

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 2, стр. 79-89
Белки семейства АВС и воспаление
А. А. Ставровская a, *, Е. Ю. Рыбалкина a, **
a Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Минздрава России
115487 Москва, Каширское шоссе, 24, Россия
* E-mail: astavrovskaya@yahoo.com
** E-mail: Kate_Rybalkina@mail.ru
Поступила в редакцию 29.06.2018
После доработки 12.11.2018
Принята к публикации 15.11.2018
Аннотация
Воспаление – важнейший общебиологический процесс – ответ васкуляризованных тканей организма на инфекцию и/или повреждение тканей. В опухолях обычно имеются очаги воспаления, влияющие на процесс канцерогенеза. Белки семейства АВС известны прежде всего, благодаря их способности выводить токсические вещества и антибиотики из клеток. За последнее десятилетие стало известно, что АВС-транспортеры могут выполнять в клетках и другие функции, в частности функции регуляторов важных биологических процессов. Они могут участвовать и в регуляции воспаления. Полученные данные показывают, что активность АВС-белков изменяется в очаге воспаления. АВС-белки могут быть необходимы для выработки медиаторов воспаления, они участвуют в транспорте из клеток цитокинов типа интерлейкина-2, интерферона-γ, фактора некроза опухолей-α. Вклад АВС-транспортеров в воспаление может определяться их экспрессией различными иммунными клетками и влиянием на повышение жизнеспособности этих клеток при медикаментозном лечении. Разбираются различные молекулярные механизмы, определяющие связь АВС-транспортеров и воспаления. В частности такие, как регуляция активности АВС-белков провоспалительными цитокинами, функции этих белков в качестве звена иммунного ответа, влияние микробиома кишечника на активность АВС-белков. Данный обзор посвящен взаимосвязям белков семейства АВС и процессов воспаления.
ВВЕДЕНИЕ
Белки семейства АВС (ATP binding cassette) принадлежат к суперсемейству, представители которого имеются у всех организмов – как у прокариот, так и у эукариот, что свидетельствует об их важнейшей роли в защите всех живых клеток. Эти белки используют энергию АТР для того, чтобы перемещать различные субстраты через клеточную мембрану [1]. Они могут определять как поступление веществ в клетки, так и выброс из клеток различных соединений, которые являются субстратами АВС-транспортеров. В суперсемейство АВС в настоящее время входят около 300 белков эукариот, переносящих самые разные соединения [1–3]. Большая часть транспортеров семейства АВС (129) обнаружена у растений [4]. У человека найдено около 50 трaнспортных белков, входящих в семейство АВС [4]. Особенно интенсивно из этих транспортеров изучали Р-гликопротеин (АВСВ1, далее обозначаемый Pgp), ВСRP (ABCG2) и MRP1(ABCC1) благодаря их участию в определении множественной лекарственной устойчивости (МЛУ) опухолевых клеток. В последнее десятилетие стало известно, что АВС-транспортеры могут выполнять в клетках и другие функции, в частности, функции регуляторов важных биологических процессов [2, 3]. Встает вопрос об их участии в регуляции процессов воспаления, ответы на который разбираются в этой работе.
ОБЩАЯ ХАРАКТЕРИСТИКА ПРОЦЕССА ВОСПАЛЕНИЯ
Воспаление – важнейший процесс – ответ васкуляризованных тканей организма на инфекцию и/или на повреждение. Важно подчеркнуть, что при воспалении происходит перемещение защитных клеток и молекул из циркулирующей крови к тем местам, где они необходимы для ликвидации повреждающего агента. Представления о воспалении как о защитном процессе возникли очень давно. Принято считать, что впервые об этом писал Гиппократ (460–377 гг. до н.э.). Естественно, за большой промежуток времени, прошедший с тех пор, результаты изучения воспаления накопились и давно стали предметом описания в учебниках. Приведем лишь самые необходимые для нашего обзора результаты этих многочисленных исследований.
Воспаление подразделяют на острое и хроническое. Острое воспаление проявляется деструкцией клеток и тканей (альтерацией), изменениями микроциркуляции и проницаемости сосудов (экссудацией), и миграцией лимфоцитов в сочетании с пролиферацией клеток тканей [5]. Таким образом, компонентами воспаления являются альтерация, экссудация и пролиферация.
Альтерация включает комплекс реакций, которые приводят к развитию хемоаттракции – привлечению в очаг воспаления клеток, продуцирующих медиаторы воспаления, которые обеспечивают химические и молекулярные связи между процессами, протекающими в очаге. Таким образом, процесс альтерации весьма важен с точки зрения связи АВС-транспортеров и воспаления.
Медиаторы воспаления подразделяют на клеточные и гуморальные [5]. Клетки, функции которых активируются при воспалении, разделяют на две группы: (1) клетки, присутствующие в ткани постоянно — дендритные клетки, макрофаги и тучные клетки; (2) клетки, поступающие в зону воспаления извне. К последним относятся гранулоциты, моноциты и лимфоциты. С помощью клеточных медиаторов включается сосудистая реакция, в результате чего в процессе воспаления начинают принимать участие гуморальные медиаторы воспаления, и в очаг повреждения поступает соответствующий экссудат, содержащий различные биологически активные вещества, включая факторы роста, стимулирующие пролиферацию не только клеток воспаления, но и других клеток, в том числе имеющих отношение к канцерогенезу. Воспаление и канцерогенез/опухолевая прогрессия тесно связаны. Эту связь отметил еще 150 лет назад Р. Вирхов. Эпидемиологические данные подтвердили это положение и показали, что примерно 25% всех опухолей возникают благодаря хронической инфекции или хроническому воспалению [6]. В настоящее время стало ясно, что микроокружение опухолевых клеток и, в первую очередь, воспалительные цитокины, факторы роста и клетки, принимающие участие в процессах воспаления, оказывают большое воздействие на процессы развития опухолей, влияя на пролиферацию, выживаемость и миграцию малигнизированных клеток, на их чувствительность к химиотерапии [6]. Хроническое воспаление может не только стимулировать, но и подавлять иммунные реакции [7].
СВЯЗЬ МЕЖДУ ВОСПАЛЕНИЕМ И БЕЛКАМИ АВС-ТРАНСПОРТЕРАМИ
Отметим, прежде всего данные, свидетельствующие о том, что АВС- транспортер Pgp может иметь прямое отношение к развитию воспалительных заболеваний кишечника, т.е. непосредственно влиять на процесс воспаления. Обнаружено, что у мышей с нокаутом гена mdr1a (mdr1a–/–) (кодирует один из двух Pgp) спонтанно развивается хронический колит [8, 9]. Повышение экспрессии Pgp в слизистой кишечника в ответ на введение фактора роста кератиноцитов или пробиотиков приводило к снижению степени воспаления кишечника у мышей с колитом, индуцированным декстрансульфатом натрия [10–13].
Обнаружено, что Pgp защищает клетки кишечного эпителия от заражения Listeria monocytogenes. Количество L. monocytogenes в клетках желудочно-кишечного тракта было значительно больше у мышей с нокаутом гена mdr1a, чем у животных дикого типа [14].
Показано также, что с полиморфизмом гена ABCB1 может быть связана частота возникновения язвенного колита у людей. Полиморфизм гена ABCB1 и, в первую очередь, полиморфизм C3435T в экзоне 26, который коррелирует с более низкой экспрессией Pgp в клетках слизистой кишечника [15], с высокой частотой встречается у больных язвенным колитом в разных популяциях [16–19].
Однако в ряде работ такая зависимость не была найдена [20–22]. Молекулярные механизмы этого феномена пока остаются неясными. Дальнейшие исследования должны дать ответы на вопрос, каким образом дефекты гена ABCB1 могут сказываться на воспалении. Это может быть связано как с функционированием Pgp в качестве мембранного насоса, так и с неканоническими активностями Pgp – с его ролью как регулятора биологических процессов, например, с тем, что АВС-транспортеры и особенно Pgp могут участвовать в регуляции комплексной системы иммунного ответа на инфекцию [23].
Изменяется ли активность АВС-белков в очагах воспаления? Полученные в последнее время данные позволяют ответить положительно на этот вопрос. Показано, что при рассеянном склерозе, известном также как множественный склероз, значительно повышена экспрессия Pgp и белков MRP-1, MRP-2 в астроцитах, присутствующих в очагах воспаления, характерных для этого заболевания [24, 25].
Однако не всегда воспаление связано с повышением экспрессии АВС-транспортеров. Englund и соавт. обнаружили, что в биопсиях, взятых у 16 больных язвенным колитом, количество мРНК ABCB1 (MDR1) и ABCG2 (BCRP) значительно ниже, чем в биопсиях, взятых у здоровых доноров [26]. Показано также, что у больных язвенным колитом снижена экспрессия Pgp в воспаленной ткани слизистой кишечника [27]. Таким образом, при воспалении наблюдается как повышение, так и снижение количества АВС-белков в клетках и тканях.
МЕХАНИЗМЫ, ОПРЕДЕЛЯЮЩИЕ СВЯЗЬ АВС-ТРАНСПОРТЕРОВ И ВОСПАЛЕНИЯ
Провоспалительные цитокины могут регулировать экспрессию АВС-белков. В культивируемых гепатоцитах крысы экспрессия Pgp или его генов mdr1a и mdr1b снижается под действием IL-1β, IL-6 или TNF-α [28, 29].
Pgp и другой АВС-транспортер, BCRP, экспрессируются на высоком уровне в эндотелии сосудов головного мозга и играют важную роль в функционировании гематоэнцефалического барьера (ГЭБ). Они защищают клетки головного мозга от токсических воздействий, но при этом ограничивают поступление лекарственных средств в клетки мозга и препятствуют фармакотерапии. Изучение действия провоспалительных цитокинов на клеточные элементы ГЭБ показало, что уровни экспрессии и активность BCRP и Pgp в этих клетках могут изменяться при воспалении [30, 31]. Так Poller и соавт. [30] исследовали влияние провоспалительных цитокинов (IL-1β, IL-6 и TNF-α) на экспрессию и активность BCRP и Pgp в клеточной линии hCMEC/D3 человека – модели ГЭБ. Выявлено значительное снижение уровня мРНК и белка BCRP под действием цитокинов IL-1β, IL-6 и TNF-α, а также существенное подавление функциональной активности BCRР. Уровни мРНК MDR1 слегка снижались при добавлении IL-6 и значительно увеличились после добавления TNF-α [30]. Эти данные свидетельствуют о том, что провоспалительные цитокины, количество которых возрастает при воспалении, могут влиять на активность АСВ-транспортеров.
Однако необходимо отметить, что регуляция экспрессии АВС-транспортеров цитокинами в ГЭБ подвержена значительным временным изменениям. Harts и соавт. выявили быструю (за минуты) и обратимую инактивацию Pgp в капиллярах головного мозга крыс (in vitro) при действии TNF-α и эндотелина-1 (ЭТ-1) [32]. Они исследовали также долгосрочные последствия непрерывного воздействия TNF-α и ЭТ-1. Оказалось, что TNF-α и ЭT-1 быстро снижали транспортную активность Pgp в капиллярах мозга, но не влияли на количество самого белка. Активность Pgp оставалась низкой в течение 2–3 ч, но затем наблюдалось резкое увеличение как экспрессии белка, так и его транспортной активности. Через 6 ч экспрессия белка-транспортера и интенсивность транспорта были вдвое выше, чем в контрольных образцах [33]. Предполагая, что подобные эффекты происходят in vivo, эти результаты могут свидетельствовать об усилении функции ГЭБ при хроническом воспалении и о возможном снижении эффективности препаратов, используемых в терапии болезней ЦНС (если они являются субстратами Pgp).
В ряде экспериментальных работ показано, что цитокины могут повышать уровень экспрессии гена MDR1 в иммунных клетках. Так, IL-12, TNF-α, INF-γ увеличивали уровень экспрессии гена MDR1 в мононуклеарных клетках периферической крови человека [34]. INF-γ повышал экспрессию MDR1 в макрофагах [35].
Введение мышам этих провоспалительных цитокинов также приводило к значительному снижению количества мРНК Pgp в печени животных [36]. В пользу связи Pgp и воспаления свидетельствуют также данные опытов с использованием с фактора роста кератиноцитов-2 (ФРК2). Введение мышам ФРК2 привело к повышению экспрессии Pgp клетками слизистой кишечника животных и к снижению степени воспалительной реакции кишечника [10, 12].
На уровень экспрессии гена MDR1, как показано, может влиять и сам патоген. На модели экспериментального туберкулеза установлено, что к 8 неделе после аэрозольного инфицирования мышей линии C57BL/6 низкими дозами вирулентного штамма H37Rv M. tuberculosis уровень экспрессии mdr1a был статистически значимо выше, чем в контрольной группе [37]. На клеточных линиях убедительно показано влияние бактериального заражения на экспрессию гена MDR1. В частности, изучено влияние микобактерий туберкулеза (Mtb) на изменение уровня экспрессии гена MDR1 в промоноцитарной линии U1 (субклон линии U937, полученный от хронически инфицированных HIV-1) [38]. Показано, что заражение Mtb приводит к повышению экспрессии гена MDR1 в макрофагах. Изучение влияния Staphylococcus aureus и Escherichia coli на экспрессию MDR1, MRP1 и BCRP в клетках HC11 (эпителий молочной железы) выявило статистически значимое повышение уровня экспрессии гена MDR1 [39]. Количество мРНК генов BCRP и MRP1 при этом не изменялось.
Микробиом кишечника может прямо влиять на функцию Pgp (в качестве насоса, выводящего ксенобиотики из клеток). Об этом свидетельствуют данные, показывающие, что в линии T84 клеток аденокарциномы человека уже через 4 ч после заражения бактерией Salmonella typhimurim увеличивается накопление родамина 123 (Rh123) – субстрата Pgp. Обработка клеток верапамилом приводила примерно к такому же результату, т.е. эффект бактериальной инфекции был значительным и сравнимым с действием верапамила, известного ингибитора функции Pgp. Введение малой интерферирующей РНК к гену MDR1 делало клетки более чувствительными к инфицированию [40].
Pgp, экспрессируемый клетками слизистой кишечника, может, в свою очередь, непосредственно влиять на процессы бактериальной инфекции. Показано, что сверхэкспрессия Pgp в клетках эпителия слизистой кишечника приводила к повышению резистентности к заражению L. monocytogenes или S. typhimurium [14, 41]. Установлено также, что транскрипция гена MDR1, кодирующего Pgp, индуцируется в линии моноцитов THP-1, инфицированной L. monocytogenes, вызывающей воспаление [42]. При этом оказалось, что Pgp необходим для активации выработки интерферона первого типа (ИФ1) в ответ на инфекцию L. monocytogenes. Подавление функциональной активности Pgp верапамилом или подавление транскрипции гена MDR1 приводило к снижению выработки ИФ1 зараженными клетками. Эта функция Pgp оказалась чувствительной именно к ИФ1, но не к другим цитокинам, вырабатываемым при заражении L. monocytogenes.
Pgp может быть важным звеном иммунного ответа на инфекции и участвовать в развитии воспаления. Исследования, проведенные в 90-х годах, доказали, что Pgp влияет на секрецию различных медиаторов воспаления и стероидов. Barnes и coавт. нашли, что Pgp участвует в переносе стероидов, причем эффективность переноса зависела от гидрофобности стероидов и степени фосфорилирования Pgp [43]. На двух типах клеток (мезангиальных клетках человека и эпителиальных клетках почки свиньи) получены данные, показывающие, что выведение из клеток фактора роста тромбоцитов модулируется ингибиторами Pgp [44, 45]. Это позволило полагать, что фактор роста тромбоцитов является субстратом Pgp. Исследования, проведенные с использованием активированных Т-клеток периферической крови человека, показали, что Pgp участвует в трансмембранном транспорте ряда цитокинов (IL-12, IL-2, IL-4, IFN-γ) [46, 47].
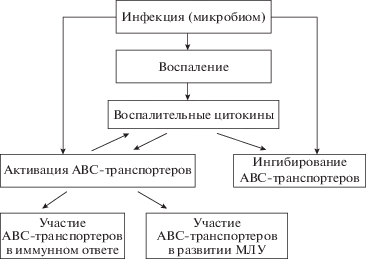
Таким образом, механизмы, определяющие связь АВС-транспортеров и воспаления, множественны и взаимосвязаны (рис. 1). Полученные данные свидетельствуют о неоднозначности результатов преодоления МЛУ путем подавления активности АВС-транспортеров. Например, подавление активности белков МЛУ (в частности, Pgp) может привести к таким нежелательным результатам, как нарушение выработки ИФ1 в ответ на бактериальную инфекцию и ускоренное развитие инфекционного процесса.
Рис. 1.
Взаимосвязи процессов воспаления и активности АВС транспортеров. Инфекция приводит к воспалению. Клетки в очаге воспаления продуцируют различные воспалительные цитокины (ВЦ), включая факторы роста. ВЦ могут как активировать, так и ингибировать транспортные белки семейства АВС. Активированные АВС-транспортеры, в свою очередь, могут принимать участие в секреции клетками воспалительных цитокинов (таких, как IL-12, IL-2, IL-4, IFN-γ). Активные АВС-транспортеры могут определять множественную лекарственную устойчивость опухолевых клеток, а также принимать участие в иммунном ответе на инфекцию.
ПУТИ СИГНАЛЬНОЙ ТРАНСДУКЦИИ И БЕЛКИ, ОПРЕДЕЛЯЮЩИЕ СВЯЗЬ ЭКСПРЕССИИ АВС-ТРАНСПОРТЕРОВ И ВОСПАЛЕНИЯ
АВС-транспортеры могут влиять на сигнальные пути, активность которых определяет воспаление.
1) Белки ABCA1 и ABCG1 подавляют эти сигнальные пути, действуя через Толл-подобные рецепторы (Toll-like receptors, от немецкого toll — замечательный, TLR) [48–50]. Напомним, что TLR — класс клеточных рецепторов с одним трансмембранным доменом. TLR активируют клеточный иммунный ответ. Они играют ключевую роль во врожденном иммунитете [51]. Макрофаги из перитонеальной жидкости мышей с генотипами и Abca1−/−Abcg1−/− характеризовались повышенной экспрессией воспалительных цитокинов и хемокинов в ответ на введение лигандов TLR2, 3 или 4 [52]. Эти данные свидетельствуют в пользу того, что АВС-транспортеры могут подавлять экспрессию воспалительных цитокинов путем взаимодействия с TLR.
2) С использованием экспериментальной модели воспалительного заболевания желудочно-кишечного тракта у крыс и мышей, индуцированного введением раствора 2,4,6-тринитробензолсульфоновой кислоты (2,4,6-trinitrobenzene sulfonic acid – ТСА), показано, что хроническое воспаление приводит к повышению экспрессии Pgp мононуклеарными клетками периферической крови в результате активации сигнального пути STAT3/NF-κВ [53]. Мононуклеарные клетки периферической крови больных животных накапливали меньше циклоспорина А (субстрата Pgp) и секретировали больше цитокинов IL-1β, IL-6, IL-17 и TNF-α, чем мононуклеарные клетки контрольных животных.
3) Стабильность и гликозилирование Pgp регулируются киназой Pim-1. Сверхэкспрессия серин/треониновой протеинкиназы Pim-1 часто отмечается при различных новообразованиях, включая острые миелоидные лейкозы, острые лимфобластные лейкозы, рак предстательной железы и желудка [54]. Киназа Pim-1 выступает в качестве промотора продвижения опухолевых клеток по клеточному циклу, миграции клеток и процессов трансляции. Pim-1 подавляет апоптоз [55]. Получены данные, свидетельствующие о том, что сверхэкспрессия киназ PIM1/2 приводит к выраженному воспалительному ответу, т.е. существует связь между экспрессией PIM1/2 и воспалением [55].
Показано, что киназа Pim-1фосфорилирует Pgp по Ser683 [53]. Подавление активности Pim-1 приводило к снижению степени фосфорилирования и стабильности Pgp [56]. Pim-1 фосфорилирует также и BCRP (ABCG2) [57]. Таким образом, киназа Pim-1 вовлечена как в регуляцию воспаления, так и в регуляцию активности по меньшей мере двух АВС-транспортеров.
4) В регуляции активности Pgp, а также в регуляции воспаления принимает участие многофункциональный белок YB-1. YB-1 входит в суперсемейство белков с эволюционно консервативным доменом холодового шока. В прокариотических и эукариотических клетках белок YB-1 связывается с ДНК и РНК и модулирует транскрипцию и трансляцию многих генов. Показано, что YB-1 является участником процессов воспаления [58, 59]. YB-1 регулирует экспрессию нескольких генов, кодирующих драйверы воспаления mTOR, STAT3, MMP-2, CD44, CCL5, CCL2 [58]. Недавно обнаружили, что YB-1 регулирует экспрессию IL-10, важного противовоспалительного цитокина [60]. Всплеск интереса к YB-1 и его внутриклеточной локализации начался с опубликованной в 1997 году работы R. Bargou и соавт. [61], которые впервые показали, что внутриклеточный YB-1 регулирует экспрессию гена MDR1. Взаимодействуют ли эти два белка (YB-1 и Pgp) в процессе воспаления? Что вносят они в процессы воспаления? Какую роль процессы воспаления в сочетании с экспрессией YB-1 и Pgp играют в канцерогенезе и опухолевой прогрессии? На эти вопросы еще предстоит дать ответы.
ЭКСПРЕССИЯ И АКТИВНОСТЬ PGP ПРИ АУТОИММУННЫХ ЗАБОЛЕВАНИЯХ
Аутоиммунные заболевания, такие как системная красная волчанка (СКВ), ревматоидный артрит (РА) и псориатический артрит (ПСА), являются хроническими воспалительными нарушениями неизвестной этиологии, которые характеризуются широким спектром аномалий иммунной системы, нарушающих функции почек, сердца, суставов, мозга, кожи. Кортикостероиды, синтетические и биологические иммунодепрессанты и противоревматические препараты (сульфасалазин, метотрексат, лефлуномид) улучшают течение аутоиммунных заболеваний. Тем не менее, значительное число пациентов изначально не реагирует на эти препараты или с течением времени становятся резистентными к ним. За последние 13 лет проведено немало исследований, в которых оценивали экспрессию Pgp лимфоцитами периферической крови пациентов с различными аутоиммунными заболеваниями. Как правило, в таких исследованиях активность Pgp определяли методом проточной цитометрии и по выбросу флуоресцентного красителя Rh123. Обнаружено повышение экспрессии Pgp и его активности в лимфоцитах периферической крови пациентов с СКВ и РА. При этом отмечена выраженная корреляция между уровнем экспрессии Pgp и активностью заболевания [62–66].
Анализ изменений экспрессии и активности Pgp у больных СКВ, получавших лечение, выявил высокий уровень Pgp у больных, резистентных к кортикостероидам [66–68]. Такие же результаты получены и у больных нефротическим синдромом, получавших стероидные препараты. Экспрессия и активность Pgp в лимфоцитах значимо снижалась у больных, которые отвечали на терапию, и увеличивались при развитии резистентности [69, 70].
В мононуклеарных клетках периферической крови больных СКВ, получавших метотрексат и ответивших на него, активность Р-гликопротеина была выше, чем у резистентных к этому препарату [71].
Противоположные результаты получены при определении экспрессии и активности Pgp лимфоцитами периферической крови больных РА, получивших метотрексат в течение 4 месяцев. У больных, которые оказались устойчивыми к терапии, как экспрессия так и активность Pgp увеличились по сравнению с исходными значениями. У больных, ответивших на терапию, эти показатели значительно снизились [72]. Определить механизмы, лежащие в основе этой резистентности, а также понять, может ли активность Pgp влиять на эффективность лечения метотрексатом, позволят дальнейшие исследования.
Японскими исследователями выявлена сверхэкспрессия Pgp периферическими лимфоцитами пациентов с высокой активностью РА. Значительная корреляция между уровнем экспрессии Pgp и активностью РА связана с активным выбросом лекарственных средств из цитоплазмы лимфоцитов и резистентностью к базовым противоревматическим препаратам. Оказалось, что препараты, которые снижают экспрессию Pgp, а также антагонисты Pgp (например, циклоспорин), могут снизить выброс кортикостероидов из лимфоцитов in vitro. Эти результаты показывают, что оба типа препаратов можно использовать для преодоления лекарственной резистентности и улучшения клинического исхода РА [73]. В ранней работе этими же исследователями было показано, что уровни дексаметазона в лимфоцитах больных РА уменьшались в соответствии с увеличением экспрессии Pgp. Такролимус, ингибитор Pgp, повышал уровень дексаметазона в лимфоцитах [64].
Расширенное исследование терапевтического действия такролимуса было проведено другой группой ученых [74]. Пациенты с рефрактерным РА (113) получали такролимус (1.5–3 мг/день) в дополнение к противоревматическим препаратам, включая метотрексат. Ответ оценивали через 2 недели. Экспрессию гена MDR1 и P-гликопротеина оценивали в мононуклеарных клетках периферической крови. Активность P-гликопротеина определяли по отношению содержания остаточного внутриклеточного меченного тритием дексаметазона к его содержанию в среде (отношение C/M). Хороший клинический ответ на такролимус отмечен через 2 недели у 22 из 113 пациентов. До введения такролимуса у всех пациентов с РА обнаружена повышенная экспрессия гена MDR-1 и P-гликопротеина и низкое отношение C/M. Реакция на такролимус коррелировала с экспрессией P-гликопротеина и отношением C/M. Соотношение С/М значимо коррелировало с экспрессией Р-гликопротеина на CD4+ лимфоцитах [74].
Трое устойчивых к метотрексату и сульфасалазину пациентов с ПСА в течение месяца получали дополнительно циклоспорин А. Функцию Pgp до и после лечения циклоспорином А оценивали по изменению выброса Rh123 лимфоцитами периферической крови обработанными верапамилом. Прием циклоспорина привел к улучшению клинических показателей, что свидетельствовало о преодолении устойчивости к метотрексату. Показано, что введение циклоспорина в схему комплексной терапии РА привело к значительному снижению функции Pgp в CD3+ и CD8+ лимфоцитах [75].
Эти немногочисленные, но убедительные данные позволяют предположить, что активируемые различными стимулами лимфоциты больных с активными аутоиммунными заболеваниями, приобретают МЛУ к противоревматическим препаратам и кортикостероидам, опосредованную Pgp. Ингибирование/уменьшение Pgp могло бы преодолеть такую лекарственную устойчивость. Уровень экспрессии и активности Pgp в лимфоцитах периферической крови можно рассматривать в качестве перспективного маркера лекарственной устойчивости и терапевтической мишени для ее предотвращения.
В связи с тем, что исследованию экспрессии Pgp при воспалительных заболеваниях придается все большее значение, определенный интерес представляют две работы, в которых оценивали количество внеклеточного (сывороточного) Pgp.
Группа исследователей под руководством Gonzalez-Lopez L. оценила связь уровня экспрессии Pgp в сыворотке крови при СКВ с активностью болезни и ответом на лечение. В исследовании участвовало 93 пациента с СКВ, все получали глюкокортикоиды в стабильных дозах в течение 6 месяцев до начала исследования. Все пациенты были разделены на две группы. В первую группу вошли пациенты с активной СКВ [индекс активности СКВ (SLEDAI) ≥ 3], во вторую – с неактивной СКВ (SLEDAI < 3) после лечения. В контрольную группу вошли 43 здоровых донора. Экспрессию Pgp измеряли с методом ELISA. У пациентов с активной СКВ, несмотря на проводимую терапию, уровни Pgp были выше, чем в группе с неактивной СКВ и в контрольной группе. Уровни Pgp коррелировали с величинами SLEDAI, т.е с активностью заболевания [76].
Выявлено также значительное повышение экспрессии Pgp в слизи носовых пазух больных хроническим риносинуситом. Высокая секреция Pgp чаще встречалась у пациентов с назальными полипами, чем у пациентов без полипов и была ассоциирована с более плохими показателями тяжести заболевания. Предполагается, что уровень экспрессии Pgp в слизи носовых пазух можно рассматривать как новый неинвазивный биомаркер хронического риносинусита и использовать его для выделения пациентов, у которых возможно применение ингибиторов Pgp [77].
В чем биологический смысл связи между изменением экспрессии АВС-белков и воспалением? Приведем лишь несколько возможных объяснений, которые представляются нам наиболее вероятными.
АВС-белки могут быть необходимы для выработки медиаторов воспаления. АВС-транспортеры могут выводить из клеток такие противовоспалительные молекулы, как лейкотриены, простагландины, цитокины (например, IL-2, IFN-γ, TNF-α, биоактивные липиды). Вклад АВС-транспортеров в воспаление может определяться их экспрессией различными иммунными клетками и влиянием активных транспортеров на повышение жизнеспособности этих клеток при медикаментозном лечении.
Список литературы
Higgins C.F. 2007. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 446 (7137), 749–757.
Gillet J.P., Gottesman M.M. 2010. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 596, 47–76.
Ставровская А.А., Генс Г.П. 2014. Некоторые новые аспекты исследований множественной лекарственной устойчивости опухолевых клеток. Успехи молекулярной онкологии. 1, 5–11.
Wilkens S. 2015. Structure and mechanism of ABC transporters. F1000Prime Rep. 7, 14.
Хитров Н.К., Саркисов Д.С., Пальцев М.А. 1999. Руководство по общей патологии человека. М.: Медицина, 728 с.
Grivennikov S.I., Greten F.R., Karin M. 2010. Immunity, inflammation, and cancer. Cell. 140(6), 883–899.
Mantovani A., Romero P., Paluka A.K., Marincola F.M. 2008. Tumor immunity: Effector response to tumor and the influence of the microenvironment. Lancet. 371, 771–783.
Panwala C.M., Jones J.C., Viney J.L.1998. A novel model of inflammatory bowel disease: Mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J. Immunol. 161, 5733–5744.
Wilk J.N., Bilsborough J., Viney J.L. 2005. The mdr1a–/– mouse model of spontaneous colitis: A relevant and appropriate animal model to study inflammatory bowel disease. Immunol. Res. 31, 151–159.
Saksena S., Priyamvada S., Kumar A., Akhtar M., Soni V., Anbazhagan A.N., Alakkam A., Alrefai W.A., Dudeja P.K., Gill R.K. 2013. Keratinocyte growth factor-2 stimulates P-glycoprotein expression and function in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G615–G622.
Saksena S., Goyal S., Raheja G., Singh V., Akhtar M., Nazir T.M., Alrefai W.A., Gill R.K., Dudeja P.K. 2011. Upregulation of P-glycoprotein by probiotics in intestinal epithelial cells and in the dextran sulfate sodium model of colitis in mice. Am. J. Physiol. Gastrointest. Liver. Physiol. 300, G1115–G1123.
Miceli R., Hubert M., Santiago G., Yao D.L., Coleman T.A., Huddleston K.A., Connolly K. 1999. Efficacy of keratinocyte growth factor-2 in dextran sulfate sodium-induced murine colitis. J. Pharmacol. Exp. Ther. 290, 464–471.
Nanda Kumar N.S., Balamurugan R., Jayakanthan K., Pulimood A., Pugazhendhi S., Ramakrishna B.S. 2008. Probiotic administration alters the gut flora and attenuates colitis in mice administered dextran sodium sulfate. J. Gastroenterol. Hepatol. 23, 1834–1839.
Neudeck B.L., Loeb J.M., Faith N.G., Czuprynski C.J. 2004. Intestinal P-glycoprotein acts as a natural defense mechanism against Listeria monocytogenes. Infect. Immun. 72, 3849–3854.
Hoffmeyer S., Burk O., von Richter O., Arnold H.P., Brockmöller J., Johne A., Cascorbi I., Gerloff T., Roots I., Eichelbaum M., Brinkmann U. 2000. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA. 97, 3473–3478.
Farnood A., Naderi N., Moghaddam S.J., Noorinayer B., Firouzi F., Aghazadeh R., Daryani N.E., Zali M.R. 2007. The frequency of C3435T MDR1 gene polymorphism in Iranian patients with ulcerative colitis. Int. J. Colorectal Dis. 22, 999–1003.
Juyal G., Midha V., Amre D., Sood A., Seidman E., Thelma B.K. 2009. Associations between common variants in the MDR1 (ABCB1) gene and ulcerative colitis among North Indians. Pharmacogenet. Genomics. 19, 77–85.
Ho G.T., Nimmo E.R., Tenesa A., Fennell J., Drummond H., Mowat C., Arnott I.D., Satsangi J. 2005. Allelic variations of the multidrug resistance gene determine susceptibility and disease behavior in ulcerative colitis. Gastroenterology. 128, 288–296.
Mijac D., Vukovic-Petrovic I., Mijac V., Perovic V., Milic N., Djuranovic S., Bojic D., Popovic D., Culafic D., Krstic M., Jankovic G., Pravica V., Markovic M. 2018. MDR1 gene polymorphisms are associated with ulcerative colitis in a cohort of Serbian patients with inflammatory bowel disease. PLoS One.15, 13(3). e0194536.
Croucher P.J., Mascheretti S., Foelsch U.R., Hampe J., Schreiber S. 2003. Lack of association between the C3435T MDR1 gene polymorphism and inflammatory bowel disease in two independent Northern European populations. Gastroenterology. 125, 1919–1920.
Oostenbrug L.E., Dijkstra G., Nolte I.M., van Dullemen H.M., Oosterom E., Faber K.N., de Jong D.J., van der Linde K., te Meerman G.J., van der Steege G., Kleibeuker J.H., Jansen P.L. 2006. Absence of association between the multidrug resistance (MDR1) gene and inflammatory bowel disease. Scand. J. Gastroenterol. 41, 1174–1182.
Fischer S., Lakatos P.L., Lakatos L., Kovacs A., Molnar T., Altorjay I., Papp M., Szilvasi A., Tulassay Z., Osztovits J., Papp J., Demeter P., Schwab R., Tordai A., Andrikovics H. 2007. ATP-binding cassette transporter ABCG2 (BCRP) and ABCB1 (MDR1) variants are not associated with disease susceptibility, disease phenotype response to medical therapy or need for surgeryin Hungarian patients with inflammatory bowel diseases. Scand. J. Gastroenterol. 42,726–733.
Ставровская А.А., Моисеева Н.И. 2016. Неканонические функции транспортного белка Р-гликопротеина. Биол. мембраны. 33 (5), 323–334.
Kooij G., Mizee M.R., van Horssen J., Reijerkerk A., Witte M.E., Drexhage J.A., van der Pol S.M., van Het Hof B., Scheffer G., Scheper R., Dijkstra C.D., van der Valk P., de Vries H.E. 2011. Adenosine triphosphate-binding cassette transporters mediate chemokine (C-C motif) ligand 2 secretion from reactive astrocytes: Relevance to multiple sclerosis pathogenesis. Brain. 134(Pt 2), 555–570.
Kooij G., van Horssen J., Bandaru V.V.R., Haughey N.J., de Vries H.E. 2012. The role of ATP-binding cassette transporters in neuro-inflammation: Relevance for bioactive lipids. Front. Pharmacol. 3, 74.
Englund G., Jacobson A., Rorsman F., Artursson P., Kindmark A., Rönnblom A. 2007. Efflux transporters in ulcerative colitis: Decreased expression of BCRP (ABCG2) and Pgp (ABCB1). Inflamm. Bowel Dis. 13, 291–297.
Gutmann H., Hruz P., Zimmermann C., Straumann A., Terracciano L., Hammann F., Lehmann F., Beglinger C., Drewe J. 2008. Breast cancer resistance protein and P-glycoprotein expression in patients with newly diagnosed and therapy-refractory ulcerative colitis compared with healthy controls. Digestion. 78, 154–162.
Sukhai M., Yong A., Kalitsky J., Piquette-Miller M. 2000. Inflammation and interleukin-6 mediate reductions in the hepatic expression and transcription of the mdr1a and mdr1b genes. Mol. Cell. Biol. Res. Commun. 4, 248–256.
Sukhai M., Yong A., Pak A., Piquette-Miller M. 2001. Decreased expression of Pgl glycoprotein in interleukin-1beta and interleukin-6 treated rat hepatocytes. Inflamm. Res 50, 362–370.
Poller B., Drewe J., Krahenbuhl S., Huwyler J., Gutmann H. 2010. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol. Neurobiol. 30, 63–70.
Iqbal M., Ho H.L., Petropoulos S., Moisiadis V.G., Gibb W., Matthews S.G. 2012. Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood-brain barrier. PLoS ONE. 7 (8), e43022.
Hartz A.M., Bauer B., Fricker G., Miller D.S. 2006. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol. Pharmacol. 69, 462–470.
Bauer B., Hartz A.M., Miller D.S. 2007. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 71, 667–675.
Liptrott N.J., Penny M., Bray P.G., Sathish J., Khoo S.H., Back D.J., Owen A. 2009. The impact of cytokines on the expression of drug transporters, cytochrome P450 enzymes and chemokine receptors in human PBMC. Br. J. Pharmacol. 156, 497–508.
Puddu P., Fais S., Luciani F., Gherardi G., Dupuis M.L., Romagnoli G., Ramoni C., Cianfriglia M., Gessani S. 1999. Interferon-gamma up-regulates expression and activity of P-glycoprotein in human peripheral blood monocyte-derived macrophages. Lab. Invest. 79, 1299–1309.
Hartmann G., Kim H., Piquette-Miller M. 2001. Regulation of the hepatic multidrug resistance gene expression by endotoxin and inflammatory cytokines in mice. Int. Immunopharmacol. 1, 189–199.
Lee S.H., Oh T., Jeon B.Y., Kwak E.-Y., Shim W.-S., Cho S.-N., Shim C.-K. 2009. Tissue-specific changes in mRNA expression of Abc and Slc transporters in murine pulmonary tuberculosis. Xenobiotica. 39 (10), 738–748.
Gollapudi S., Reddy M., Gangadharam P., Tsuruo T., Gupta S. 1994. Mycobacterium tuberculosis induces expression of P-glycoprotein in promonocytic U1 cells chronically infected with HIV type 1. Biochem. Biophys. Res. Commun. 199 (3), 1181–1187.
Yagdiran Y., Tallkvist J., Artursson K., Oskarsson A. 2016. Staphylococcus aureus and lipopolysaccharide modulate gene expressions of drug transporters in mouse mammary epithelial cells correlation to inflammatory biomarkers. PLoS ONE. 11 (9), e0161346.
Haslam I.S., Jones K., Coleman T., Simmons N.L. 2008. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem. Pharmacol. 76 (7), 850–861.
Siccardi D., Mumy K.L., Wall D.M., Bien J.D., McCormick B.A. 2008. Salmonella enterica serovar Typhimurium modulates P-glycoprotein in the intestinal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1392–G1400
Sigal N., Kaplan Zeevi M., Weinstein S., Peer D., Herskovits A.A. 2015. The human P-glycoprotein transporter enhances the type I interferon response to Listeria monocytogenes infection. Infect. Immun. 83 (6), 2358–2368.
Barnes K.M., Dickstein B., Cutler G.B., Jr, Fojo T., Bates S.E. 1996. Steroid treatment, accumulation, and antagonism of P-glycoprotein in multidrugresistant cells. Biochemistry. 35, 4820–4827.
Ernest S., Bello-Reuss E. 1999. Secretion of platelet-activating factor is mediated by MDR1 P-glycoprotein in cultured human mesangial cells. J. Am. Soc. Nephrol. 10, 2306–2313.
Raggers R.J., Vogels I., van Meer G. 2001. Multidrug-resistance P-glycoprotein (MDR1) secretes platelet-activating factor. Biochem. J. 357, 859– 865.
Frank M.H., Denton M.D., Alexander S.I., Khoury S.J., Sayegh M.H., Briscoe D.M. 2001. Specific MDR1 P-glycoprotein blockade inhibits human alloimmune T cell activation in vitro. J. Immunol. 166, 2451–2459.
Drach J., Gsur A., Hamilton G., Zhao S., Angerler J., Fiegl M., Zojer N., Raderer M., Haberl I., Andreeff M., Huber H. 1996. Involvement of P-glycoprotein in the transmembrane transport of interleukin-2 (IL-2), IL-4, and interferon-gamma in normal human T lymphocytes. Blood. 88, 1747–1754.
Yvan-Charvet L., Welch C., Pagler T.A., Ranalletta M., Lamkanfi M., Han S., Ishibashi M., Li R., Wang N., Tall A.R. 2008. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via Toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 118, 1837–1847.
Koseki M., Hirano K., Masuda D., Ikegami C., Tanaka M., Ota A., Sandoval J.C., Nakagawa-Toyama Y., Sato S.B., Kobayashi T., Shimada Y., Ohno- Iwashita Y., Matsuura F., Shimomura I., Yamashita S. 2007. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-alpha secretion in Abca1-deficient macrophages. J. Lipid Res. 48, 299–306.
Zhu X., Lee J.Y., Timmins J.M., Brown J.M., Boudyguina E., Mulya A., Gebre A.K., Willingham M.C., Hiltbold E.M., Mishra N., Maeda N., Parks J.S. 2008. Increased cellular free cholesterol in macrophage-specific Abca1 knockout mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 283, 22930–22941.
Rifkin I.R., Leadbetter E.A., Busconi L., Viglianti G., Marshak-Rothstein A. 2005. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 204, 27–42.
Westerterp M., Bochem A.E., Yvan-Charvet L., Murphy A.J., Wang N., Tall A.R. 2014. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 114 (1), 157–170.
Liu J., Zhou F., Chen Q., Kang A., Lu M., Liu W., Zang X., Wang G., Zhang J. 2015. Chronic inflammation up-regulates Pgp in peripheral mononuclear blood cells via the STAT3/NF-κB pathway in 2,4,6-trinitrobenzene sulfonic acid-induced colitis mice. Sci. Rep. 5, 13558. doi 10.1038/srep13558
Bachmann M., Moroy T. 2005. The serine/threonine kinase Pim-1. Internat. J. Biochem. Cell Biol. 37 (4), 726–730.
Jiménez-García M.-P., Lucena-Cacace A., Robles-Frías M.-J., Ferrer I., Narlik-Grassow M., Blanco-Aparicio C., Carnero A. 2017. Inflammation and stem markers association to PIM1/PIM2 kinase-induced tumors in breast and uterus. Oncotarget, 8 (35), 58 872–58 886.
Xie Y., Burcu M., Linn D.E., Qiu Y., Baer M.R. 2010. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol. Pharmacol. 78, 2, 310–318.
Xie Y., Xu K., Linn D.E., Yang X., Guo Z., Shimelis H., Nakanishi T., Ross D.D.,Chen H., Fazli L., Gleave M.E., Qiu Y. 2008. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J. Biol. Chem. 283, 6, 3349–3356.
Елисеева И.А., Ким Е.Р., Гурьянов С.Г., Овчинников Л.П., Лябин Д.Н. 2011. Y-бокс-связывающий белок (YB-1) и его функции. Усп. Биол. Хим. 51, 65–132.
Lasham A., Print C.G., Woolley A.G., Dunn S.E., Braithwaite A.W. 2013. YB-1: Oncoprotein, prognostic marker and therapeutic target? Biochem. J. 449 (1), 11–23.
Wang J., Djudjaj S., Gibbert L., Lennartz V., Breitkopf D.M., Rauen T., Hermert D., Martin I.V., Boor P., Braun G.S., Floege J., Ostendorf T., Raffetseder U. 2017. YB-1 orchestrates onset and resolution of renal inflammation via IL10 gene regulation. J. Cell. Mol. Med. 21 (12), 3494–3505.
Bargou R.C., Jurchott K., Wagener C., Bergmann S., Metzner S., Bommert K., Mapara M.Y., Winzer K.J., Dietel M., Dorken B., Royer H.D. 1997. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat. Med. 3 (4), 447–450.
Tsujimura S., Saito K., Nakayamada S., Nakano K., Tanaka Y. 2005. Clinical relevance of the expression of P-glycoprotein on peripheral blood lymphocytes to steroid resistance in patients with systemic lupus erythematosus. Arthritis Rheum. 52 (6), 1676–1683.
Tsujimura S., Saito K., Nakayamada S., Tanaka Y. 2007. Relevance of multidrug resistance 1 and P-glycoprotein to drug resistance in patients with systemic lupus erythematosus. Histol. Histopathol. 22 (4), 465–468.
Tsujimura S., Saito K., Nawata M., Nakayamada S., Tanaka Y. 2008. Overcoming drug resistance induced by P-glycoprotein on lymphocytes in patients with refractory rheumatoid arthritis. Ann. Rheum. Dis. 67 (3), 380–388.
Zhang B., Shi Y., Lei T.C. 2012. Detection of active P-glycoprotein in systemic lupus erythematosus patients with poor disease control. Exp. Ther. Med. 4 (4), 705–710.
Tsujimura S., Adachi T., Saito K., Tanaka Y. 2017. Role of P-glycoprotein on CD69+CD4+ cells in the pathogenesis of proliferative lupus nephritis and non-responsiveness to immunosuppressive therapy. RMD Open. 3 (1), e000423.
Henmi K., Yoshida M., Yoshikawa N., Hirano T. 2008. P-glycoprotein functions in peripheral-blood CD4+ cells of patients with systemic lupus erythematosus. Biol. Pharm. Bull. 31(5), 873–878.
Kansal A., Tripathi D., Rai M.K., Agarwal V. 2016. Persistent expression and function of P-glycoprotein on peripheral blood lymphocytes identifies corticosteroid resistance in patients with systemic lupus erythematosus. Clin. Rheumatol. 35(2), 341–349.
Badr H.S., El-Hawy M.A., Helwa M.A. 2016. P-glycoprotein activity in steroid-responsive vs. steroid-resistant nephrotic syndrome. Indian J. Pediatr. 83(11), 1222–1226
Prasad N., Jaiswal A.K., Agarwal V., Yadav B., Sharma R.K., Rai M., Singh H., Chaturvedi S., Singh A. 2015. Differential alteration in peripheral T-regulatory and T-effector cells with change in P-glycoprotein expression in childhood nephrotic syndrome: A longitudinal study. Cytokine. 72(2), 190–196.
García-Carrasco M., Mendoza-Pinto C., Macías-Díaz S., Etchegaray-Morales I., Méndez-Martínez S., Soto-Santillán P., Pérez-Romano B., Jiménez-Herrera E.A., Guzmán-Ruiz O., Ruiz-Argüelles A. 2017. Clinical relevance of P-glycoprotein activity on peripheral blood mononuclear cells and polymorphonuclear neutrophils to methotrexate in systemic lupus erythematosus patients. Clin. Rheumatol. 36 (10), 2267–2272.
Prasad S., Tripathi D., Rai M.K., Aggarwal S., Mittal B., Agarwal V. 2014. Multidrug resistance protein-1 expression, function and polymorphisms in patients with rheumatoid arthritis not responding to methotrexate. Int. J. Rheum. Dis. 17 (8), 878–886.
Tsujimura S., Tanaka Y. 2015. Disease control by regulation of P-glycoprotein on lymphocytes in patients with rheumatoid arthritis. World J. Exp. Med. 5 (4), 225–231.
Suzuki K., Saito K., Tsujimura S., Nakayamada S., Yamaoka K., Sawamukai N., Iwata S., Nawata M., Nakano K., Tanaka Y. 2010. Tacrolimus, a calcineurin inhibitor, overcomes treatment unresponsiveness mediated by P-glycoprotein on lymphocytes in refractory rheumatoid arthritis. J. Rheumatol. 37 (3), 512–520.
Diamanti A.P., Rosado M., Germano V., Scarsella M., Giorda E., Podestà E., D’Amelio R., Carsetti R., La-ganà B. 2011. Reversion of resistance to immunosuppressive agents in threepatients with psoriatic arthritis by cyclosporine A: Modulation of P-glycoprotein function. Clin. Immunol. 138 (1), 9–13.
Perez-Guerrero E.E., Gamez-Nava J.I., Muñoz-Valle J.F., Cardona-Muñoz E.G.,, Bonilla-Lara D.,, Fajardo-Robledo N.S., Nava-Zavala A.H., Garcia-Cobian T.A., Rincón-Sánchez A.R., Murillo-Vazquez J.D., Cardona-Müller D., Vazquez-Villegas M.L., Totsuka-Sutto S.E., Gonzalez-Lopez L. 2018. Serum levels of P-glycoprotein and persistence of disease activity despite treatment in patients with systemic lupus erythematosus. Clin. Exp. Med. 18 (1), 109–117.
Nocera A.L., Meurer A.T., Miyake M.M., Sadow P.M., Han X., Bleier B.S. 2017. Secreted P-glycoprotein is a noninvasive biomarker of chronic rhinosinusitis. Laryngoscope. 127 (1), E1–E4.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии