

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 6, стр. 409-416
Роль митохондриальной сигнальной системы NO/cGMP/PKG в активации и ингибировании дыхания L-аргинином и донорами NO
В. В. Дынник a, *, Е. В. Гришина a, Н. И. Федотчева a
a Институт теоретической и экспериментальной биофизики РАН
142290 г. Пущино, Московской обл., ул. Институтская, 3, Россия
* E-mail: dynnik@rambler.ru
Поступила в редакцию 19.04.2019
После доработки 17.05.2019
Принята к публикации 17.05.2019
Аннотация
Участие митохондриальной кальций-зависимой NO-синтазы (mtNOS) в регуляции митохондриального дыхания изучено недостаточно. Более того, возможное функционирование сигнальной системы с участием mtNOS/гуанилатциклазы (GC)/киназы G (PKG) и влияние этой системы (mtNOS/GC/PKG–SS) на дыхание митохондрий еще не проанализировано. Для исследования этого вопроса мы провели эксперименты на изолированных митохондриях печени крыс и сердца с применением специфических ингибиторов NOS, GC и PKG. Высокая скорость митохондриального дыхания поддерживалась пируватом и глутаматом или сукцинатом в присутствии гексокиназы, глюкозы и ADP. Показано, что L-аргинин и донор NO SNP оказывают дозозависимое действие на скорость дыхания митохондрий. При низких концентрациях L-аргинин (до 200 мкМ) и SNP (до 100 мкМ) активируют дыхание митохондрий. Ингибиторы NOS, GC и PKG устраняют этот эффект, что указывает на участие mtNOS/GC/PKG–SS в активации дыхания. В высоких концентрациях L-аргинин и SNP, наоборот, ингибируют дыхание. В этих условиях ингибиторы GC и PKG вызывают обратный эффект, усиливая ингибирование дыхания, что указывает на противоположное влияние избытка NO и PKG на митохондриальное дыхание. На основании полученных результатов можно предположить, что функционирование кальций-зависимой mtNOS/GC/PKG–SS может обеспечивать активацию дыхания при малых концентрациях L-аргинина или SNP в среде.
ВВЕДЕНИЕ
Исследованиями последних десятилетий убедительно показано, что экзогенный NO, а также NO, продуцируемый цитоплазматическими NO-синтазами (NOS), приводят к подавлению дыхания митохондрий, ингибируя цитохром-с-оксидазу (COX) и комплексы I и II цепи переноса электронов [1–6]. Ингибирование COX посредством NO обратимо и конкурентно по отношению к кислороду [1]. В экспериментах, выполненных на клетках НЕК в условиях сверхэкспрессии индуцибельной синтазы оксида азота (iNOS), показано, что увеличение концентрации L-аргинина в среде от 1 до 10 мкМ приводит к восстановлению цитохромов, накоплению NO и подавлению потребления кислорода [6]. Еще одним источником NO является локализованная на внутренней мембране митохондрий изоформа mtNOS, которая согласно данным, полученным на митохондриях печени и сердца, образуется в результате сплайсинга мРНК нейрональной nNOSa [7–9]. Активация кальций-зависимой mtNOS ее субстратом L-аргинином и Са2+ также вызывает ингибирование дыхания митохондрий, сопряженное с резким ростом NO [10]. Как и цитоплазматические формы, mtNOS, является кислород- и NADPH-зависимым ферментом, поэтому продукция митохондриального NO максимальна при высоких значениях мембранного потенциала ΔΨm и низких скоростях дыхания митохондрий (в состоянии покоя – в состоянии 4) [10]. NO является источником нитрозотиолов, пероксинитрита, АФК и др., мишенями которых являются многочисленные белки митохондриального метаболизма. Поэтому митохондрия зачастую рассматривается как генератор и мишень действия NO [11] вне связи с регуляторным действием Са2+. Глобальный характер действия Са2+ на метаболизм митохондрий, включающий активацию белков дыхательной цепи и цикла Кребса [12], активацию синтеза NO, индукцию митохондриальной поры [13, 14], свидетельствует о наличии регуляторного процесса, основанного на взаимном влиянии NO и Са2+, который в настоящее время недостаточно изучен.
Учитывая сложный и малоисследованный характер взаимодействия NO и Са2+, нами проанализировано влияние доноров NO и L-аргинина на регуляцию дыхания митохондрий. Мы предполагаем, что регуляция дыхания митохондрий с участием Са2+-зависимой mtNOS не исчерпывается только нитрозилированием белков митохондрий или ингибированием ферментов дыхательной цепи посредством NO, но может включать также функционирование митохондриальной сигнальной системы с участием mtNOS , гуанилатциклазы (GC) и протеинкиназы G (PKG). Влияние этой системы на метаболизм и дыхание митохондрий недостаточно изучено. Задача настоящей работы заключалась в том, чтобы с помощью ингибиторного анализа показать, что L-аргинин и доноры NO, активируя митохондриальную Ca2+→mtNOS→NO→mtGC→cGMP→mtPKG–сигнальную систему (mtNOS/GC/PKG–SS), могут участвовать в регуляции скорости дыхания митохондрий.
МАТЕРИАЛЫ И МЕТОДЫ
Все процедуры на животных выполнялись в соответствии с директивой ЕС 86/609/EEC и были одобрены комитетом по этике Института теоретической и экспериментальной биофизики РАН. Самцов крыс линии Вистар 6–8 недель содержали в одинаковых условиях в кондиционированных и проветриваемых помещениях при температуре 20–22°С (свет/темнота = 12 ч/12 ч). Эксперименты проводили при 26°С. Митохондрии печени выделяли с использованием стандартных методик дифференциального центрифугирования в среде, содержащей 300 мМ сахарозы, 1 мМ EGTA и 10 мМ Трис-HCl (рН 7.4). Митохондриальные препараты дважды промывали средой выделения, не содержащей EGTA, ресуспендировали в среде того же состава и хранили на льду. Инкубационная среда для митохондрий содержала: 125 мМ KCl, 3 мМ KH2PO4, 10 мМ HEPES (pH 7.4), 1 мМ MgCl2. Содержание митохондриального белка определяли методом Лоури с бычьим сывороточным альбумином в качестве стандарта. Потребление кислорода в митохондриальной суспензии определяли полярографическим методом с помощью O2-электрода Кларка в закрытой камере объемом 1 мл, содержащей 1.0 мг белка, при постоянном перемешивании. В качестве субстратов дыхания использовали L‑глутамат (10 мМ) и пируват (0.5–1 мМ) для поддержания полного оборота цикла Кребса и сохранения субстратного фосфорилирования в цикле. В некоторых экспериментах дыхание поддерживалось сукцинатом (5 мМ) в присутствии ротенона (1–2 мкМ) или пируватом (1 мМ) и L‑малатом (5 мМ). Все эксперименты выполняли в присутствии 10 ед. гексокиназы, 5 мМ глюкозы и 1 мМ MgCl. Последующие добавки 0.75 мМ ADP обеспечивали высокую стационарную скорость дыхания (VO2ss = d[O2]/dt), близкую к максимальной скорости дыхания в состоянии 3 (80–85% от VO2max). Чтобы оценить статистическую значимость результатов, VO2ss измеряли в стационарном состоянии при [O2] = 270‒300 нг-атом О/мл на линейных участках соответствующих полярографических кривых в серии из пяти‒шести экспериментов. Разность электрических потенциалов (ΔΨm) на внутренней мембране митохондрий определяли по распределению липофильного катиона тетрафенилфосфония (ТРР+), концентрацию которого во внешней среде [ТРР+]out регистрировали с помощью ТРР+–селективного электрода. В работе использованы реактивы фирмы Sigma (США). Статистический анализ экспериментальных данных проводили с использованием t-критерия Сигма-Плот 11 для сравнения пар величин. Данные представлены как среднее значение ± S.E.M. n = 4‒6 независимых экспериментов. За уровень значимости принято p <0.05.
РЕЗУЛЬТАТЫ
Активация и ингибирование дыхания митохондрий SNP и L-аргинином. На рис. 1 представлены полярографические кривые, характеризующие потребление кислорода митохондриями печени крыс при активации скорости дыхания добавками 0.75 мМ АDP в среду. В качестве субстратов окисления использованы пируват (1 мМ) и L-глутамат (10 мМ). Стационарную скорость дыхания поддерживали добавлением гексокиназы и глюкозы в среду инкубации. На рис. 1 представлена также соответствующая динамика митохондриального потециала ΔΨm (TPP+). Преинкубация митохондрий с низкими концентрациями SNP (20 мкМ; пунктирная кривая на рис. 1а) или L-аргинина (20 мкМ; пунктирная кривая на рис. 1б) вызывает увеличение скорости дыхания митохондрий, что характеризуется увеличением наклона соответствующих кривых.
Рис. 1.
Влияние SNP (слева) и L-аргинина (справа) на потребление кислорода (линии O2) и митохондриальный потециал ΔΨm (линии TPP+) зависит от концентраций этих источников NO в среде. Представлены репрезентативные эксперименты. Черные кривые соответствуют контрольным экспериментам. Пунктирные кривые соответствуют низким концентрациям SNP (рис. 1а, 10 мкМ) и L-аргинина (рис. 1б, 100 мкМ) в инкубационной среде. Серые линии соответствуют высоким концентрациям SNP (рис. 1а, 500 мкМ) и L-аргинина (рис. 1б, 1000 мкМ). Митохондрии инкубировали в закрытой камере объемом 1 мл. Среда инкубации включала 1 мМ пирувата, 10 мМ L-глутамата, а также глюкозу и гексокиназу. в, г – Зависимости средней скорости дыхания в стационарном состоянии VO2ss от концентраций SNP (в) и L-аргинина (г) в среде. Количество независимых экспериментов n = 5. Данные представляют собой среднее ± S.E.M.
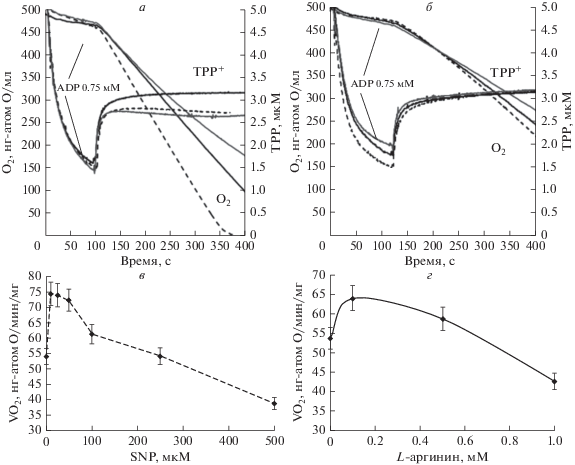
Высокие концентрации SNP (400 мкМ; серая кривая на рис. 1а) и L-аргинина (1000 мкМ; серая кривая на рис. 1б), наоборот, вызывают ингибирование митохондриального дыхания до значений ниже контрольных величин. Наблюдаемое ингибирование дыхания SNP и L-аргинином хорошо известно [1–6].
Такое подавление дыхания может быть вызвано известным ингибированием COX и других белков дыхательной цепи избытком NO, выделяющегося из SNP или продуцируемого mtNOS из L-аргинина [1, 4, 6]. В отличие от ингибирующего эффекта, активация дыхания малыми концентрациями доноров NO.или L-аргинина обнаружена впервые и наблюдается при разных комбинациях субстратов на разных типах митохондрий. На рис. 2а, 2б (серые линии) показано, что преинкубация митохондрий печени крыс с малыми концентрациями SNP и L-аргинина (20 мкМ) при окислении сукцината (5 мМ) в присутствии ротенона (2 мкМ) также вызывает увеличение стационарной скорости дыхания в сравнении с контролем.
Рис. 2.
Активация потребления кислорода при инкубации митохондрий с малыми концентрациями SNP (а, 5 мкМ) и L-аргинина (б, 20 мкМ). Устранение этой активации ингибитором PKG KT5823 (KT). Представлены репрезентативные эксперименты. Митохондрии (1.0–1.2 мг) инкубировали в закрытой камере объемом 1 мл. Инкубационная среда включала 5 мМ сукцината, 2 мкМ ротенона и гексокиназу. Остальные условия как на рис. 1. Черные линии соответствуют контрольным экспериментам. Серые линии соответствуют низким (активирующим) концентрациям SNP (рис. 2а, 5 мкМ) и L-аргинина (рис. 2б, 20 мкМ). Пунктирные линии показывают устранение активирующего действия SNP и L-аргинина ингибитором PKG KT (1.2 мкМ).
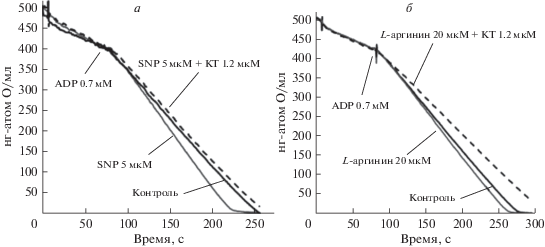
Сходный эффект наблюдается и на митохондриях сердца крыс, преинкубированных с малыми концентрациями SNP и L-аргинина (2–10 мкМ) в присутствии субстратов пирувата (1 мМ) и L-глутамата (10 мМ). Добавление в среду инкубации 5 мкМ L-аргинина вызывало увеличение скорости митохондриального дыхания на 20–25% по сравнению с контролем. Высокие концентрации SNP и L-аргинина также вызывали ингибирование дыхания (не показано).
Влияние ингибиторов mtNOS/GC/PKG–SS на активацию дыхания SNP и L-аргинином. Обнаруженная активация дыхания SNP и L-аргинином (рис. 1, 2) может указывать на вовлечение новых сигнальных механизмов, не связанных с прямым действием NO, нитрозотиолов, пероксинитрита и др. Мы предположили, что сигнальная система mtNOS/GC/PKG–SS может участвовать в регуляции дыхания митохондрий, подобно тому, как это имеет место для митохондриальной сигнальной системы с участием аденилатциклазы и протеинкиназы А (AC/PKA) [15–17]. Результаты, представленные на рис. 2, показывают, что активирующие эффекты 5 мкМ SNP или 20 мкМ L-аргинина (серые кривые) устраняются при наличии в среде инкубации 1.2 мкМ ингибитора PKG KT5823 (КТ). Этот результат свидетельствует об участии mtNOS/GC/PKG–SS и ее финального медиатора PKG в регуляции скорости дыхания митохондрий.
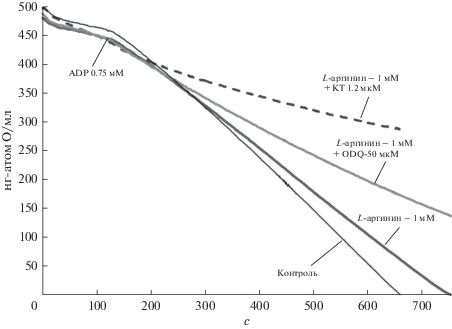
Влияние ингибиторов mtNOS/GC/PKG–SS на торможение дыхания высокими концентрациями SNP и L-аргинина. На рис. 3 показано, что ингибирование дыхания и потребления кислорода, вызываемое преинкубацией митохондрий с большими концентрациями L-аргинина (1 мМ), наоборот, усиливается в присутствии ингибиторов GC (пунктирная кривая [ODQ] = 50 мкМ) и PKG (пунктирная кривая [KT] = 1.2 мкМ). В контроле в состоянии дыхания 4 скорость дыхания митохондрий составляет 14–15 нг-атом О/мин/мг/белка. При достижении максимальной стационарной скорости дыхания, задаваемой ADP-гексокиназной системой, скорость дыхания повышается до 56 нг-атом О/мин/мг/белка В присутствии 1 мМ L-аргинина скорость дыхания уменьшается до 46 нг-атом О/мин/мг/белка. При преинкубации митохондрий с ингибиторами GC (ODQ) и PKG (KТ), происходит дальнейшее торможение дыхания до 28 и 16 нг-атом О/мин/мг/белка, соответственно. На основании этих результатов можно предположить, что защитный эффект сигнальной цепочки mtNOS/GC/PKG–SS, связанный с активацией белков дыхательной цепи посредством PKG, устраняется в присутствии ингибиторов ODQ или KT и доминирующим становится ингибирование дыхания избытком NO, продуцируемого mtNOS.
Рис. 3.
Усиление ингибирования потребления кислорода ингибиторами сигнальной цепочки mtNOS/GC/PKG–SS при инкубации митохондрий с большими концентрациями L-аргинина (1 мМ). Представлены репрезентативные данные. Условия экспериментов такие же, как и на рис. 1. Концентрации ингибиторов ODQ и KT приведены в мкМ.
Влияние модуляции активности mtNOS/GC/PKG–SS на скорость дыхания митохондрий. В сериях из четырех–шести экспериментов были измерены значения стационарной скорости митохондриального дыхания (VO2ss) при [O] = = 300–320 нг-атом О/мл на линейных участках соответствующих полярографических кривых. Рассчитанные средние значения VO2ss изображены на рис. 4 в виде столбиков. Левые серые столбики соответствуют контрольным экспериментам с VO2ss = 91.8 ± 2.7 нг-атом О/мин /мг/белка. Три следующих набора столбиков характеризуют влияние различных концентраций SNP (рис. 4а) и L-аргинина (рис. 4в) на средние значения VO2ss. Представленные данные показывают, что введение в среду инкубации 5–50 мкМ SNP вызывает увеличение скорости митохондриального дыхания (VO2ss) в сравнении с контролем. Максимальный эффект 27–28% наблюдается при 10 мкМ SNP (рис. 4а, первый и третий столбики; р < 0.05). Преинкубация митохондрий с ингибиторами ODQ и KT устраняет активацию дыхания, вызываемую 10 мкМ SNP (рис. 4а, столбики 6 и 7; р <0.05).
Рис. 4.
Влияние SNP, L-аргинина и ингибиторов mtNOS/GC/PKG–SS на стационарную скорость потребления кислорода (VO2ss) при инкубации митохондрий с сукцинатом (5 мМ) и ротеноном (2 мкМ). Все остальные условия, как на рис. 1. Влияние SNP (а) и L-аргинина (б) на величины VO2ss. Левые столбики представляют контрольные величины. Последующие четыре и три столбика на рис. 4а и 4б, соответственно, характеризуют дозозависимые эффекты SNP (а) и L-аргинина (б) на VO2ss. Правые столбики описывают влияние ингибиторов 7-NI, ODQ, KT5823 (KT) и 1400W сигнальной системы mtNOS/GC/PKG–SS на VO2ss. Видно, что преинкубация митохондрий с ингибиторами 7-NI, ODQ и KT приводит к устранению активации дыхания SNP и L-аргинином. Преинкубация митохондрий с 50–100 нМ специфического ингибитора индуцибельной iNOS 1400W не влияет на исследуемые эффекты (рис. 4б, правый столбик). Все концентрации приведены в мкМ. Данные представлены, как средние ± S.E.M. На рис. 4а n = 4 для столбиков 1–5 и n = 6 для столбиков 6 и 7. На рис. 4б: n = 4 для столбиков 1–4 и n = 5 для столбиков 5–8. Символы *, ** указывают на статистически значимую разницу между сравниваемыми величинами VO2ss; *, ** p < 0.05.
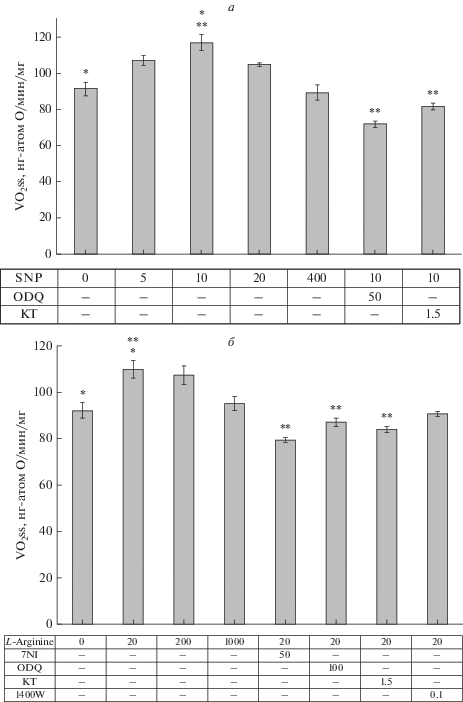
Подобно действию SNP, L-аргинин (20 мкМ) увеличивает VO2ss в сравнении с контролем на 15–16% (рис. 4б, первый и второй столбики; р < 0.05). Ингибиторы NOS (7-NI), GC (ODQ) и PKG (KT) устраняют активацию дыхания, вызываемую L-аргинином (рис. 4б, 5–7 столбики; р < 0.05), что указывает на функционирование всех трех белков сигнальной цепочки mtNOS/GC/PKG-SS. Отсутствие влияния специфического ингибитора 1400W (100 нМ) кальций-независимой iNOS на величину скорости дыхания митохондрий (рис. 4в, столбик 8; р < 0.05) свидетельствует о том, что этот фермент не участвует в продукции NO митохондриальной сигнальной системой mtNOS/GC/PKG-SS в условиях данного эксперимента.
ЗАКЛЮЧЕНИЕ
Полученные результаты показывают, что PKG может участвовать в регуляции окислительного фосфорилирования. Возможно, это происходит за счет фосфорилирования некоторых митохондрильных белков, подобно механизму, обеспечиваемому каскадом митохондриальной AC/PKA [15–17]. Как следует из наших данных, активация mtNOS/GC/PKG–SS L-аргинином может обеспечить двунаправленный контроль дыхания, который включает активацию дыхания с участием PKG и прямое ингибирование цитохромов избытком NO. Известно, что цитоплазматические изоформы GC имеют примерно в 10 раз более высокое сродство к NO, чем COX [18]. Допуская сопоставимую чувствительность к NO митохондриальной и цитозольных GC, мы можем предположить, что при низких концентрациях L-аргинина (или NO) активация дыхания с помощью PKG, реализуемая при активации mtNOS/GC/PKG–SS L‑аргинином и кальцием, будет доминировать над хорошо известным ингибирующим эффектом NO [1–6].
Функционирование сигнальной цепочки mtNOS/GC/PKG–SS возможно только при наличии и постоянном ресинтезе коферментов NADPH и GTP в матриксе митохондрий, осуществляемом с участием нескольких ферментов. Отсутствие NADPH и GTP в среде инкубации митоходрий исключает возможность функционирования внемитохордриальной цепочки, например, при загрязнении митохондриальной фракции цитоплазматическими белками, включая GC, PKG, дикиназу и др.
Известно, что существуют специальные механизмы, обеспечивающие импорт десятков различных киназ и фосфатаз в митохондрии, включая протеинкиназу В [19, 20]. Можно предполагать, что GC и PKG транспортируются в митохондрии с использованием таких механизмов. Однако прямые экспериментальные данные, свидетельствующие о транслокации PKG и GC в митохондрии, в настоящее время отсутствуют. Это может быть задачей последующих исследований. Тем не менее, представленные результаты, полученные с использованием специфических ингибиторов белков сигнальной системы mtNOS/GC/PKG–SS, указывают на ее возможное функционирование в митохондриях.
Работа выполнена при поддержке РФФИ (проекты № 14-04-01695 ВД; №14-04-01597 ЕГ). Авторы благодарят М.А. Симонову, М.Х. Галимову и А.И. Сергеева за техническую поддержку.
Список литературы
Cleeter M.W., Cooper J.M., Darley-Usmar V.M.; Moncada S., Schapira A.H. 1994. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 345 (1), 50–54. https://doi.org/10.1016/0014-5793(94)00424-2
Giulivi C. 1998. Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Biochem. J. 332, 673–679.
Brown G.C. 2001. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys.Acta. 1504 (1), 46–57. https://doi.org/10.1016/S0005-2728(00)00238-3
Parihara M.S., Nazarewicza R.R., Kincaida E., Bringoldb U., Ghafourifara P. 2008. Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem. Biophys. Res. Commun. 366 (1), 23–28. https://doi.org/10.1016/j.bbrc.2007.11.056
Giulivi C., Kato K., Cooper C.E. 2006. Nitric oxide regulation of mitochondrial oxygen consumption I: Cellular physiology. Am. J. Physiol. Cell. Physiol. 291 (6), C1225–C1231. https://doi.org/10.1152/ajpcell.00307.2006
Palacios-Callender M., Hollis V., Frakich N., Mateo J., Moncada S. 2007. Cytochrome c oxidase maintains mitochondrial respiration during partial inhibition by nitric oxide. J. Cell Sci. 120 (Pt 1), 160–165. https://doi.org/10.1242/jcs.03308
Kanai A., Epperly M., Pearce L., Birder L., Zeidel M., Meyers S., Greenberger J., de Groat W., Apodaca G., Peterson J. 2004. Differing roles of mitochondrial nitric oxide synthase in cardiomyocytes and urothelial cells. Am. J. Physiol. Heart Circ. Physiol. 286 (1), H13–H21. Erratum in: Am. J. Physiol. Heart Circ. Physiol. 296 (2), H536.
Elfering S.L., Sarkela T.M., Giulivi C. 2004. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 277, 38 079–38 086.
Kanai A.J., Pearce L.L, Clemens P.R., Birder L.A., VanBibber M.M., Choi S.Y., de Groat W.C., Peterson J. 2004. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA. 98, 14 126–14 131.
Boveris A., Valdez L.B., Zaobornyj T., Bustamante J. 2006. Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim. Biophys. Acta. 1757 (5–6), 535–542. doi 10.101`6/j.bbabio.2006.02.010
Giulivi C. 2007. Mitochondria as generators and targets of nitric oxide. Novartis Fdn Symp. 287, 92–100.
Traaseth N., Elfering S, Solien J., Haynes V., Giulivi C. 2004. Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta. 1658 (1–2), 64–71. https://doi.org/10.1016/j.bbabio.2004.04.015
Lemasters J.J., Tom P., Theruvath I., Zhong Z., Nieminen A.-L. 2009. Mitochondrial calcium and the permeability transition in cell death, Biochim. Biophys. Acta. 1787, 1395–1401. https://doi.org/10.1016/j.bbabio.2009.06.009
Halestrap A.P. 2010. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38 (4), 841–860. .https://doi.org/10.1042/BST0380841
Sardanelli A.M., Technikova-Dobrova Z., Scacco S.C., Speranza F., Papa S. 1995. Characterization of proteins phosphorylated by the cAMP-dependent protein kinase of bovine heart mitochondria. FEBS Lett. 377 (3), 470–474. https://doi.org/10.1016/0014-5793(95)01407-1
Di Benedetto G., Pendin D., Greotti E., Pizzo P., Pozzan T. 2014. Ca2+ and cAMP cross-talk in mitochondria. J. Physiol. 592 (2), 305–312. https://doi.org/10.1113/jphysiol.2013.259135
Ould Amer Y., Hebert-Chatelain E. 2018. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. Biophys. Acta. 1859 (9), 868–877. https://doi.org/10.1016/j.bbabio.2018.04.005
Rodríguez-Juárez F., Aguirre E., Cadenas S. 2007. Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration. Biochem. J. 405, 223–231. doi 101042/BJ20070033
Lim S., Smith K.R., Lim S.‑T. S., Tian R., Lu J., Tan M. 2016. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 6 (25). eCollection 2016. https://doi.org/10.1186/s13578-016-0089-3
Su C.C., Yang J.Y., Leu H.B., Chen, Y., Wang P.H. 2012. Mitochondrial Akt-regulated mitochondrial apoptosis signaling in cardiac muscle cells. Am. J. Physiol. Heart. Circ. Physiol. 302 (3), H716–H723, https://doi.org/10.1152/ajpheart.00455.2011
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии