97 (м, 2H, CH2NH2), 3.29–3.79 (м, 7H, 3OCH2, OCH).
13C-ЯМР (75 МГц): 14.05, 15.60, 22.63, 26.08, 29.28, 29.43, 29.53, 29.56, 29.62, 31.86, 43.33, 65.48, 71.23, 71.72, 79.27.
HRMS-ESI, m/z: [M + H]+ рассчитано для C15H34NO2 260.2584, найдено 260.2557.
rac-1-Децилокси-3-формамидо-2-этоксипропан (XVII). К раствору соединения (XVI) (2.73 г, 11.0 ммоль) и DIPEA (1.87 мл, 11.0 ммоль) в DCM (42 мл) добавляли (формилокси)ацетонитрил (0.8 мл, 11.0 ммоль) и перемешивали 5 ч. Затем реакционную смесь концентрировали в вакууме, хроматографировали на силикагеле, элюируя системой DCM–MeOH (50 : 1). Получили 2.14 г (71%) соединения (XVII) в виде бесцветного масла.
1H-ЯМР (300 МГц, основной ротамер): 0.85 (т, 3Н, J 6.8, (CH2)7CH3), 1.11–1.36 (м, 17H, (CH2)7CH3, CH2CH3), 1.53 (п, 2H, J 6.0, OCH2CH2), 3.19–3.73 (м, 9H, 3OCH2, CHO, CH2N), 6.17 (c, 1H, NH), 8.16 (c, 1H, NHCHO).
13C-ЯМР (75 МГц): δ 14.11, 15.50, 22.67, 26.07, 29.31, 29.43, 29.54, 29.56, 29.58, 31.88, 39.37, 65.38, 71.35, 71.90, 76.06, 161.46.
HRMS-ESI, m/z: [M + H]+ рассчитано для C16H34NO3 288.2533, найдено 288.2533.
HRMS-ESI, m/z: [M + Na]+ рассчитано для C16H33NO3Na 310.2353, найдено 310.2351.
rac-1-Децилокси-3-изоциано-2-этоксипропан (XVIII). К 0.1 М раствору соединения (XVII) (287 мг, 1.0 ммоль) в DCM добавляли Ph3P (393 мг, 1.5 ммоль), I2 (380 мг, 1.5 ммоль) и TEA (0.42 мл, 3.0 ммоль). Реакционную смесь перемешивали в течение 48 ч при 24°С, концентрировали, выделяли колоночной хроматографией на силикагеле, элюируя системой петролейный эфир–этилацетат (8 : 2). Получили 202 мг (72%) соединения (XVIII).
1H-ЯМР (400 МГц): δ 0.90 (т, 3H, J 6.8, (CH2)7CH3), 1.20–1.39 (м, 17H, (CH2)7CH3, CH2CH3), 1.58 (п, 2H, J 6.9, OCH2CH2), 3.40–3.72 (м, 9H, 3OCH2, CHO, CH2N).
13C-ЯМР (101 МГц): δ 14.09, 15.37, 22.65, 26.03, 29.29, 29.40, 29.50, 29.53, 29.56, 31.87, 42.96 (т, J 7.0, CH2NC), 66.05, 69.23, 71.87, 75.79, 157.35 (т, J 5.5, NC).
MS-ESI, m/z: [M + NH4]+ рассчитано для C16H35N2O2 287.27, найдено: 287.42.
MS-ESI, m/z: [M + Na]+ рассчитано для C16H31NNaO2 292.23, найдено 293.08.
1-Азидо-1-дезокси-2,3,4,6-тетра-O-ацетил-β-D-глюкопираноза (XX). К раствору бромида (XIX) (27 г, 67.0 ммоль) в DMF (113 мл) добавляли NaN3 (6 г, 0.1 моль) и перемешивали 3 ч при 90°С. К реакционной смеси добавляли CHCl3 (150 мл) и H2O (150 мл). Экстрагировали CHCl3 (4 × 100 мл), объединенный органический экстракт промывали H2O (3 × 100 мл). Сушили безводным Na2SO4, фильтровали, упаривали, остаток хроматографировали на силикагеле, элюируя системой толуол–этилацетат (1 : 1). Получили 22 г (90%) соединения (XX) в виде желтого кристаллизующегося масла.
1H-ЯМР (300 МГц): δ 2.00, 2.02, 2.07, 2.09 (все с по 3H, 4OAc), 3.79 (ддд, 1H, J 2.4, 4.7, 9.9, H-5), 4.16 (дд, 1H, J 2.4, 12.5, Н-6), 4.27 (дд, 1H, J 4.7, 12.5, H-6), 4.64 (д, 1H, J 8.9, H-1), 4.95 (дд, 1H, J 8.9, 9.5, H-2), 5.09 (дд, 1H, J 9.4, 9.9, H-4), 5.21 (дд, 1H, J 9.4, 9.5, H-3).
13C-ЯМР (75 МГц): δ 20.62, 20.77, 61.73, 67.94, 70.69, 72.66, 74.07, 87.96, 169.29, 169.39, 170.19, 170.69.
MS-ESI, m/z: [M + Na]+ рассчитано для C14H19NaO9 396.10, найдено 396.78.
1-Амино-1-дезокси-2,3,4,6-тетра-O-ацетил-β-D-глюкопираноза (XXI). Суспензию соединения (XX) (20 г, 0.05 моль) и 10% Pd/C (57 мг, 0.5 ммоль) в этилацетате (107 мл) вакуумировали 5 мин, затем выдерживали в атмосфере водорода при перемешивании в течение 12 ч. Растворитель удаляли на роторном испарителе, хроматографировали в системе толуол–этилацетат (6 : 3). Получили 17 г (91%) соединения (XXI) в виде желтого кристаллизующегося масла.
1H-ЯМР (300 МГц): δ 2.00, 2.01, 2.07, 2.08 (все с по 3H, 4OAc), 2.98 (уш. c, 2H, NH2), 3.70 (ддд, 1H, J 2.4, 4.8, 10.0, H-5), 4.05–4.15 (м, 1H, H-6), 4.17–4.29 (м, 2H, H-1, H-6), 4.86 (дд, 1H, J 9.5, 9.7, H-2), 5.04 (дд, 1H, J 9.4, 10.0, H-4), 5.23 (дд, 1H, J 9.4, 9.5, H-3).
13C-ЯМР (75 МГц): δ 20.61, 20.78, 20.86, 62.17, 68.64, 71.84, 72.91, 73.08, 84.50, 169.53, 170.18, 170.27, 170.73.
MS-ESI, m/z: [M + H]+ рассчитано для C14H22NO9 348.1289, найдено 348.1298.
2,3,4,6-Тетра-O-ацетил-1-дезокси-1-формамидо-β-D-глюкопираноза (XXII). К охлажденному до 0°С раствору уксусного ангидрида (6 мл, 0.06 моль) добавляли муравьиную кислоту (3 мл, 0.9 моль) и перемешивали при 60°С в течение 2 ч. Затем раствор охлаждали до 24°С и добавляли к соединению (XXI) (1 г, 3.0 ммоль), далее добавляли DIPEA (2 мл, 0.01 моль). Реакционную массу перемешивали 3 ч, переносили в делительную воронку, приливали этилацетат (50 мл) и 25% NH3 · H2O (25 мл). Экстрагировали этилацетатом (3 × 25 мл), органический остаток промывали водой (3 × 25 мл). Сушили Na2SO4, фильтровали, упаривали. Получили 0.9 г (82%) соединения (XXII) в виде желтого кристаллизующегося масла.
1H-ЯМР (300 МГц): δ 2.01, 2.03, 2.05, 2.07 (все с по 3H, 4 OAc), 3.83 (ддд, 1H, J 2.2, 4.5, 10.2, H-5), 4.04–4.18 (м, 2H, H-1, H-6), 4.30 (дд, 1H, J 4.5, 12.5, H-6), 4.94 (дд, 1H, J 9.5, 9.6, H-2), 5.06 (дд, 1H, J 9.4, 10.2, H-4), 5.30 (дд, 1H, J 9.4, 9.6, H-3), 6.50 (д, 1H, J 9.3, NH), 8.21 (c, 1H, CHO).
13C-ЯМР (75 МГц): δ 20.69 (2CH3, +), 20.78 (CH3, +), 20.84 (CH3, +), 61.73 (CH2, –), 68.18 (CH, +), 70.54 (CH, +), 72.71 (CH, +), 73.86 (CH, +), 169.68 (C, –), 169.98 (C, –), 170.13 (C, –), 170.74 (C, –), 171.11 (C, –).
HRMS-ESI, m/z: [M + H]+ рассчитано для C15H22NO10 376.1238, найдено 376.1239.
1-Дезокси-1-изоциано-2,3,4,6-тетра-O-ацетил-β-D-глюкопираноза (XXIII). Способ А: к раствору соединения (XXII) (1 г, 3.0 ммоль), Ph3P (2 г, 8.0 ммоль) и TEA (2 мл, 11.0 ммоль) в DCM (27 мл) добавляли CBr4 (3 г, 8.0 ммоль) и перемешивали 3 ч при 24°С. Растворитель удаляли в вакууме, остаток хроматографировали, элюируя смесью толуол–этилацетат (7 : 3). Получили 0.7 г (69%) соединения (XXIII) в виде желтого кристаллизующегося масла.
Способ Б: к раствору соединения (XXII) (1 г, 3.0 ммоль), Ph3P (2 г, 8.0 ммоль) и TEA (2 мл, 11.0 ммоль) в DCM (27 мл) добавляли I2 (3 г, 8.0 ммоль) и перемешивали 3 ч при 24°С. Растворитель удаляли в вакууме, остаток хроматографировали, элюируя системой толуол–этилацетат (7 : 3). Получили 0.6 г (60%) соединения (XXIII) в виде желтого кристаллизующегося масла.
1H-ЯМР (600 МГц): δ 2.02, 2.03, 2.11, 2.12 (все с по 3H, 4 OAc), 3.74 (ддд, 1H, J 2.2, 4.8, 10.0, 5-CH), 4.15 (дд, 1H, J 2.2, 12.6, 6-HaHb), 4.25 (дд, 1H, J 4.8, 12.6, 6-HaHb), 4.85 (ABCD, 1H, J 9.3 Гц, Н-1), 5.14 (ABCD, 1H, J 9.1, 10.0, 4-СН), 5.20 (ABCD, 1H, J 9.3, 9.5, H-2) 5.22 (ABCD, 1H, J 9.1, 9.5, Н-3).
13C-ЯМР (151 МГц): δ 20.44, 20.50, 20.66, 61.70, 67.95, 71.64, 72.50, 75.08, 79.74, 165.85, 168.87, 169.17, 169.97, 170.43.
HRMS-ESI, m/z: [M + H]+ рассчитано для C15H20NO9 358.1133, найдено 357.1152.
ЗАКЛЮЧЕНИЕ
Проведен синтез изонитрильных производных углеводов и диглицерида, содержащего протяженный алкильный заместитель при атоме С-1 и этильный – при атоме С-2 глицерина. Впервые установлены и описаны точные значения КССВ для изонитрильного производного D-глюкозы, что позволяет говорить о пространственной геометрии молекулы. Полученные соединения планируется в дальнейшем использовать в качестве строительных блоков в многокомпонентных реакциях Уги для получения липофильных полиаминов.
Список литературы
Giovenzana G.B., Tron G.C., Di Paola S., Menegotto I.G., Pirali T. // Angewandte Chem. 2006. V. 118. P. 1117–1120. https://doi.org/10.1002/ange.200503095
Giustiniano M., Basso A., Mercalli V., Massarotti A., Novellino E., Tron G.C., Zhu J. // Chem. Soc. Rev. 2017. V. 46. P. 1295–1357. https://doi.org/10.1039/c6cs00444j
Mossetti R., Pirali T., Saggiorato D., Tron G.C. // Chem. Commun. 2011. V. 47. P. 6966–6968. https://doi.org/10.1039/c1cc12067k
Hofmann A.W. // Justus Liebigs Annalen der Chem. 1867. V. 144. P. 114–120. https://doi.org/10.1002/jlac.18671440116
Ugi I., Meyr R. // Angewandte Chem. 1958. V. 70. P. 702–703. https://doi.org/10.1002/ange.19580702213
Josien H., Ko S.B., Bom D., Curran D.P. // Chem. Europ. J. 1998. V. 4. P. 67–83. https://doi.org/10.1002/(SICI)1521-3765(199801)4: 1<67::AID-CHEM67>3.0.CO;2-F
Wang X., Wang Q.G., Luo Q.L. // Synthesis. 2015. V. 47. P. 49–54. https://doi.org/10.1055/s-0034-1379111
Gautier A. // Justus Liebigs Annalen der Chem. 1867. V. 142. P. 289–294. https://doi.org/10.1002/jlac.18671420304
El Kaim L., Grimaud L., Schiltz A. // Org. Biomol. Chem. 2009. V. 7. P. 3024–3026. https://doi.org/10.1039/b908541f
Gassman P.G., Guggenheim T.L. // J. Am. Chem. Soc. 1982. V. 104. P. 5849–5850. https://doi.org/10.1021/ja00385a078
Kitano Y., Chiba K., Tada M. // Synlett. 1999. V. 1999. P. 288–290. https://doi.org/10.1055/s-1999-2615
Kitano Y., Chiba K., Tada M. // Synthesis. 2001. V. 2001. P. 0437–0443. https://doi.org/10.1055/s-2001-11423
Пучков П.А., Перевощикова К.А., Карташова И.А., Лунева А.С., Кабилова Т.О., Морозова Н.Г., Зенкова М.А., Маслов М.А. // Биоорг. химия. 2017. Т. 43. С. 543–552. [Puchkov P.A., Perevoshchikova K.A., Kartashova I.A., Luneva A.S., Kabilova T.O., Morozova N.G., Zenkova M.A., Maslov M.A. // Russ. J. Bioorg. Chem. 2017. V. 43. P. 561–569.] https://doi.org/10.1134/S1068162017050107
Petukhov I.A. Maslov M.A. Morozova N.G., Serebrennikova G.A. // Russ. Chem. Bull. 2010. V. 59. P. 260–268. https://doi.org/10.1007/s11172-010-0071-x
Perevoshchikova K.A., Nichugovskiy A.I., Isagulieva A.K., Morozova N.G., Ivanov I.V., Maslov M.A., Shtil A.A. // Mend. Comm. 2019. V. 29. P. 616–618. https://doi.org/10.1016/j.mencom.2019.11.003
Morozova N.G., Timofeev G.A., Timakova A.A., Shmendel E.V., Kubasova T.S., Tyutyunnik L.L., Markova A.A., Maslov M.A., Shtil A.A. // Mend. Comm. 2015. V. 25. P. 248–249. https://doi.org/10.1016/j.mencom.2015.07.003
Morozova N.G., Shmendel E.V., Timofeev G.A., Ivanov I.V., Kubasova T.S., Plyavnik N.V., Markova A.A., Maslov M.A., Shtil A.A. // Mend. Comm. 2019. V. 29. P. 166–168. https://doi.org/10.1016/j.mencom.2019.03.016
Shmendel E.V., Perevoshchikova K.A., Shishova D.K., Kubasova T.S., Tyutyunnik L.L., Maslov M.A., Morozova N.G., Shtil A.A. //Russ. Chem. Bull. 2015. V. 64. P. 1648–1654. https://doi.org/10.1007/s11172-015-1055-7
Lashin W.H., Nassar I.F., El Farargy A.F., Abdelhamid A.O. // Russ. J. Bioorg. Chem. 2020. V. 46. P. 1074–1086. https://doi.org/10.1134/S1068162020060163
Tolan H.E., Radwan M.A., Soliman H.A., Awad H.M., El-Sayed W.A. // Russ. J. Bioorg. Chem. 2020. V. 46. P. 1136–1147. https://doi.org/10.1134/S1068162020060345
Горюнова О.В., Захарчук Г.М., Жукова О.С., Фетисова Л.В., Кузьмина Н.Е. // Биоорг. химия. 2014. Т. 40. С. 12–19. [Goryunova O.V., Zakharchuk G.M., Zhukova O.S., Fetisova L.V., Kuzmina N.E. // Russ. J. Bioorg. Chem. 2014. V. 40. P. 9–15.] https://doi.org/10.1134/S106816201401004X
Морозова Н.Г., Маслов М.А., Мягченков В.В., Серебренникова Г.А. // Биоорг. химия. 2010. Т. 36. С. 714–720. [Morozova N.G., Maslov M.A., Myagchenkov V.V., Serebrennikova G.A. // Russ. J. Bioorg. Chem. 2010. V. 36. P. 657–662.] https://doi.org/10.1134/S1068162010050146
La Spisa F., Feo A., Mossetti R., Tron G.C. // Org. Lett. 2012. V. 14. P. 6044–6047. https://doi.org/10.1021/ol302935y
Zheng Q., Tang S., Fu X., Chen Z., Ye Y., Lan X., Jiang L., Huang Y., Ding J., Geng M., Huang M., Wan H. // Bioorg. Med. Chem. Lett. 2017. V. 27. P. 5262–5266. https://doi.org/10.1016/j.bmcl.2017.10.029
Lemieux R.U., Brice C. // Canad. J. Chem. 1952. V. 30. P. 295–310. https://doi.org/10.1139/v52-041
Polisar J.G., Li L., Norton J.R. // Tetrahedron Lett. 2011. V. 52. P. 2933–2934. https://doi.org/10.1016/j.tetlet.2011.03.122
Appel R. // Angewandte Chem. Int. Ed. Engl. 1975. V. 14. P. 801–811. https://doi.org/10.1002/anie.197508011
Arezzini B., Ferrali M., Ferrari E., Frassineti C., Lazzari S., Marverti G., Spagnolo F., Saladini M. // Eur. J. Med. Chem. 2008. V. 43. P. 2549–2556. https://doi.org/10.1016/j.ejmech.2008.02.045
Hu G., Li W., Hu Y., Xu A., Yan J., Liu L., Zhang X., Liu K., Zhang A. // Macromol. 2013. V. 46. P. 1124–1132. https://doi.org/10.1021/ma302536t
Cheshkov D.A., Sheberstov K.F., Sinitsyn D.O., Chertkov V.A. // Magnet. Reson. Chem. 2018. V. 56. P. 449–457. https://doi.org/10.1002/mrc.4689
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия