

Биологические мембраны: Журнал мембранной и клеточной биологии, 2020, T. 37, № 6, стр. 426-433
Влияние L-аргинина и доноров оксида азота на индукцию митохондриальной поры ионами кальция и пальмитоилкарнитином. Возможное участие митохондриальной NO/cGMP/PKG сигнальной системы
В. В. Дынник a, *, Е. В. Гришина a, Н. И. Федотчева a
a Институт теоретической и экспериментальной биофизики РАН
142290 Московская обл., Пущино, Россия
* E-mail: dynnik@rambler.ru
Поступила в редакцию 10.05.2019
После доработки 07.07.2020
Принята к публикации 09.07.2020
Аннотация
Работа посвящена исследованию механизмов защитного действия L-аргинина и донора оксида азота SNAP при индукции циклоспорин А-зависимой митохондриальной поры (МРТР) избытком Ca2+ или D,L-пальмитоилкарнитина (PC). Нами было проанализировано возможное вовлечение в регуляцию МРТР сигнальной системы с участием митохондриальных ферментов NO-синтазы (mtNOS), гуанилатциклазы (GC) и cGMP-зависимой протеинкиназы G (PKG) (mtNOS/GC/PKG-SS). Эксперименты были проведены на изолированных митохондриях печени с использованием ингибиторного анализа. Результаты исследований показали, что L-аргинин и SNAP оказывают дозозависимое влияние на открытие MPTP. При низких концентрациях L-аргинин и SNAP (5–100 мкМ) активировали защитные механизмы, обеспечивая увеличение кальциевой буферной емкости митохондрий (CRC) и критической (пороговой) концентрации пальмитоилкарнитина (PC*), необходимой для индукции MPTP. При более высоких концентрациях L-аргинин (более 500 мкМ) и SNAP (более 100 мкМ) вызывали противоположный эффект, подавляя дыхание и способствуя открытию поры. Ингибиторы NOS, GC и PKG устраняли защитные эффекты, наблюдавшиеся при малых концентрациях L-аргинина и SNAP. Эксперименты, проведенные с использованием специфического ингибитора индуцибельной NOS (W1400), показали, что этот кальций-независимый фермент не участвует в регуляции MPTP в условиях наших экспериментов. На основании полученных результатов можно предположить, что кальций-зависимая митохондриальная сигнальная система mtNOS/GC/PKG-SS может быть вовлечена в регуляцию MPTP, обеспечивая увеличение CRC и PC*. При высоких концентрациях L-аргинина или SNAP избыток NO преодолевает защиту, обеспечиваемую mtPKG, и способствует открытию поры.
ВВЕДЕНИЕ
Влияние L-аргинина, доноров NO и ингибиторов Ca2+-зависимой изоформы митохондриальной NO-синтазы (mtNOS) на кальциевую емкость митохондрий (CRC), индукцию митохондриальной поры (MPTP), высвобождение цитохрома C (CytC) изучается более 25 лет. Полученные за этот период результаты исследований противоречивы. Так, еще в 1999 году было показано, что активация mtNOS Ca2+ и L-аргинином вызывает высвобождение CytC, в то время как ингибирование mtNOS уменьшает выход CytC, повышает митохондриальный потенциал (ΔΨm) и CRC [1]. Позже было обнаружено, что NO вызывает концентрационно-зависимые эффекты [2]. Было показано, что очень низкие или высокие концентрации NO-доноров способствуют набуханию митохондрий, высвобождению CytC и открытию MPTP, индуцированному избытком ионов кальция. И наоборот, промежуточные концентрации доноров NO обеспечивают защитные эффекты, уменьшая набухание митохондрий и вероятность открытия поры. Защитные и неблагоприятные воздействия доноров NO объясняли возможным действием S-нитрозотиолов и пероксинитрита, соответственно [1, 2].
Неоднозначные эффекты доноров NO на открытие MPTP также наблюдались в экспериментах, выполненных на клетках. Было показано, что малые концентрации доноров NO уменьшают накопление митохондриального Cа2+, а высокие концентрации, наоборот, способствуют выходу Cа2+ и открытию MPTP [3]. В этих экспериментах добавленный L-аргинин ингибировал поглощение Ca2+ митохондриями и предотвращал открытие MPTP [2, 4]. Авторы предположили, что внутримитохондриальный NO может быть вовлечен в отрицательную обратную связь, предотвращающую открытие MPTP за счет ингибирования транспорта Ca2+ [3]. Согласно другой точке зрения, уменьшение поглощения Ca2+ может быть обусловлено деполяризацией митохондрий, вызываемой NO [5].
Представленные выше противоречивые эффекты доноров NO и L-аргинина на ΔΨm и контроль MPTP не могут быть объяснены действием механизмов, основанных только на редокс-регуляции митохондриальных процессов S-нитрозилированием [6, 7] и S-глутатионированием [8–11] различных белков. Потенциальные механизмы защиты могут включать внутримитохондриальные сигнальные цепи, учитывающие взаимодействие кальция и NO и другие, пока неизвестные обратные связи. Недавно Seya и соавторы обнаружили, что митохондриальная фракция сердца обладает активностью cGMP-зависимой протеинкиназы G (PKG) [12]. Продукция cGMP митохондриальной гуанилатциклазой (GC) также была продемонстрирована этой группой [13]. Было показано, что SNAP и 8-Bromo-cGMP способствуют кальций-зависимому высвобождению CytC из митохондрий сердца, вовлекая потенциал-зависимый анионный канал (VDAC) в качестве конечной мишени сигнального пути. Наблюдаемые эффекты предотвращались применением ингибиторов NOS, GC, PKG и VDAC. Гидролиз cGMP митохондриальной фосфодиэстеразой циклических нуклеотидов PDE2A также был продемонстрирован в митохондриях мозга и печени в независимых экспериментах [14]. Эти данные указывают на то, что митохондрии содержат все элементы автономной Ca2+-зависимой сигнальной системы: Ca2+ → mtNOS → NO → → GC → cGMP → PKG (mtNOS/GC/PKG-SS), которая может участвовать в регуляции митохондриального дыхания [15] и MPTP.
В данной работе были проведены эксперименты на изолированных митохондриях печени крысы, в которых анализировалось влияние агонистов и ингибиторов mtNOS/GC/PKG-SS на диссипацию ΔΨm и индукцию МРТР, вызываемые избытком Ca2+ или пальмитоилкарнитина (PC). Предварительные результаты были опубликованы ранее [16].
МАТЕРИАЛЫ И МЕТОДЫ
Все процедуры на животных выполнялись в соответствии с директивой ЕС 86/609/EEC и были одобрены комитетом по этике Института теоретической и экспериментальной биофизики РАН. Самцов крыс линии Вистар (6–8 недель) содержали в одинаковых условиях в кондиционированных и проветриваемых помещениях при температуре 20–22°C (свет/темнота = 12 ч/12 ч). Эксперименты проводили при 26°C. Митохондрии печени выделяли с использованием стандартных методик дифференциального центрифугирования в среде, содержащей 300 мМ сахарозы, 1 мМ EGTA и 10 мМ Трис-HCl (pH 7.4). Митохондриальные препараты дважды промывали средой выделения, не содержащей EGTA, ресуспендировали в среде того же состава и хранили на льду. Инкубационная среда для митохондрий содержала: 125 мМ KCl, 3 мМ KH2PO4, 10 мМ HEPES (pH 7.4), 1 мМ MgCl2. Содержание митохондриального белка определяли методом Лоури с бычьим сывороточным альбумином в качестве стандарта. Разность электрических потенциалов (ΔΨm) на внутренней мембране митохондрий определяли по распределению липофильного катиона тетрафенилфосфония (ТРР+), концентрацию которого во внешней среде [ТРР+]out регистрировали с помощью ТРР+-селективного электрода. Измерение концентрации кальция в среде производили с использованием Ca2+-селективного электрода (Nico, Москва, Россия). Все эксперименты проводили в закрытой или открытой кювете объемом 1 мл, содержащей 1.0 мг мит. белка, при постоянном перемешивании. В качестве субстратов дыхания использовали L-глутамат (10 мМ) и пируват (1 мМ) для поддержания полного оборота цикла Кребса и сохранения субстратного фосфорилирования в цикле. В некоторых экспериментах дыхание митохондрий поддерживалось окислением сукцината (5 мМ) в присутствии ротенона (1–2 мкМ) или пирувата (1 мМ) и L-малата (5 мМ). Все эксперименты выполнялись в присутствии 10 ед. гексокиназы, 10 мМ глюкозы и 1 мМ MgCl2. Последующие добавки 0.75 мМ ADP обеспечивали высокую стационарную скорость дыхания (VO2SS), которая была близка к максимальной скорости дыхания, наблюдающейся в Состоянии 3 (VO2SS = 80–85% от VO2max (V3)). Часть экспериментов была выполнена при низкой скорости дыхания митохондрий, в отсутствие гексокиназы и ADP в среде (Состояние 4, V4). Скорость набухания митохондрий (VSW = Δ o.d./мин) определялась в открытой кювете объемом 2 мл при 0.35 мг. мит. белка на 1 мл с использованием спектрофотометра Ocean Optics USB4000. Открытие MPTP регистрировали как: выход кальция из митохондрий в инкубационную среду; диссипацию митохондриального потенциала ΔΨm; набухание митохондрий. Индукция MPTP достигалась путем последовательных добавок в инкубационную среду 20 мкМ Ca2+ (CaCl2) или 20 мкМ PC. Критические концентрации Ca2+ или PC, необходимые для индукции поры, представляют митохондриальную кальциевую буферную емкость (CRC) и пороговую концентрацию PC (PC*). Величины этих параметров характеризуют чувствительность комплекса МРТР к действию различных модуляторов поры. Влияние mtNOS/PKG-SS на открытие MPTP оценивали путем определения значений CRC, PC* и VSW, соответственно. Активацию mtNOS-SS обеспечивали введением в среду инкубации L-аргинина, NO доноров или Ca2+. 7-NI, ODQ и KT5823 были использованы для ингибирования mtNOS, GC и PKG соответственно. В работе использованы реактивы фирмы “Sigma” (США). Статистический анализ экспериментальных данных проводился с использованием t-критерия с использованием SigmaPlot 11. Данные представлены как среднее значение ± S.E.M. n = 4–6 независимых экспериментов. За уровень значимости было принято p < 0.05.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Индукция МРТР избытком кальция и защитные эффекты L-аргинина. На рис. 1а показано, что при последовательных добавках 20 мкМ Ca2+ в контроле CRC составляет 100 мкМ (нижняя кривая). Преинкубация митохондрий с 300 мкМ L-аргинина приводит к увеличению CRC до 180 мкМ. Введение в среду инкубации ингибитора NOS 7NI (100 мкМ) уменьшает величину CRC до 80 мкМ, при достижении которой происходит диссипация ΔΨm (рост ТРР+ в среде, верхняя кривая). Преинкубация митохондрий с селективным ингибитором iNOS 1400W (50–100 нМ) не влияла на величину CRC, что свидетельствует об отсутствии iNOS в митохондриях в исследуемых условиях.
Рис. 1.
Диссипация ΔΨm и индукция МРТР ионами Ca2+. Защитный эффект L-аргинина (300 мкМ) и его устранение ингибиторами белков системы mtNOS/GC/PKG-SS: 7NI (а, 100 мкМ), ODQ (б, 100 мкМ) и KT5823 (в, 1.5 мкМ, KT), соответственно. Представлены репрезентативные эксперименты. Митохондрии инкубировали в открытой камере объемом 1 мл. Среда инкубации (см. Материалы и методы) включала 1 мМ пирувата, 10 мМ L-глутамата и гексокиназу. Добавки Ca2+ и PC (по 20 мкМ), а также ADP показаны стрелками.
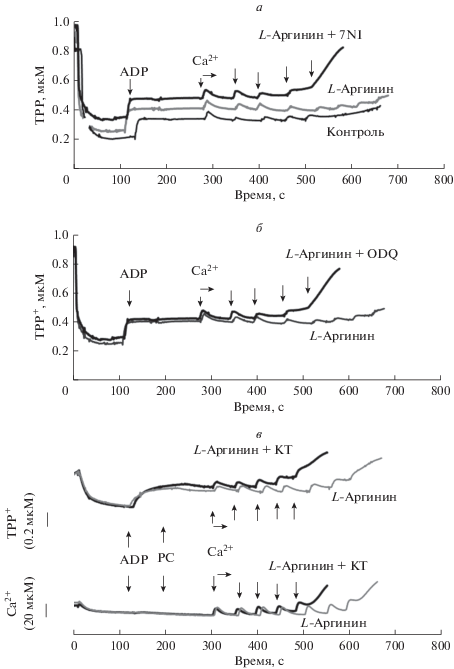
Защитный эффект L-аргинина также устраняется ингибиторами GC (100 мкМ ODQ, рис. 1б, верхняя кривая) и PKG (1.5 мкМ KT5823, рис. 1в, верхние кривые). Из рис. 1в видно, что уменьшение ΔΨm (верхняя кривая) сопряжено с выбросом Ca2+ из митохондрий (нижние кривые). Видно также, что преинкубация митохондрий с KT5823, селективным ингибитором PKG, уменьшает CRC от 180 до 120 мкм.
Эти данные свидетельствуют о том, что защитный эффект L-аргинина, направленный на уменьшение вероятности индукции поры избытком Ca2+, может быть обусловлен функционированием mtNOS/GC/PKG-SS, эффектором которой является PKG. Можно предположить, что наблюдаемый защитный эффект L-аргинина связан с фосфорилированием структурных элементов комплекса белков МРТР с участием PKG.
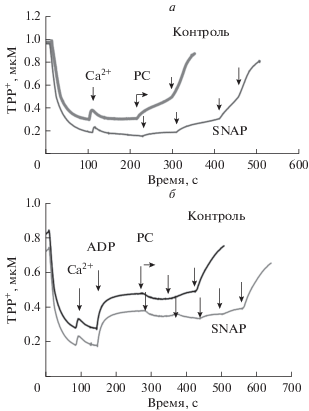
Влияние скорости дыхания митохондрий на PC-индуцированное открытие MPTP и защитный эффект SNAP. Известно, что скорость продукции NO mtNOS экспоненциально зависит от величины ΔΨm и максимальна в Состоянии 4 [17, 18]. Кроме того, хорошо известно, что вероятность открытия поры различными агентами сильно уменьшается при наличии аденилатов, гексокиназы и Mg2+ в среде инкубации [19, 20]. Последнее может быть связано со стабилизацией конформации элементов макрокомплекса МРТР, включая транслокатор аденилатов, ATP-синтазу и др. В наших условиях, в присутствии избытка гексокиназы и глюкозы в среде, добавка 0.75 мМ ADP увеличивает скорость дыхания митохондрий от VO2 = 20–24 нг-атом О/мин/мг (V4, Состояние 4, ADP = 0) до стационарных значений VO2SS = 70–77 нг-атом О/мин/мг (VO2 = 80–90% от скорости дыхания в Состоянии 3). Результаты, представленные на рис. 2, показывают, что в митохондриях, находящихся в состоянии активного дыхании (VO2SS), критические величины PC* в контроле и в присутствии SNAP выше, чем в Состоянии 4 (V4). Сравнение верхних кривых, представленных на рис. 2, показывает, что в митохондриях, находящихся в Состоянии 4 (верхняя линия на рис. 2а), 40 мкм PC вызывает диссипацию ΔΨm, тогда как в состоянии с высокой скоростью дыхания (верхняя линия на рис. 2б), критическая концентрация PC* превышает 60 мкМ. В присутствии 20 мкМ SNAP в Состоянии 4 величина PC* увеличивается до 80 мкМ, в то время как при высокой скорости дыхания (VO2SS) величина PC* составляет 100 мкМ и выше (нижние линии на рис. 2а, 2б).
Рис. 2.
Влияние скорости дыхания митохондрий на диссипацию ΔΨm избытком PC и на защитный эффект SNAP. Представлены репрезентативные эксперименты. Инкубационная среда включала 10 мМ L-глутамата и 1 мМ пирувата, а также гексокиназу и глюкозу. Эксперименты, представленные на панели а, проведены в отсутствие ADP в среде (Состояние 4). На панели б высокая скорость дыхания (VO2 = 80–90% от скорости дыхания в Состоянии 3) обеспечивалась добавками 0.75 мМ ADP. На панелях а и б черные (верхние) линии соответствуют контрольным экспериментам ([SNAP] = 0), а серые линии – опытам, в которых инкубационная среда содержала 20 мкМ SNAP. Добавки Ca2+ и PC (по 20 мкМ), а также ADP показаны стрелками.
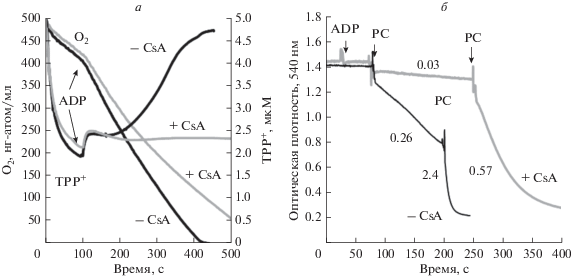
Диссипация ΔΨm, ингибирование дыхания и набухание митохондрий, вызываемые PC. Защитный эффект циклоспорина А. Преинкубация митохондрий с пируватом, L-глутаматом и низкими концентрациями PC (20 мкМ) повышает ΔΨm и увеличивает стационарную скорость митохондриального дыхания на 35–40% по сравнению со значениями, достигнаемыми при дыхании митохондрий в присутствии пирувата и L-глутамата (VO2SS = 85–88 нг-атом О/мин/мг). Напротив, высокие дозы PC (выше 50 мкМ) вызывают быструю диссипацию ΔΨm увеличение [TPP+] и прогрессирующее угнетение дыхания. Этот процесс развивается вскоре после добавки ADP (рис. 3а, черные линии). Преинкубация митохондрий с CsA предотвращает падение ΔΨm, несмотря на сильное подавление дыхания (рис. 3а, серые линии). Помимо диссипации ΔΨm и подавления дыхания, PC вызывает набухание митохондрий. Как показано на рис. 3б, низкие концентрации PC (20 мкМ) вызывают набухание митохондрий, которое характеризуется скоростью набухания VSW = 0.26 Δ o.d./мин (черная кривая). Преинкубация митохондрий с CsA (2 мкМ) предотвращает этот эффект. Вторая добавка 20 мкМ PC приводит к быстрому митохондриальному набуханию, характеризующемуся VSW = 2.4 Δ o.d./мин (черная кривая). Однако в этом случае CsA обеспечивает ограниченный защитный эффект, уменьшая VSW в четыре раза до 0.57 Δ o.d./мин и не оказывая влияния на амплитуду набухания (серая кривая). Таким образом, наряду с кальцием PC является важным агентом, вызывающим индукцию MPTP.
Рис. 3.
Избыток PC вызывает диссипацию ΔΨm и подавление дыхания (а), а также набухание митохондрий (б). Присутствие циклоспорина A (CsA, 1.5 мкМ) в среде препятствует диссипации ΔΨm, набуханию митохондрий и индукции поры (б). Представлены репрезентативные эксперименты. а – Влияние избытка PC (50 мкМ) и CsA (1.5 мкМ) на митохондриальное дыхание (линии O2) и ΔΨm (линии TPP+). Инкубационная среда включала 10 мМ L-глутамата и 1 мМ пирувата. Митохондрии инкубировали с PC (черные линии) или с PC и CsA (1.5 мкМ, серые линии). Все остальные условия как на рис. 1. б – Набухание митохондрий, вызываемое добавлением PC в отсутствие (нижние черные линии) и в присутствии CsA (2 мкМ, верхние серые линии) в среде. Максимальные значения скорости набухания (VSW, выраженные в изменениях ед. оптической плотности/мин; VSW = Δ o.d./мин) указаны на рисунке. 0.75 мМ ADP добавляли за 1 мин до первого введения PC. Инкубационная среда включала 5 мМ сукцината и 2 мкМ ротенона. Все остальные условия как на рис. 1.
Кальций и активные формы кислорода (АФК) обычно считаются ключевыми медиаторами, участвующими в индукции MPTP и гибели клеток [20, 21]. Однако ишемия/реперфузия и некоторые другие патологические процессы, помимо роста Cа2+ и АФК, характеризуются накоплением длинноцепочечных жирных кислот [22, 23] и их карнитиновых производных [23], которые окисляются в митохондриях в виде ацил-КоА. Хорошо известно, что длинноцепочечные ацил-КоА (С14 и выше) ингибируют различные NAD(P)H-зависимые дегидрогеназы [24]. Наши предварительные результаты показали, что PC индуцирует CsA-зависимую диссипацию ΔΨm в митохондриях печени [16]. Поэтому в настоящем исследовании для изучения влияния mtNOS/GC/PKG-SS на MPTP в качестве параметров “чувствительности” поры к токсинам были использованы величины CRC и PC*, определяемые при нагрузке митохондрий кальцием и PC.
Влияние модуляции активности mtNOS/GC/ PKG-SS на PC*. Чтобы оценить статистическую достоверность полученных результатов, были определены средние величины PC* в сериях из четырех–шести экспериментов. Рассчитанные средние значения PC* представлены на рис. 4 в виде столбиков. Первый серый столбик соответствует контрольным экспериментам. Влияние SNAP и L-аргинина, а также ингибиторов 7-NI (100 мкМ), ODQ (100 мкМ), KT5823 (1.5 мкМ) и 1400W (100 нМ) на средние величины PC* показано на рис. 4. Видно, что увеличение концентрации SNAP до 100 мкМ (2 и 3 столбики) увеличивает критическое значение PC* до 109.2 ± 8.1 мкМ, что на 28% выше величины PC* в контроле (84.3 ± ± 7.8 мкМ (1 столбик). Сходный защитный эффект вызывает преинкубация митохондрий с 20 мкМ L-аргинина (6 столбик).
Рис. 4.
Влияние модуляции активности mtNOS/GC/PKG-SS на величину PC*. Столбики представляют средние значения величин PC* ± S.E.M, полученные в 4–6 экспериментах при последовательных добавках 20 мкМ PC, подобно тому, как это показано на рис. 1 для Са2+. Инкубационная среда такая же, как и на рис. 1. 20 мкМ Ca2+ добавляли через 1 мин после активации дыхания 0.75 мМ ADP, затем митохондрии нагружали PC. Концентрации SNAP, L-аргинина (Arg), 7-NI, ODQ, KT5823 (KT) и 1400W приведены в мкМ. n = 4 (столбики 1–3), n = 6 (столбики 4–9). Линии над столбиками показывают сравниваемые пары величин PC*. * p < 0.05.
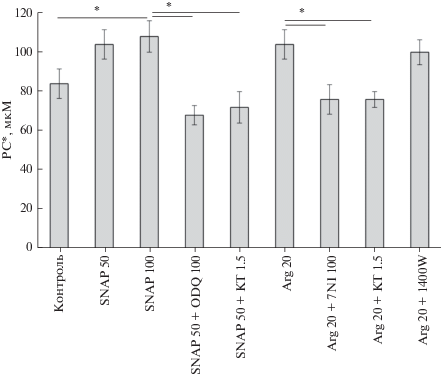
Видно, что ингибиторы mtNOS/GC/PKG-SS 7-NI, ODQ и KT5823 устраняют защитные эффекты L-аргинина и SNAP, уменьшая критические (пороговые) концентрации PC*, необходимые для индукции поры (рис. 4, столбики 4, 5 и 7, 8), до величин ниже контрольных.
ЗАКЛЮЧЕНИЕ
Таким образом, основываясь на результатах, полученных с помощью ингибиторного анализа, можно предполагать участие митохондриальной mtNOS/GC/PKG-SS в регуляции МРТР. Следует отметить, что mtNOS/GC/PKG-SS нельзя рассматривать как избыточный элемент в многоуровневом контроле окислительного фосфорилирования и индукции MPTP. Будучи зависимой от кальция, эта сигнальная система может участвовать в функционировании нескольких петель обратной связи, две из которых направлены на активацию митохондриального дыхания и на уменьшение вероятности индукции MPTP. В этом случае митохондриальная PKG, наряду с цитозольной PKG1, может рассматриваться как конечный эффектор защиты, участвующий в контроле MPTP.
Вклад авторов: ВВД координировал проект и написал рукопись. НИФ и ЕВГ провели эксперименты, проанализировали и обсудили данные. НИФ приняла участие в доработке статьи.
Финансирование. Работа выполнена при поддержке Российского фонда фундаментальных исследований (проект № 14-04-01695 ВД).
Благодарности. Авторы благодарят М.А. Симонову, М.Х. Галимову и А.И. Сергеева за техническую поддержку.
Конфликт интересов. У авторов нет конфликта интересов.
Список литературы
Ghafourifar P., Schenk U., Klein S.D., Richter C. 1999. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J. Biol. Chem. 274 (44), 31 185–31 188. https://doi.org/10.1074/jbc.274.44.31185
Brookes P.S., Salinas E.P., Darley-Usmar K., Eiserich J.P., Freeman B.A, Darley-Usmar V.M., Anderson P.G. 2000. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J. Biol. Chem. 275 (27), 20 474–20 479. https://doi.org/10.1074/jbc.M00.10.77200
Dedkova E.N., Blatter L.A. 2005. Modulation of mitochondrial Ca2+ by nitric oxide in cultured bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 289 (4), C836–C845. https://doi.org/10.1152/ajpcell.00011.2005
Dedkova E.N., Blatter L.A. 2009. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J. Physiol. 587 (4), 851–872. https://doi.org/10.1113/jphysiol.2008.165423
Davidson S.M., Duchen M.R. 2006. Effects of NO on mitochondrial function in cardiomyocytes: Pathophysiological relevance. Cardiovasc. Res. 71 (1), 10–21. https://doi.org/10.1016/jcardiores.2006.01.019
Piantadosi C.A. 2012. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta. 1820 (6), 712–721. https://doi.org/10.1016/j.bbagen.2011.03.008
Chang A.H., Sancheti H., Garcia J., Kaplowitz N., Cadenas E., Han D. 2014. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 27 (5), 794–804. https://doi.org/10.1021/tx400462r
Queiroga C.S., Almeida A.S., Martel C., Brenner C., Alves P.M., Vieira H.L. 2010. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 285 (22), 17 077–17 088. https://doi.org/10.1074/jbc.M109.065052
Yap L.P., Garcia J.V., Han D.S., Cadenas E. 2010. Role of nitric oxide-mediated glutathionylation in neuronal function: potential regulation of energy utilization. Biochem. J. 428 (1), 85–93. https://doi.org/10.1042/BJ20100164
Mailloux R.J., Willmore W.G. 2014. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2, 68. https://doi.org/10.3389/fcell.2014.00068
Mailloux R.J., Treberg J.R. 2016. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol. 8, 110–118. https://doi.org/10.1016/j.redox.2015.12.010
Seya K., Ono K., Fujisawa S., Okumura K., Motomura S., Furukawa K. 2013. Cytosolic Ca2+-induced apoptosis in rat cardiomyocytes via mitochondrial NO-cGMP-protein kinase G pathway. J. Pharmacol. Exp. Ther. 344 (1), 77–84. https://doi.org/10.1124/jpet.112.198176
Seya K., Motomura S., Furukawa K. 2007. Cardiac mitochondrial cGMP stimulates cytochrome c release. Clin. Sci. (Lond.). 112 (2), 113–121.https://doi.org/10.1042/CS20060144
Acin-Perez R., Russwurm M., Günnewig K., Gertz M., Zoidl G., Ramos L., Buck J., Levin L.R., Rassow J., Manfredi G., Steegborn C. 2011. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J. Biol. Chem. 286 (35), 30423–3032. https://doi.org/10.1074/jbc.M111.266379
Dynnik V.V., Grishina E.V., Fedotcheva N.I. 2019. Implication of mitochondrial NO/cGMP/PKG signaling system in the activation and inhibition of mitochondrial respiration by L-arginine and NO donors. Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology. 13 (4), 334–340. https://doi.org/10.1134/S1990747819040056
Grishina E.V., Galimova M.H., Djafarov R.H., Sergeev A.I., Fedotcheva N.I., Dynnik V.V. 2016. Induction of cyclosporine-sensitive mitochondrial permeability transition pore by substrates forming acetyl-CoA under normal conditions and in type 2 diabetes. Biochemistry (Moscow). Supplement Series A.Membrane and Cell Biology. 10 (1)¸ 11–18. https://doi.org/10.1134/S1990747815050049
Boveris A., Valdez L.B., Zaobornyj T., Bustamante J. 2006. Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim. Biophys. Acta. 1757 (5–6), 535–542. https://doi.org/10.1016/j.bbabio.2006.02.010
Valdez L.B., Zaobornyj T., Boveris A. 2006. Mitochondrial metabolic states and membrane potential modulate mtNOS activity. Biochim. Biophys. Acta. 1757 (3), 166–172. https://doi.org/10.1016/j.bbabio.2006.02.013
Giorgio V., Guo L., Bassot C., Petronilli V., Bernardi P. 2018. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium. 70, 56–63. https://doi.org/10.1016/j.ceca.2017.05.004
Lemasters J.J., Theruvath T.P., Zhong Z., Nieminen A.L. 2009. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta. 1787, 1395–1401. https://doi.org/10.1016/j.bbabio.2009.06.009
Halestrap A.P. 2010. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38 (4), 841–860. https://doi.org/10.1042/BST0380841
Yamada K.A., McHowat J., Yan G.X., Donahue K., Peirick J., Cleber A.G. Corr P.B. 1994. Cellular uncoupling induced by accumulation of long-chain acylcarnitine during ischemia. Circ. Res. 74 (1), 83–95. https://doi.org/10.1161/01.res.74.183
De Windt L.J., Willems J., Roemen T.H., Coumans W.A., Reneman R.S., Van Den Vusse G.J., Bilsen M.V. 2001. Ischemic-reperfused isolated working mouse hearts: Membrane damage and type IIA phospholipase A2. Am. J. Physiol. Heart. Circ. Physiol. 280 (6), H2572–H2580.https://doi.org/10.1152/ajpheart.2001.280.6.H2572
Lai J.C., Cooper A.J. 1991. Neurotoxicity of ammonia and fatty acids: Differential inhibition of mitochondrial dehydrogenases by ammonia and fatty acyl coenzyme A derivatives. Neurochem. Res. 16 (7), 795–803.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии