

Биологические мембраны: Журнал мембранной и клеточной биологии, 2020, T. 37, № 6, стр. 403-425
Вклад одновалентных (Na+ и K+) и двухвалентных (Ca2+) ионов в механизмы синаптической пластичности
Л. В. Смольянинова a, *, А. А. Шиян a, Г. В. Максимов a, С. Н. Орлов a
a Московский государственный университет им. М.В. Ломоносова,
биологический факультет
119992 Моcква, Россия
* E-mail: smolyaninovalarisa1@gmail.com
Поступила в редакцию 17.06.2020
После доработки 12.07.2020
Принята к публикации 14.07.2020
Аннотация
В обзоре представлены механизмы участия ионов (Na+, K+, Ca2+) в процессах синаптической пластичности в постсинаптическом нейроне при длительной потенциации и длительной депрессии. Предполагается, что основными участниками являются AMPA- и NMDA-рецепторы, потенциал-зависимые Na+-, K+-, Ca2+-каналы, Ca2+- и Na+-активируемые K+-каналы, ATP-чувствительные K+-каналы, Ca2+-каналы эндоплазматического ретикулума. В обзоре приводится их молекулярная характеристика и обсуждается роль при длительной потенциации и длительной депрессии. Впервые рассматривается значение изменения внутриклеточного соотношения [Na+]i/[K+]i и Са2+-зависимый механизм от формирования сигнала до уровня экспрессии генов. Мы считаем, что необходимы дополнительные исследования для идентификации подмножества нейрональных генов, дифференциальная экспрессия которых способствует синаптической пластичности, реализуемой с участием [Na+]i/[K+]i-чувствительного, Ca2+-независимого механизма сопряжения “возбуждение–транскрипция”.
ВВЕДЕНИЕ
Синаптическую пластичность можно определить как способность к изменению силы взаимодействия между пре- и постсинаптическими нейронами, которая усиливается или ослабевает в течение времени в ответ на изменение их активности. Стимуляция синапсов, вызванная изменением мембранного потенциала (МП), нейромедиаторами, модуляторами, цитокинами влечет за собой клеточный ответ как в пресинаптическом, так и постсинаптическом нейронах [1, 2]. Изменение состояния синапсов может осуществляться на короткие, средние и долгие периоды (кратковременная, среднесрочная и длительная пластичность). Кратковременная пластичность длится секунды и осуществляется с помощью пост-трансляционной модификации белков [3–6], среднесрочная пластичность происходит при участии новосинтезированных белков и длится от минут до нескольких часов, а длительная пластичность занимает часы и больше и требует экспрессии генов [7]. Длительная пластичность может выражаться в длительной депрессии (long-term depression, LTD) или в длительной потенциации (long-term potentiation, LTP) синапсов (рис. 1а, [8]) [9]. Эффект LTP был впервые открыт Тимоти Блиссом и Тери Ломом в 1973 году и позволил прояснить механизмы длительной пластичности (рис. 1б). Показано, что LTP инициируется входом Ca2+ в постсинаптический нейрон в ответ на сильную синаптическую активность [10–12]. Изменение внутриклеточной концентрации Ca2+ ([Ca2+]i) приводит к синапс-специфическим изменениям, включающим вставку и удаление субъединиц глутаматных рецепторов из плазматической мембраны, изменению функции синаптических белков за счет пост-трансляционной модификации, стимуляции трансляции или деградации белков, а также инициирует сигнальные каскады, приводящие к активации генной экспрессии [13–15]. Несмотря на то, что Ca2+ играет ключевую роль во внутриклеточной передаче сигнала при LTP и LTD, показано, что увеличению [Ca2+]i предшествует изменение соотношения [Na+]i/[K+]i. Известно, что для генерации потенциала действия (ПД) и проведения серии ПД различной частоты ключевую роль играют потенциал-образующие ионы Na+ и K+. Установлено, что в ряде клеток изменение соотношения [Na+]i/[K+]i может влиять на экспрессию генов Ca2+-независимым путем [16, 17]. В связи с этим в данном обзоре представлен анализ участия потенциал- и лиганд-зависимых каналов, транспортеров и ATP-аз в формировании комплексного ответа нейронов при LTP и LTD. В обзоре представлены общие характеристики ионных каналов, транспортеров и ATP-аз в нейронах мозга, механизмы участия одно- и двухвалентных ионов в синаптической пластичности и описана роль изменения соотношения [Na+]i/[K+]i в экспрессии генов.
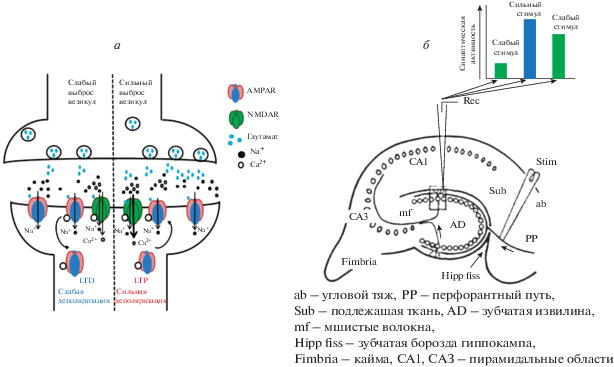
Рис. 1.
а – Длительная депрессия (LTD) и длительная потенциация (LTP) в постсинаптическом нейроне. Слабая активация пресинаптического нейрона приводит к слабой деполяризации постсинаптического нейрона и входу кальция через NMDA-рецепторы. Это приводит к активации фосфатаз и дефосфорилированию AMPA-рецепторов, что вызывает их эндоцитоз. Сильная активация вызывает сильную деполяризацию, что приводит к фосфорилированию АМРА-рецепторов и их экзоцитозу. б – Схема синаптического пути в экспериментах Блисса и Ломо при открытии LTP [8]. Stim – стимулирующий электрод; Rec – записывающий электрод. Впервые была использована предварительная слабая стимуляция перфорантного пути перед сильной стимуляцией и измерена синаптическая активность в зубчатой извилине. Повторная слабая стимуляция, по сравнению с первой слабой стимуляцией вызывала усиление синаптической активности, получившей название длительная потенциация (LTP).
1. ВАЖНЕЙШИЕ ИОННЫЕ КАНАЛЫ И ТРАНСПОРТЕРЫ, ВОВЛЕЧЕННЫЕ В ТРАНСПОРТ ИОНОВ (Na+, K+, Ca2+) ПРИ LTP И LTD В НЕЙРОНАХ
AMPA-рецепторы
Установлено, что глутаматные ионотропные AMPA-рецепторы (AMPAR) участвуют в индукции и формировании LTP и LTD. (рис. 2). Пирамидальные нейроны коры и гиппокампа содержат порядка 10 000 синапсов, и в каждом синапсе AMPAR независимо регулируют синаптическую активность. Известно 4 гена (GRIA1–GRIA4), кодирующие субъединицы AMPAR. Молекула рецептора представляет собой гомо- или гетеромерные комплексы субъединиц GluA1–GluA4. Субъединицы состоят из доменов – ATD (участвует в сборке, доставке рецепторов), LBD (связывает агонисты/антагонисты и активирует воротный механизм), TMD (формирует ионный канал) и CTD (регулирует, локализует рецепторы) [18]. В состав комплекса обычно включена GluA2 субъединица, которая выполняет важную роль в процессе селективного и потенциал-зависимого транспорта ионов через канал AMPAR. После транскрипции эта субъединица подвергается редактированию РНК, в результате чего участок РНК, кодирующий кодон для аминокислотного остатка глутамина, локализованного в области поры канала, заменяется на РНК кодон, кодирующий аминокислотный остаток аргинина. Наличие аминокислотного остатка аргинина в области поры канала ограничивает токи ионов Na+ (внутрь) и K+ (наружу) и блокирует вход двухвалентных ионов в клетку. Известно, что входящий ток через AMPAR осуществляется при отрицательных величинах, а выходящий ток – при положительном МП. Установлено, что при отрицательном МП входящий ток обусловлен Na+ (Erm ~ ~ EK ⪡ ENa), в то время как при высокоамплитудной деполяризации нейронов через AMPAR выходит K+ (Eam ~ ENa ⪢ EK) [19]. По сравнению с одновалентными катионами, у AMPAR многих нейронов выявлена низкая Ca2+-проницаемость, PCa/PNa = 3 в культивируемых эмбриональных клетках мозжечка мышей [20]. Известно, что в головном мозге существуют AMPAR не имеющие в составе комплексов GluA2 субъединицу. В поре канала таких AMPAR вместо аминокислотного остатка аргинина локализован аминокислотный остаток глутамина, что увеличивает проницаемость для Na+ и даже для Ca2+ [21]. Эти каналы получили название Ca2+-проницаемые AMPA-рецепторы (CP-AMPAR). Доказано, что у рецепторов с GluA2, вольт-амперная характеристика линейна, а у рецепторов без субъединицы GluA2 нелинейна: при положительных значениях МП CP-AMPAR с большей эффективностью переносят ионы в клетку, чем из клетки [21–23]. Интересно, что на ранних стадиях эмбрионального развития нейроны больше экспрессируют CP-AMPAR, в то время как Ca2+-непроницаемые AMPAR преобладают в зрелом мозге [24]. Liu и Cull-Candy установили, что повторяемая высокочаcтотная электростимуляция нервных клеток вызывает быстрое снижение проницаемости Ca2+ через CP-AMPAR за счет активации AMPAR [25].
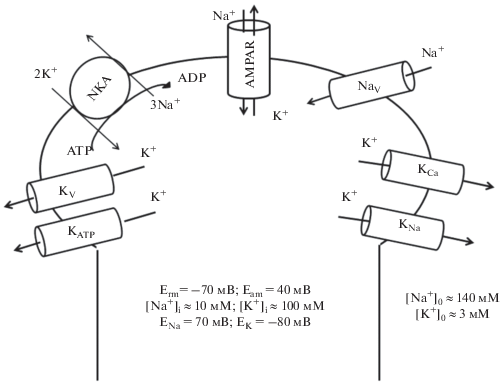
Рис. 2.
Ионные каналы и транспортеры, вовлеченные в регуляцию внутриклеточной концентрации одновалентных ионов в постсинаптическом нейроне. NKA – Na+,K+-ATP-аза; AMPAR – глутаматные ионотропные AMPA-рецепторы, проницаемые для одновалентных катионов; Nav и Kv – потенциал-зависимые Na+- и K+-каналы; KCa – Ca2+-активируемые K+-каналы; KNa – Na+-активируемые К+-каналы; KATP – ATP-зависимые K+-каналы; Erm и Eam – потенциал покоя и потенциал действия соответственно; ENa и EK – равновесный потенциал Нернста для Na+ и K+ соответственно.
NMDA-рецепторы
Известно, что активность глутаматных ионотропных NMDA-рецепторов (NMDAR) важна для осуществления синаптической пластичности при LTP и LTD (рис. 3). Установлено, что NMDAR важны на стадии индукции LTP и LTD. NMDAR представляют собой гетеротетрамерные комплексы субъединиц GluN1, GluN2A-D и GluN3A-B. Обычно комплексы состоят из пар GluN1 (1 из 8 видов) и GluN2 (1 из 4 подтипов, A–D) субъединиц, связывающих глицин или D-серин и глутамат, соответственно. Субъединицы NMDAR включают несколько доменов – ATD (контролирует вероятность открытия ионного канала и скорость деактивации ионного канала, а также содержит участки для связывания аллостерических модуляторов [26–28]), LBD (связывает агонисты/антагонисты, контролирующие активацию ионного канала), TMD (формирует ионный канал), CTD (связывает постсинаптические белки, что обеспечивает передачу внутриклеточных сигналов) и NTD [29]. NMDAR обладают высокой селективностью к катионам и низкой к анионам. Проводимость NMDAR, как и у AMPAR, зависит от их состава. Проницаемость для Ca2+ и Na+ составляет PCa/PNa ~ 8–10 [30]. Вольт-амперные характеристики NMDAR более сложные, чем у AMPAR: при отрицательном МП, близком к потенциалу покоя (ПП), ионы Mg2+ входят в пору NMDAR, блокируя прохождение всех других ионов. При деполяризации мембраны и увеличении положительного заряда на цитоплазматической поверхности мембраны Mg2+ выталкивается из поры, позволяя Na+ и Ca2+ проникать в клетку. При положительном МП NMDAR характеризуется максимальной проницаемостью: в клетку входит Na+ и Са2+, а выходит K+. NMDAR имеют значительно более медленную кинетику изменений мембранного тока, чем AMPAR. В синапсах после экзоцитоза глутамата NMDAR активируются медленнее, а максимальная проводимость наблюдается значительно позже, чем для AMPAR. NMDAR могут оставаться в активном состоянии в течение сотен миллисекунд, а AMPAR – в течение всего нескольких миллисекунд. Важно отметить, что NMDAR активируются только при деполяризации мембраны, обусловленной активацией Na+-каналов, AMPAR или/и потенциал-зависимых Na+-каналов, и при условии, что Mg2+ не блокирует прохождение ионов через канал NMDAR.
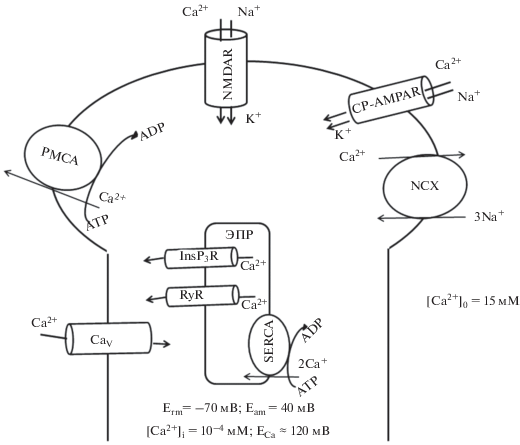
Рис. 3.
Ионные каналы и транспортеры, вовлеченные в регуляцию внутриклеточной концентрации кальция в постсинаптическом нейроне. NMDAR – глутаматные ионотропные NMDA-рецепторы, проницаемые для одно- и двухвалентных катионов; CP-AMPAR – Ca2+-проницаемые AMPA-рецепторы; CaV – потенциал-зависимые Ca2+-каналы; NCХ – Na+/Ca2+-обменник; PMCA – Ca2+-ATP-аза плазматической мембраны; SERCA – Ca2+-ATP-аза эндоплазматического ретикулума; RyR и InsP3R – Ca2+-проницаемые рианодиновые и инозитол-3-фосфатные рецепторы соответственно; Erm и Eam – потенциал покоя и потенциал действия соответственно; ECa – равновесный потенциал Нернста для Ca2+.
Потенциал-зависимые Na+-каналы
Потенциал-зависимые Na+-каналы (NaV) формируют входящий Na+-ток, который участвует в генерации и проведении ПД в нейронах (рис. 2) [31]. Известно, что NaV являются гетеромерами, которые состоят из потенциал-чувствительной α-субъединицы (~260 кДа), ассоциированной с одной или несколькими регуляторными β-субъединицами (~35 кДа), кодируемые SCN1A–SCN11A и SCN1B–SCN4B генами, соответственно. Известно, что α-субъединица образует пору канала, β‑субъединица контролирует активность канала и его потенциал-зависимые характеристики. Каждая α-субъединица состоит из гомологичных доменов, включающих шесть трансмембранных сегментов (S1–S6), из которых S1–S4 формируют сенсор потенциала, а S5 и S6 и их связующая P-петля формируют пору канала. Сегмент S4 содержат высокую концентрацию положительных зарядов (за счет аминокислотного остатка аргинина) и функционирует как сенсор потенциала, ответственный за активацию канала. Сегмент S6 формирует внутреннюю поверхность поры, а Р-петля формирует узкий ион-селективный фильтр, который контролирует ионную селективность канала и его проницаемость с внеклеточной стороны поры [32–34]. В клетках млекопитающих выявлено 10 изоформ α-субъединицы Nav (NaV1.1–NaV1.9 и NaX), из которых только 7 экспрессируются в нервной системе. В ЦНС взрослых животных преимущественно экспрессируются NaV1.1, NaV1.2 и NaV1.6 [35]. Известно, что NaV образуют кластеры высокой плотности в начальных сегментах аксона (НСА) и перехватов Ранвье, NaV1.1 локализованы в проксимальной, а NaV1.6 в дистальной частях НСА [36–38]. Экспрессия NaV изменяется в зависимости от стадии развития, показано, что Nav1.2 в НСА со временем заменяется на Nav1.6 [39]. Было обнаружено, что спайки в дистальных апикальных дендритах, происходящие при участии NaV-каналов, необходимы для формирования LTP в пирамидальных нейронах гиппокампа [40].
Потенциал-зависимые K+-каналы
Потенциал-зависимые K+-каналы (KV) формируют выходящие K+-токи, которые участвуют в реполяризации мембраны и восстановлении ее потенциала до уровня ПП после деполяризации, вызванной ПД (рис. 2). Известно, что KV-каналы – тетрамеры, состоящие из четырех α-субъединиц, формирующих ионную пору. Каждая α-субъединица состоит из шести α-спиральных трансмембранных сегментов (S1–S6), Р-петли, повторно входящей в мембрану между S5 и S6 сегментами, и цитоплазматическими N- и C-концами белка. Сегменты S1–S4 контролируют открывание и закрывание каналов, а S4 функционирует как сенсор потенциала. При изменении мембранного потенциала происходит конформационная перестройка в области S4 сегмента, вызывающая активацию канала [41]. У млекопитающих выявлено 12 подсемейств KV-каналов (KV1–KV12), у которых полумаксимальная активация и инактивация происходят при величинах мембранного потенциала от –30 до +20 мВ и от –25 до –90 мВ. Известно, что KV нейронов человека кодируют 40 генов, обеспечивая разнообразие формирования возбудимости (ритмического ответа) нейронов. Например, KV1.4 и KV4s активируются при низком потенциале, а KV3.4 – при высоком потенциале [42]. KV обнаружены в дендритных шипиках, НСА, соме нейронов, перехватах Ранвье и пресинаптических терминалях. В нейронах гиппокампа и стриатуме в СА1 регионе обнаружена высокая экспрессия дополнительной субъединицы KVb1. Показано, что у животных с нокаутом по гену Kvb1 синаптическая пластичность сохраняется, но обнаруживаются нарушения при выполнении некоторых тестов по исследованию памяти [43].
Потенциал-зависимые Ca2+-каналы
В настоящее время в нейрональных клетках выявлены три подтипа потенциал-зависимых Ca2+-каналов (VDCC или CaV): L-тип (CaV1.1–1.4), T-тип (CaV3.1–3.3) и N-тип (CaV2.1–2.3) (рис. 3). Каналы Т-типа получили свое название в связи с малой амплитудой проводимости и ее быстрой инактивацией, тогда как каналы L-типа характеризуются высокой амплитудой проводимости (5–9 пСм) и медленной кинетикой инактивации. Эти каналы блокируются производными 1,4-дигидропиридина. Каналы N-типа впервые были обнаружены в нейрональных клетках и нечувствительны к производным 1,4-дигидропиридина. Каналы N-типа разделяют на подтипы по их чувствительности к пептидным токсинам, выделенным из беспозвоночных. Каналы N-типа чувствительны к ω-конотоксину GVIA, P/Q-типа чувствительны к ω-Aga токсину IVA, R-типа нечувствительны к этим токсинам. Каналы CaV являются олигомерами и состоят из α1-субъединицы, образующую пору, через которую Ca2+ проходит в клетку, внутриклеточной β-субъединицы, трансмембранной γ-субъединицы и α2δ-комплекса, образованного за счет дисульфидной связи внеклеточной α2-субъединицы и трансмембранной δ-субъединицы. Обнаружено 10 генов для α1-субъединиц, четыре гена для β-субъединиц, четыре гена для α2δ-комплекса и восемь генов для γ-субъединицы [44]. α1-Субъединица состоит из четырех гомологичных доменов, включающих шесть α-спиральных трансмембранных сегментов (S1–S6), из которых S1–S4 являются сенсором потенциала, а S5–S6 образуют пору. Внутриклеточные петли связывающие домены регулируются Ca2+ и Ca2+-связывающими белками. Активация CaV Т-типа происходит при положительных значениях МП (до –70 мВ), для L-типа и N-типа при положительных значениях МП (до –20 мВ). Известно, что изменения трансмембранного потенциала, а также вход Ca2+ в клетку переводит CaV L-типа, N-типа, P/Q-типа в инактивированное состояние [45]. В нейронах CaV L-типа обеспечивают до ~80% транспорта ионов, опосредованных CaV. Многочисленные исследования показали, что наряду со входом Ca2+ в клетку CaV L-типа служат в качестве сенсоров напряжения, которые могут вызывать изменения МП для регуляции внутриклеточных процессов, включая выход Ca2+ из эндоплазматического ретикулума (ЭПР). Среди четырех изоформ CaV L-типа (Cav1.1–1.4) в возбудимых нейронах экспрессируются Cav1.2 и Cav1.3 [46]. Эти каналы участвуют в Ca2+-зависимых процессах, вовлеченных в контроль экспрессии генов, а также процесс сопряжения “возбуждение–транскрипция” [47]. Известно, что CaV1.2 и CaV1.3 в постсинаптическом нейроне обнаруживаются в дендритных шипиках и соме нейронов [48, 49]. Показано, что преимущественно CaV1.2, а не CaV1.3 участвуют в LTP и в процессе формирования пространственной памяти генетически модифицированных мышей [50–53].
Ca2+-активируемые K+-каналы
В нейронах обнаружены три типа Ca2+-активируемых K+-каналов (KCa): каналы с высокой проводимостью (ВК-каналы, KCa1.1), низкой проводимостью (SK-каналы, KCa2.1–2.3) и средней проводимостью (IK-каналы, KCa3.1) (рис. 2). BK-каналы активируются при деполяризации мембраны и (или) изменении [Ca2+]i, их проводимость составляет 200–400 пСм. При увеличении [Ca2+]i через ВК-каналы К+ выходит из клетки, что приводит к быстрой реполяризации мембраны, которая блокирует вход Ca2+ через CaV. Известно, что BK-каналы могут быть открыты в отсутствие Ca2+, что указывает на то, что влияние Ca2+ и деполяризации на вероятность открытия каналов являются практически независимыми процессами, и их действие на ВК кооперативно [54]. Полумаксимальная эффективная концентрация Ca2+, необходимая для активации ВК-каналов, при 30 мВ составляет >10 мкM [55], что значительно выше, чем [Ca2+]i в покое и при возбуждении. Установлено, что ВК-каналы образуют макромолекулярный комплекс с CaV1.2, CaV2.1 и CaV2.2 и активируются Ca2+, проникающим через эти каналы [56, 57]. Инактивация ВК-каналов, локализованных в пресинаптических терминалях, приводит к продолжительному выбросу нейротрансмиттера в синаптическую щель [58]. В постсинаптическом нейроне возможны межбелковые взаимодействия между ВК-каналами и NMDAR [59]. ВК-каналы представляют собой гомотетрамеры, состоящие из четырех идентичных α-субъединиц (BKα, кодируемая KCNMA1), образующих пору, либо α-субъединица образует комплекс с регуляторными β- или γ-субъединицами [60]. ВК-каналы могут быть расположены не только в плазматической мембране, но и в ядерной оболочке, что, вероятно, свидетельствует об их прямом влиянии на транскрипцию генов [61]. Установлено, что активность BK-каналов снижается при LTP [62]. В отличие от BK-каналов, активация SK- и IK-каналов зависит только от [Ca2+]i. SK- и IK-каналы обладают большой аффинностью к Ca2+ EC50 ∼ 0.3–0.5 мкM [63]. Проводимость SK-каналов составляет ~10 пСм [64, 65], а для IK-каналов 33–42 пСм [66, 67] при эквивалентной концентрации К+ (свыше 100 мМ) по обе стороны мембраны. SK- и IK-каналы структурно сходны с KV. В структуре SK- и IK-каналов в С-концевой области присутствует кальмодулин-связывающий домен, с которым постоянно связан кальмодулин (СаМ). Кальций, связываясь в СаМ, вызывает конформационные перестройки в структуре SK- и IK-каналов и открывает К+-проницаемую пору [68]. Активация данных каналов влияет на процесс генерации ПД, а также на пороговую величину для ритмического возбуждения. В CA1 нейронах гиппокампа SK- и IK-каналы участвуют в следовой гиперполяризации и влияют на возбудимость нейронов [69]. Выход К+ после активации данных каналов способен снижать постсинаптический потенциал [70]. Для SK-каналов была обнаружена функциональная связь с CaV L-типа и NMDAR в СА1 нейронах гиппокампа [71, 72]. Реполяризация постсинаптической мембраны способствует Mg2+-зависимому блоку NMDAR, что ограничивает вход Са2+ в клетку. Таким образом, SK-каналы могут являться компонентом/элементом отрицательной обратной связи для ослабления синаптической передачи [72]. Установлено, что блокирование SK-каналов проводит к усилению LTP в гиппокампе, а повышенная экспрессия SK-каналов снижает пространственную память и условно-рефлекторное замирание [73]. Роль IK-каналов в LTP/LTD пока не установлена.
Na+-активируемые K+-каналы
Na+-активируемые K+-каналы (KNa) характеризуются высокой проводимостью и активируются при связывании Na+ в области цитоплазматических доменов канала (рис. 2) [74]. Впервые их обнаружили в кардиомиоцитах и отнесли к новому типу ВК-каналов, для активации которых требовалось не менее 20 мМ Na+, но не требовался Ca2+ или ATP [75]. Важно, что для активации KNa необходима внутриклеточная концентрация Na+ ([Na]i), значительно превышающая ту, которая обычно имеется в цитоплазме. Предположено, что KNa играют важную роль в регуляции возбуждения в нейронах и активируются в условиях существенного повышения [Na]i при гипоксии и ишемии [76–78]. Отметим, что в норме у некоторых нейронов KNa обеспечивают выход K+ в зависимости от входа Na+, но не от [Na+]i [79]. KNa обнаружены во всех нейронах мозга и кодируются генами Slack (Slo2.2, KCNT1) и Slick (Slo2.1, KCNT2). Термин Slack происходит от названия “sequence like A calcium-activated K channel”, поскольку часть сегментов поры и последующий S6 сегмент идентичны на 7% каналу ВК (Slo1). Полумаксимальная активация канала (проводимость ~180 пСм) осуществляется при ~40 мМ [Na+]i. Активность Slack-канала может зависеть от протеинкиназ С и А, а также от эндогенных модуляторов (NAD+, эстрадиол и фосфатидилинозитол-4,5-бисфосфат) [80]. Установлено, что Slack-каналы непосредственно взаимодействуют с белком FMRP, который регулирует их активность. При отсутствии взаимодействия FMRP со Slack-каналами развивается Fragile X синдром, распространенная форма наследственной дисфункции интеллекта и аутизма у человека [81]. Таким образом, в настоящее время постулируется важная роль KNa в синаптической пластичности и интеллектуальной функции.
ATP-чувствительные К+-каналы
ATP-чувствительные K+-каналы (KATP) осуществляют выход K+, который ингибируется внутриклеточной концентрацией ATP, который является лигандом этих каналов. В связи с тем, что KATP регулируются нуклеотидами, считается, что они связывают энергетический статус клетки с электрической активность плазматической мембраны (рис. 2) [82–84]. Повышение ADP/ATP активирует KATP-каналы, что приводит к выходу К+ из клетки и гиперполяризации, а уменьшение соотношения ADP/ATP наоборот закрывает KATP-каналы. Установлено, что проводимость KATP составляет 44 пСм при концентрации K+ 100 мМ по обе стороны аксолеммы. Отметим, что вероятность активации данного канала возрастает после серии ПД [85]. Таким образом, активация KATP влияет на кинетику развития ПД и процессы восстановления после проведения ритмического возбуждения. KATP формируют гетерооктамерные комплексы, состоящие из четырех идентичных Kir6.1- либо Kir6.2-субъединиц, формирующих пору канала, и четырех идентичных SUR-субъединиц (одной из SUR1, SUR2A или SUR2B). Субъединицы Kir состоят из двух трансмембранных регионов, М1 и М2, соединенных внеклеточным регионом, содержащим K+ селективный мотив (глицин-тирозин-глицин). Регуляторные субъединицы SUR принадлежат к классу АВС-транспортеров и содержат два внутриклеточных домена для связывания ATP и других нуклеотидов. Комбинация разных Kir- и SUR-субъединиц формирует KATP-каналы с различной чувствительностью к нуклеотидам. В нейронах экспрессируются обе субъединицы Kir6.1 и Kir6.2, с доминирующей экспрессией Kir6.2. Например, в пирамидальных нейронах гиппокампа обнаружены KATP в такой комбинации субъединиц: SUR1-Kir6.1, SUR1-Kir6.2 и SUR2-Kir6.2 [86]. Установлено, что у мышей с нокаутом только по гену Kir6.2, но не Kir6.1 наблюдаются нарушения пространственной памяти и LTP [87, 88].
Кальциевые каналы ЭПР
Известно, что повышение [Ca2+]i может быть усилено за счет выхода Ca2+ из ЭПР и других органелл (митохондрии, лизосомы). Этот процесс получил название индуцируемый кальцием кальциевый выброс (CICR). При работе Ca2+-ATP-азы ЭПР (SERCA) концентрация Ca2+ внутри ЭПР составляет 10–3 М [89]. Выход Ca2+ из ЭПР происходит при участии двух основных рецепторов – рианодиновых (RyR) и инозитол-3-фосфатных (InsP3R) рецепторов (рис. 3) [90–92].
RyR – внутриклеточные Ca2+-каналы ЭПР, которые контролируют выход Ca2+ из ЭПР в цитоплазму. RyR состоят из четырех идентичных протомеров (~565 кДа), которые формируют центральную трансмембранную пору и большой цитоплазматический ансамбль, который участвует в образовании открытого и закрытого состояния канала за счет конформационных переходов [93, 94]. Ионный канал RyR обладает высокой проводимостью (100–500 пСм) для одновалентных и двухвалентных ионов (PCa/PK = 6–7) и быстрой кинетикой активации. RyR регулируются Ca2+, Mg2+, ATP, СаМ, сAMP, NAADP, протеинкиназами, фосфатазами и активными формами кислорода [95–97]. Изоформы RyR (RyR1–3) экспрессируются в нейронах разных отделов мозга (мозжечок, гиппокамп, обонятельный регион, базальные ганглии, кора больших полушарий), причем доминирует экспрессия RyR2-изоформы. Известно, что RyR включаются в CIRC при входе Ca2+ через NMDAR, а также через CaV L-типа в постсинаптических нейронах [98]. Отметим, что формирование LTP снижается при отсутствии выхода Ca2+ из ЭПР через RyR [99–101]. В CA1 нейронах гиппокампа при нокауте RyR3-изоформы после сильной тетанической стимуляции (3 серии по 100 Гц, 100 пульсов) инициация LTP не нарушается, но нарушается при слабой стимуляции (100 Гц, 21 пульс), сопровождаемой снижением пространственной памяти в водном тесте Морриса [102].
InsP3R имеют структурную гомологию с RyR (около 40%) и состоят из N-концевого инозитол-3-фосфат-связывающего домена, С-концевого домена, образующего Ca2+-канал, и связывающего домена, содержащего большинство регуляторных сайтов [103]. InsP3R, как и RyR, являются Ca2+-каналами, проницаемыми для одновалентных и двухвалентных ионов (PCa/PK = 4–5) [104–107]. Активность InsP3R регулируется Ca2+, инозитол-3-фосфатом, протеинкиназами, адениновыми нуклеотидами, рН. Известны три гена, кодирующие изоформы InsP3R1–InsP3R3. Экспрессия изоформ тканеспецифична, в ЦНС экспрессия InsP3R1 изоформы является доминирующей. Важно, что InsP3R также участвуют в инициации LTP, чаще всего при четырех, а не восьми сериях тета-взрыва (100 Гц, 5 пульсов с 200 мс интервалом), что соответствует более слабой стимуляции [100, 101]. У животных с нокаутом InsP3R1 наблюдается усиление индукции LTP, вызванной тетанической стимуляцией (100 Гц, 100 пульсов) [108]. До сих пор остается неясным, при каких условиях выход Ca2+ из ЭПР, опосредованный InsP3R, будет тормозить или стимулировать индукцию LTP.
Na+/Ca2+-обменник
Na+/Ca2+-обменник (NCX) – низкоаффинный антипортер, осуществляющий перенос трех ионов Na+ в клетку в обмен на один ион Ca2+ из клетки против его градиента (рис. 3). Работа NCX полностью обратима, т.е. движение транспортируемых ионов зависит от электрохимического градиента Na+ и Ca2+ и количества ионов, связанных с NCX. Структура NCX включает трансмембранный домен, образованный α-спиралями, и большую цитоплазматическую петлю [109, 110]. На участке цитоплазматической петли между аминокислотными остатками 371–509 расположен первый Ca2+-связывающий домен (CBD1), обладающий более высокой аффиностью к Ca2+ (Kd = 140–400 нМ), чем второй Ca2+-связывающий домен (CBD2). CBD2 расположен на участке между аминокислотными остатками 501–650 и связывает до трех ионов Ca2+ с аффинностью в 5–50 раз ниже, чем CBD1 [111]. Известно, что цитоплазматические Na+ и Сa2+ могут регулировать активность NCX. Сa2+, связываясь с цитоплазматической петлей, активирует NCX, в то время как Na+ деактивирует NCX [89]. В нейронах млекопитающих обнаружено три гена из семейства SLC8, кодирующих изоформы NCX1–NCX3 [112, 113]. Для генов NCX1 и NCX3 обнаружено 17 и 3 сплайсинг-вариантов белков, NCX2 не подвергается альтернативному сплайсингу [114]. Известно, что регуляцию изоформ осуществляют не только внутриклеточные ионы Na+ и Са2+, но и ионы H+, фосфатидилинозитол-4,5-бисфосфат, протеинкиназы [115]. NCX наряду с Ca2+-АТР-азой плазматической мембраны (PMCA) участвует в поддержании Ca2+-гомеостаза клеток в условиях повышения [Ca2+]i при формировании ритмического ответа нейронов, синаптической передаче и везикулярном транспорте. NCX имеет более низкую аффинность к Ca2+ (Kd = 1000 нМ), чем высокоаффинный РМСА (Kd = 100 нМ), но высокую скорость переноса Ca2+ (2000–5000 с–1 против 30–250 с–1), что позволяет быстрее переносить ионы Ca2+ [116]. При высокой концентрации [Ca2+]i активность РМСА снижается за счет быстрого насыщения Са2+, и в таких условиях выход Са2+ из клетки осуществляется за счет NCX. В нейронах мышей с дефицитом изоформы NCX2 при деполяризации восстановление [Ca2+]i происходит медленнее [117]. При нокауте NCX3 в нейронах наблюдается повышенный уровень [Ca2+]i в покое и задержка в снижении [Ca2+]i после деполяризации, а также снижение пространственной памяти при обучении в лабиринте Барнс и формирование памяти при анализе контекстно-зависимого состояния страха [118].
Na+,K+-ATP-аза
В нейронах низкая 5–10 мМ [Na+]i и высокая 140 мM [К+]i поддерживается в результате работы Na+,K+-ATP-азы (рис. 2). Na+,K+-ATP-аза осуществляет перенос трех ионов Na+ из клетки в обмен на два иона K+ против их электрохимических градиентов за счет гидролиза ATP. Na+,K+-ATP-аза представляет собой олигомер и состоит из каталитической α-субъединицы и регуляторных β- и γ-субъединиц. Гидролиз ATP приводит к фосфорилированию α-субъединицы Na+,K+-ATP-азы по аминокислотному остатку Asp369, что сопровождается конформационным переходом E1–E2 белка, связанным с электрогенным транспортом ионов, со скоростью 60–80 циклов фосфорилирования–дефосфорилирования в секунду. β-Субъединица необходима для доставки αβ-комплекса к плазматической мембране, а γ-субъединица изменяет чувствительность к ионам Na+, К+ и ATP при ассоциации с αβ-комплексом [119, 120]. α1-Субъединица Na+,K+-ATP-азы обнаружена во всех типах клеток, а другие α2–α4-субъединицы экспрессируются тканеспецифично. В отличие от большинства других клеток, пирамидальные и зубчатые нейроны коры больших полушарий и гиппокампа [121, 122], а также гранулярные клетки мозжечка крыс [123, 124] экспрессируют α3-субъединицу Na+,K+-ATP-азы. Показано, что α1- и α2-субъединицы обладают аффинностью к Na+ на 2–3 порядка выше, чем α3-субъединицы [125, 126]. В связи с этим считается, что α1-субъединица Na+,K+-ATP-азы играет ключевую роль в поддержании низкого соотношения [Na+]i/[K+]i в покое, в то время как α3-субъединица Na+,K+-ATP-азы вовлечена в снижение избытка [Na+]i при поддерживаемом возбуждении в нейронах [127–129]. Доказано, что α3-субъединица Na+,K+-ATP-азы выполняет ключевую роль в развитии пост-тетанической гиперполяризации, таким образом, регулируя генерацию ПД и формирование ритмического ответа отдельной клетки (“краткосрочная память”) [130]. Активность Na+,K+-ATP-азы уменьшается в нейронах в первые 15 мин после LTP и увеличивается спустя 30 мин [131]. Ингибирование Na+,K+-ATP-азы дигидроуабаином ассоциировано с новой формой LTD [132].
Ca2+-ATP-азы
Ca2+-АТР-аза плазматической мембраны (PMCA) представляет собой ATP-зависимый ионный насос, который осуществляет перенос одного иона Ca2+ из цитоплазмы клетки за счет гидролиза одной молекулы ATP (рис. 3). РМСА осуществляет восстановление концентрации [Ca2+]i (≈100–200 нM) после его повышения при функционировании нейронов. Известно, что транспорт Ca2+ связан с поглощением H+, но существенно не зависит от концентраций K+ и Na+ и МП [133, 134]. Структурно РМСА состоит из одной полипептидной цепи, включающей 10 трансмембранных сегментов и двух цитоплазматических петель, образующих четыре домена: А, P, N и СаМ-связывающий домены [135]. В состоянии покоя нейрона PMCA находится в автоингибиторном состоянии, которое реализуется за счет блокирования ATP-связывающего сайта на цитоплазматической петле (между 4 и 5 трансмембранными сегментами) С-концевым участком белка, где располагается СаМ-связывающий сайт. При повышении [Ca2+]i и связывании с CaM, С-концевой участок белка экранируется СаМ и открывается сайт для связывания ATP. Выявлены четыре гена, кодирующих изоформы РМСА1–PMCA4. Распределение изоформ неоднородно в разных тканях: PMCA1 и PMCA4 экспрессируются во всех тканях, PMCA2 и PMCA3 преимущественно в мышцах и нервной системе [136, 137]. В нейронах мозга выявлены все четыре изоформы РМСА [138]. Исследования кинетики активации изоформ РМСА доказали значительные различия в их способности отвечать на увеличение [Ca2+]i. Например, PMCA2 характеризуется максимальной аффинностью к Ca2+ и отвечает на увеличение его концентрации даже в отсутствие СаМ [139]. Известно, что изоформы РМСА взаимодействуют с различными регуляторными белками в клетках, что определяет их специфическую роль в регуляции Ca2+ в нервной клетке [140]. PMCA взаимодействует с Cav L-типа, NMDAR, nNOS и цитохром-b5-редуктазой, что обеспечивает регуляцию Ca2+-гомеостаза и сигнализацию [141]. В мозжечке крыс PMCA2 и PMCA3 изоформы обычно располагаются в пост- и пресинаптических терминалях, в соме и дендритах клеток Пуркинье, и, вероятнее всего, уровень их экспрессии коррелирует с созреванием нейронов и формированием синапсов [142–144].
Ca2+-АТР-аза эндоплазматического ретикулума (SERCA) представляет собой ATP-зависимый ионный насос, который осуществляет перенос двух ионов Ca2+ из цитоплазмы клетки в ЭПР за счет гидролиза одной молекулы ATP (рис. 3). Основная роль SERCA – поддержание Ca2+-гомеостаза. Фермент включает три домена (A, N и P), одну петлю, расположенную в области люмен ЭПР, и 10 трансмембранных α-спиралей (М1–М10). Связывание Ca2+ происходит в области А-домена, связывание ATP – на N-домене, а фосфорилированный интермедиат формируется в области Р-домена. Трансмембранные α-спирали M2, M5, M6 и M8 формируют Ca2+-канал, M4–M6 повышают эффективность переноса Ca2+ через мембрану ЭПР. SERCA экспрессируется в виде 12 изоформ, кодируемых тремя генами SERCA1–SERCA3. Все изоформы SERCA ингибируются тапсигаргином и циклопиазоновой кислотой [145]. Известно, что изоформы SERCA различаются по сродству к Ca2+, например, SERCA2b имеет в 2 раза большую чувствительность к Ca2+, чем SERCA2a [146]. Белок SERCA2b был обнаружен в большинстве нейронов, главным образом в клетках Пуркинье и пирамидальных нейронах гиппокампа. Известно, что SERCA вовлечена в механизм переноса Ca2+ из цитоплазмы в ЭПР при генерации ПД в постсинаптических дендритных шипиках [147, 148].
2. МЕХАНИЗМЫ УЧАСТИЯ ОДНОВАЛЕНТНЫХ И ДВУХВАЛЕНТНЫХ ИОНОВ В СИНАПТИЧЕСКОЙ ПЛАСТИЧНОСТИ
Установлено, что для индукции LTP в CA1 пирамидальных нейронах гиппокампа требуется повышение [Ca2+]i. При исследовании синаптической пластичности Линч и коллеги обнаружили, что в нейронах гиппокампа крыс внутриклеточный Ca2+-хелатор EGTA блокировал развитие LTP [10]. Как отмечалось выше вход Ca2+ в нейроны происходит при участии NMDAR, CaV и CP-AMPAR. Известно, что действие селективного антагониста NMDAR аминофосфовалериановой кислоты (APV) до тетанической стимуляции нейронов приводит к блокированию LTP, в то время как после тетанической стимуляции APV не вызывает аналогичного эффекта [149]. Вероятно, NMDAR выполняют важную роль при индукции LTP, а активация AMPAR важна как для индукции, так и для формирования LTP. Действительно, присутствие антагониста AMPAR цианхиноксалина (CNQX), блокировало как индукцию, так и формирование LTP [150, 151]. Установлено также, что ингибитор SERCA и антагонисты InsP3R и RyR приводят к снижению LTP и LTD [150, 152]. Доказано, что за счет высокого электрохимического градиента Ca2+ активация Ca2+-каналов и транспортеров приводит к быстрому увеличению [Ca2+]i от ~0.1 до 1 мкM. С помощью фотоактивного Ca2+-буфера, diazo-4, выявлен интервал времени, в течение которого после ритмического раздражения нейрона концентрация Ca2+ возрастает и формируется LTP. Известно, что сродство diazo-4 к Ca2+ возрастает в 1600 раз при импульсном фотолизе, что позволяет в эксперименте нивелировать повышение [Ca2+]i и доказать, что повышение постсинаптического [Ca2+]i в течение 2–2.5 с после ритмического раздражения нейрона достаточно для индукции LTP, а снижение или увеличение длительности возбуждения приводит к краткосрочной потенциации [153]. При увеличении [Ca2+]i происходит взаимодействие Ca2+ с CaM и кальциневрином, которые, в свою очередь, влияют на сопряжение “возбуждение–транскрипция” (excitation-transcription coupling, ETC) [16, 154, 155], в том числе экспрессию генов раннего ответа c-Fos, c-Jun и Egr1 (immediate early genes, IEG) [14, 156, 157]. Cole и коллеги обнаружили, что в нейронах гиппокампа крыс частота и амплитуда электростимуляции, необходимые для повышения уровня мРНК Egr1 аналогичны тем, что необходимы для индуцирования LTP в постсинаптическом нейроне. Кроме того, как и при LTP, экспрессия Egr1 не наблюдалась в присутствии антагонистов NMDAR [158]. В экспериментах Fleischmann и коллеги использовали нейроны мышей с нокаутом c-Fos и наблюдали нормальное общее и эмоциональное поведение животных, хотя были выявлены нарушения при выполнении задач пространственного и ассоциативного обучения, зависящих от гиппокампа. Эти эффекты коррелировали со снижением LTP в синапсах CA3–CA1 гиппокампа, свидетельствуя об участии экспрессии IEG в формировании молекулярных механизмов обучения и памяти [159, 160]. Greenberg и коллеги, используя модель (клеточную линию феохромацитомы PC12), обнаружили, что экспрессия c-Fos, вызванная K+-деполяризацией клеток, не происходит в присутствии 3 мМ EGTA, что может быть связано с повышением Ca2+ или, возможно, с неспецифическим повышением Na+ и K+ [161]. В наших экспериментах показано, что добавление EGTA приводило к увеличению проницаемости плазматической мембраны для одновалентных катионов и повышению отношения [Na+]i/[K+]i в сосудистых клетках гладких мышц [162, 163].
Важно, что экспрессия генов, запускаемая при возбуждении нейронов, в основном блокируется ингибиторами CaV L-типа, нежели антагонистами ионотропных глутаматных рецепторов [47]. Установлено, что LTP сопровождается транслокацией CaM в ядро [164, 165], а введение в клетку кальмидозолия, ингибитора CaM-зависимого сигналинга, блокируют LTP [166]. Отметим, что изменение [Ca2+]i влияет на транскрипцию за счет следующих процессов:
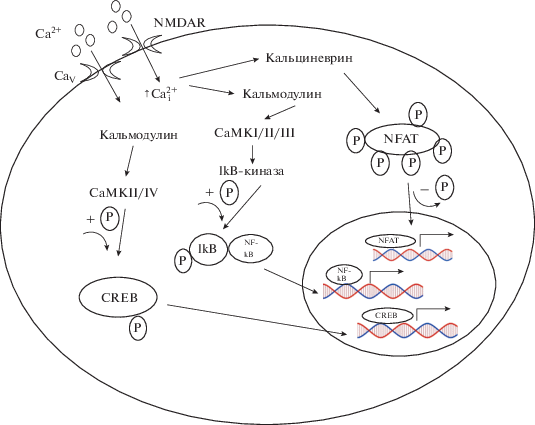
1) повышение [Ca2+]i способствует перемещению из цитозоля в ядро фактора транскрипции NF-kB. Этот процесс связан с активацией Ca2+/ кальмодулин-чувствительной протеинкиназы (CaMKI, II или III), которая запускает фосфорилирование ингибитора kВ (IkB) фосфорилированной IkB-киназой. Фосфорилированный IkB отсоединяется от NF-kB, далее NF-kB перемещается в ядро;
2) повышение [Ca2+]i способствует транспорту из цитозоля в ядро NFAT, в результате дефосфорилирования NFAT кальциневрином, (Ca2+/кальмодулин)-зависимой фосфатазой [167];
3) повышение [Ca2+]i в цитоплазме и ядре приводит к фосфорилированию CREB Ca2+/кальмодулин-чувствительными протеинкиназами – CaMKII и CaMKIV, соответственно. Фосфорилированный CREB и его коактиватор CREB-связывающий белок регулируют транскрипцию за счет связывания с CRE-последовательностями ДНК [14, 168] (рис. 4).
Рис. 4.
Механизмы вовлечения кальция в регуляцию транскрипции в нейронах. NMDAR – глутаматные ионотропные NMDA-рецепторы; CaV – потенциал-зависимые Ca2+-каналы.
Tsien и коллеги доказали, что фосфорилирование CREB зависит от активации CaV при деполяризации мембраны нейрона [16]. Помимо регуляции транскрипции, которая, вероятно, участвует в формировании долгосрочной памяти посредством дифференциальной экспрессии генов, стимулирующей рост дендритов и развитие синапса, Ca2+-зависимые белки влияют на быстрые изменения в синапсах вместе с включением или удалением субъединиц глутаматных рецепторов из плазматической мембраны и изменения функций синаптических белков посредством их посттрансляционных модификаций (фосфорилирование) [14, 24, 169]. Так, на проводимость AMPAR влияют CaMKII и Ca2+-активируемая протеинкиназа A (РКА) [170, 171]. В нейронах гиппокампа мышей CaMKII фосфорилирует GluA1-субъединицу AMPAR по аминокислотному остатку Ser831, что способствует входу Ca2+ и Na+ и приводит к временнóму увеличению концентрации этих ионов. Фосфорилирование PKA GluA1 в положении Ser845 увеличивает вероятность активации GluA1 канала в 1.5 раза [172, 173]. РКА фосфорилирует не только АМРАR, но и NMDAR, регулируя таким образом проницаемость мембраны нейрона для Ca2+ [174–176]. В отличие от CaMKII, роль которой важна на стадии индукции LTP, активация протеинкиназы С (РКС) необходима для формирования LTP. Последнее подтверждается экспериментами с применением ингибиторов РКС полимиксина В, Н-7, сфингозина и К-252b [166, 177–180]. Доказано, что ингибирование пептидами или нокаут CaMKII предотвращали поддержание LTP.
В процессе LTP происходит синтез новых AMPAR и доставка их к поверхности плазматической мембраны нейрона. Этот процесс связан с реорганизацией актинового цитоскелета. Многочисленные исследования продемонстрировали, что активация GTP-связывающих белков (Rho GTP-аз), способствующих полимеризации актина, является обязательным элементом как структурной, так и функциональной регуляции LTP [181, 182]. Murakoshi с коллегами показали, что ингибирование NMDAR APV инактивирует Rho GTP-азы (RhoA и Cdc42) и повышает плотность шипиков в CA1 гиппокампа. Ингибитор CaMKII частично блокировал активацию RhoA и Cdc42 в процессе временных и постоянных фаз LTP [183]. Показано, что высокочастотная стимуляция нейронов, используемая для индукции LTP, приводит к активации кальпаинов, деградации спектринов и тем самым к разборке актина и облегчению доставки новых AMPAR к поверхности мембраны [184]. Amini с коллегами показал, что при удалении кальпаина-1 и -2 только из ЦНС происходит снижение ответа LTP на 30–40% [185].
Как отмечалось выше, активация Na+-проницаемых АМРАR приводит к деполяризации постсинаптической мембраны, в результате чего происходит удаление Mg2+-блока с NMDAR и их активация [13]. Эти данные позволили предположить, что деполяризация представляет собой единственный и достаточный механизм вовлечения потоков Na+ в индукцию и пролонгацию синаптической пластичности [182]. Однако, учитывая, что активация AMPAR важна как для индукции LTP, так и для ее развития, можно предположить, что возбуждение нейронов связано с высокой концентрацией Na+ или повышением соотношения [Na+]i/[K+]i. На рис. 5 представлена кинетика изменений [Na+]i и [Ca2+]i при электростимуляции нейронов гиппокампа [186]. Изменения Ca2+ являются быстрыми, в то время как изменения [Na+]i длятся несколько минут. Очевидно, что при активации синаптической передачи транспорт Na+ и K+ осуществляется не только AMPAR, но и NMDAR, что регулирует [Na+]i/[K+]i. Действительно, на первичных культурах гранулярных клеток мозжечка крыс с применением Na+-чувствительного флуоресцентного красителя SBFI было показано, что активация NMDAR повышает [Na+]i от 5 до 60 мМ [187]. В CA1 пирамидальных клетках мышей при электрической стимуляции было обнаружено ~10-кратное повышение [Na+]i в присутствии ингибитора Nav тетродотоксина и антагониста AMPAR CNQX [188]. На тонких срезах ткани мозжечка мышей с флуоресцентным Na+-красителем обнаружено, что при действии глутамата в дендритах нейронов Пуркинье [Na+]i обратимо повышается на 5–10 мМ [91]. Установлено, что длительное возбуждение нейронов сопровождается уменьшением [K+]/[Na+] в цереброспинальной жидкости [17, 189–191], что также свидетельствуют о том, что наряду с повышением [Ca2+]i возбуждение приводит к изменению трансмембранных градиентов одновалентных катионов и повышению внутриклеточного соотношения [Na+]i/[K+]i. Linden с коллегами, используя культуру тканей с моделью LTD, установили, что при замене экстраклеточного Na+ на N-метил-D-глюкамин, а также на Li+ или Cs+ LTD не возникает. Все это свидетельствует о том, что постсинаптический поток Na+ и повышение [Na+]i необходимы для индукции LTD и что LTD запускается не только за счет входа Ca2+ [19].
Рис. 5.
Изменения внутриклеточной концентрации Na+ и Ca2+ в нейронах гиппокампа при действии электрической стимуляции [186].
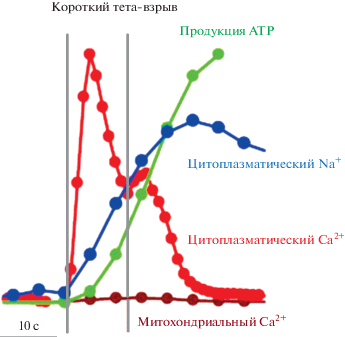
В связи с этим важно отметить, что входу Ca2+ в дендриты предшествует генерация Na+-зависимых ПД [90]. Callaway и коллеги обнаружили, что при стимуляции нейронов повышение [Na+]i частично блокируется ингибиторами NaV [192]. Показано, что активация NaV вератридином меняет процесс индукции LTD [19]. Исследований о связи AMPAR и NMDAR с повышением [Na+]i в нейронах в настоящее время явно не достаточно. В дендритных шипиках Miyazaki и Ross, используя методику одновременной визуализации изменений содержания Na+ и Ca2+, обнаружили, что в одном дендритном шипике пирамидальных нейронов гиппокампа крыс после электрической стимуляции основное количество Na+ входит через AMPAR и малое количество через NMDAR и NaV [193]. Кроме того, блокирование Ca2+-транспорта и Ca2+-опосредованного сигналинга приводили к частичному, а не полному подавлению LTP и LTD. Так, Behnisch и Reymann установили, что в CA1 нейронах гиппокампа крыс блокирование SERCA тапсигаргином снижает индукцию LTP только при слабой стимуляции, но не при тетаническом возбуждении [194]. Установлено, что у животных с нокаутом всех изоформ CREB (Ca2+-чувствительного интермедиата ETC) не было обнаружено изменений в формировании LTP и LTD в нейронах гиппокампа и выявлено нормальное условно-рефлекторное замирание [195]. Вероятно, это свидетельствует о Ca2+-независимом механизме сопряжения “возбуждение–транскрипция”.
Итак, мы считаем, что наряду с Ca2+-опосредованной синаптической пластичностью и долгосрочной памятью, связанной с транскриптомными изменениями, в организме может реализовываться Ca2+-независимый процесс, опосредованный изменениями внутриклеточной концентрации одновалентных ионов [196].
3. РОЛЬ ОДНОВАЛЕНТНЫХ КАТИОНОВ В ЭКСПРЕССИИ ГЕНОВ, ВОВЛЕЧЕННЫХ В СИНАПТИЧЕСКУЮ ПЛАСТИЧНОСТЬ
Известно, что повышение экспрессии IEG, а именно c-Fos, происходит спустя 30 мин после индукции LTP в CA1 нейронах гиппокампа [197]. Уабаин, вызывающий повышение соотношения [Na+]i/[K+]i, также приводит к резко повышенной экспрессии IEG c-Fos и c-Jun в клетках VSMC и HeLa [198, 199]. Увеличение c-Fos обнаруживается через 30 мин, когда [Na+]i увеличена в ~5 раз, а [K+]i снижена на 10–15%. Установлено, что повышение экспрессии c-Fos, вызванной уабаином, сохраняется в среде, свободной от Ca2+, и в присутствии внеклеточных (EGTA) и внутриклеточных (BAPTA) Ca2+-хелаторов. Очевидно, что увеличение [Na+]i, а не снижение [K+]i генерирует сигнал, который приводит к экспрессии c-Fos в данных типах клеток. Вероятно, что изменение экспрессии c-Fos вызвано повышением соотношения [Na+]i/[K+]i [200]. Уабаин и бескалиевая среда, вызывающие повышение соотношения [Na+]i/[K+]i, изменяли экспрессию 80 одинаковых [Na+]i/[K+]i-чувствительных генов независимо от присутствия Ca2+-хелаторов в трех типах клеток HUVEC, HeLa, RVSMC [196]. Почти половина [Na+]i/[K+]i-чувствительных генов были генами раннего ответа, участвующими в регуляции транскрипции, среди которых также были обнаружены Fos, Jun, увеличение экспрессии которых является маркером долговременной памяти.
В нейронах мозжечка установлено, что уабаин, увеличивающий соотношение [Na+]i/[K+]i, приводит к повышению экспрессии таких регуляторов транскрипции, как Fosb, Junb, а также Ptgs2, Rgs2, Rnd3, Vgf [201], для которых показано участие в синаптической пластичности. Известно, что при активации NMDAR происходит выброс арахидоновой кислоты из культивируемых нейронов [150]. PTGS (простагландин-эндопероксидсинтаза или циклооксигеназа, СОХ) – это фермент, лимитирующий скорость превращения арахидоновой кислоты в простагландины. У млекопитающих были идентифицированы два изофермента СОХ: COX-1 и COX-2, участвующие в заживлении ран и воспалительной реакции. COX-2 обнаруживается в дискретных популяциях нейронов и в большом количестве в коре, гиппокампе, в областях мозга, играющих ключевую роль в обучении и хранении памяти [202, 203]. Известно, что COX-2 участвует в синаптической передаче. Yamagata с коллегами показали, что COX-2 локализован в дендритных шипиках нейронов и его экспрессия вызывается NMDAR при высокочастотной стимуляции нейронов, связанной с индукцией LTP [203]. Показано, что предварительная обработка селективным ингибитором COX-2 NS398 значительно уменьшала LTP в перфорантном пути в срезах гиппокампа [204]. Участие COX-2 в синаптической пластичности гиппокампа было подтверждено исследованиями, демонстрирующими, что ингибирование COX-2 подавляет как LTP, так и LTD и экспериментами in vivo, в которых введение ингибиторов COX-2 препятствует запоминанию и консолидации памяти. Воздействие COX-2 проявляется в усилении выработки простагландина E2 (PGE2) в постсинаптических терминалях. Далее PGE2 взаимодействует с EP2 рецепторами в пресинаптических терминалях, что, в свою очередь, приводит к активации аденилатциклазы, повышению сAMP, PKA-опосредованной активации CaV L-типа, входу Ca2+ и усилению выброса нейротрансмиттеров [202, 205, 206]. Механизмы, приводящие к увеличению активности COX-2 в постсинаптических терминалях во время их возбуждения, остаются плохо изученными. Мы предполагаем, что это явление может быть вызвано усиленной экспрессией COX-2, вызванной диссипацией трансмембранных градиентов одновалентных ионов. Действительно, повышение соотношения [Na+]i/[K+]i в нейронах мозжечка после действия уабаина сопровождалось повышением содержания мРНК Ptgs2 в ~18 раз [201]. Воздействие высоких доз уабаина на нейроны мозжечка приводит к повышению содержания мРНК Rgs2. Белок RGS2 принадлежит к большому семейству белков, активирующих GTP-азы, которые ингибируют сигнальные каскады с участием G-белков [207]. Ingi и коллеги показали, что RGS2 быстро увеличивается в нейронах гиппокампа, коры и стриатума в ответ на стимулы, которые влияют на синаптическую пластичность [208]. Известно, что RGS2 влияет на краткосрочную пластичность путем понижения Gi-опосредованной пресинаптической сигнализации, что приводит к увеличению концентрации Ca2+ и повышению вероятности высвобождения нейротрансмиттера. Мутантные мыши, не имеющие Rgs2, демонстрируют нормальное поведение при выполнении задач обучения и памяти, однако их CA1 пирамидальные нейроны гиппокампа проявляют более слабую связь, измеряемую как уменьшенные количества шипиков [209]. Помимо Ptgs2 и Rgs2 уабаин повышает экспрессию Rnd3. Белок RND3 из Rho семейства Ras суперсемейства GTP-аз, регулирует организацию актинового цитоскелета в ответ на внеклеточные стимулы. Исследования продемонстрировали вовлечение актинового цитоскелета в синаптическую пластичность в дендритах [210] и экспрессию Nav в НСА [211]. Данные, полученные как в бесклеточных экспериментах [212], так и с использованием клеток, после воздействия на них ингибиторов Na+,K+-ATP-азы [213–215], убедительно свидетельствуют о том, что на организацию актинового цитоскелета влияет изменение соотношения [Na+]i/[K+]i. Увеличение соотношения [Na+]i/[K+]i, индуцированное уабаином, сопровождалось также повышением экспрессии Vgf. Ранее было показано, что VGF регулирует синаптическую пластичность гиппокампа [216] и его экспрессия увеличивается в ~2.5 раза в гиппокампе после острых электроконвульсивных припадков [217].
Важно отметить, что удаление внеклеточного и внутриклеточного Ca2+ с помощью Ca2+-хелаторов увеличивало, а не уменьшало число [Na+]i/[K+]i-чувствительных генов. Повышение числа [Na+]i/[K+]i-чувствительных генов при использовании внеклеточного хелатора Ca2+ (EGTA) может быть связано c его возможностью увеличивать проницаемость плазматической мембраны для одновалентных катионов и приводить к повышению отношения [Na+]i/[K+]i [162, 163]. В связи с этим требуется использовать другие подходы для оценки механизма регуляции экспрессии [Na+]i/[K+]i-чувствительных генов, например, ингибиторы Са2+-регулируемых белков. В экспериментах, выполненных на клетках RVSMC, применение антагонистов CaV L-типа (никардипина) и кальмодулина (A-7 и W-7) совместно с уабаином снижало экспрессию Ptgs2 и Nr4a1, но не влияло на экспрессию Egr1 и Atf3 [218] (рис.6). Экспрессия Ptgs2 также не зависела от присутствия антагониста CaV L-типа в клетках C2C12 [219].
Рис. 6.
Механизмы вовлечения одновалентных катионов и Ca2+ в регуляцию экспрессии генов в не-нейрональных клетках. NKA – Na+,K+-ATP-аза; CaV – потенциал-зависимые Ca2+-каналы; NCХ – Na+/Ca2+-обменник; NaREB – неидентифицированный сенсор изменения [Na+]i, активирующий NaRE; NaRE – Na+-чувствительный элемент, регулирующий транскрипцию генов Egr1, Atf3. Остальные сокращения приведены в тексте.
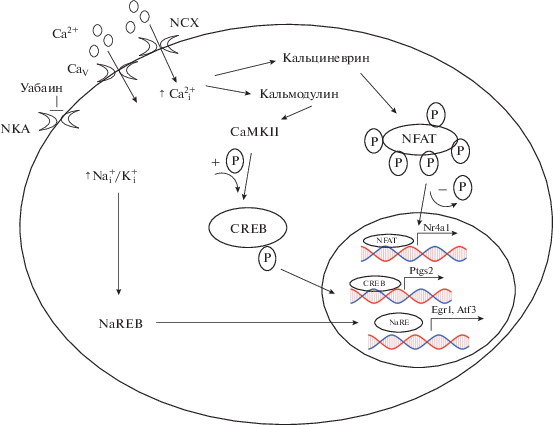
Итак, ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-независимые механизмы сопряжения “возбуждение–транскрипция” способствуют экспрессии генов за счет увеличения соотношения [Na+]i/[K+]i. Однако необходимо отметить, что воздействие ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-опосредованного и ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-независимого сигналинга в общие транскриптомные изменения, по-видимому, является специфическим для разного типа клеток. Таким образом, роль ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-опосредованного и ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-независимого сигналинга в регуляции механизмов экспрессии [Na+]i/[K+]i-чувствительных генов, вовлеченных в синаптическую пластичность нейронов, требует дальнейших исследований.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-14-50358. Funding: The reported study was funded by RFBR, project no. 19-14-50358.
Список литературы
Bailey C.H., Kandel E.R., Harris K.M. 2015. Structural components of synaptic plasticity and memory consolidation. Cold Spring Harb. Perspect. Biol. 7 (7), 1–29.
Kojima M., Klein R.L., Hatanaka H. 2002. Pre- and post-synaptic modification by neurotrophins. Neurosci. Res. 43 (3), 193–199.
Ghanbari A., Malyshev A., Volgushev M., Stevenson I.H. 2017. Estimating short-term synaptic plasticity from pre- and postsynaptic spiking. PLoS Comput. Biol. 13 (9), 1–28.
Castro-Alamancos M.A., Connors B.W. 1997. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc. Natl. Acad. Sci. USA. 94 (8), 4161–4166.
Tallent M.K., Varghis N., Skorobogatko Y., Hernandez-Cuebas L., Whelan K., Vocadlo D.J., Vosseller K. 2009. In vivo modulation of O-GIcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. J. Biol. Chem. 284 (1), 174–181.
Hafner A.-S., Donlin-Asp P.G., Leitch B., Herzog E., Schuman E.M. 2019. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 364 (6441), eaau3644.
Kukushkin N.V., Carew T.J. 2017. Memory takes time. Neuron. 95 (2), 259–279.
Bliss T.V.P., Collingridge G.L., Morris R.G.M., Reymann K.G. 2018. Long-term potentiation in the hippocampus: Discovery, mechanisms and function. Neuroforum. 24 (3), A103–A120.
Artola A., Singer W. 1993. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 16 (11), 480–487.
Lynch G., Larson J., Kelso S., Barrionuevo G., Schottler F. 1983. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 305 (5936), 719–721.
Eccles J.C. 1983. Calcium in long-term potentiation as a model for memory. Neuroscience. 10 (4), 1071–1081.
Malenka R.C. 1991. The role of postsynaptic calcium in the induction of long-term potentiation. Mol. Neurobiol. 5 (2–4), 289–295.
Lüscher C., Malenka R.C. 2012. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 4 (6), a005710–a005710.
Flavell S.W., Greenberg M.E. 2008. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci. 31 (1), 563–590.
Tatsuo S. 1994. Protein kinases involved in the expression of long-term potentiation. Int. J. Biochem. 26 (6), 735–744.
Ma H., Groth R.D., Wheeler D.G., Barrett C.F., Tsien R.W. 2011. Excitation–transcription coupling in sympathetic neurons and the molecular mechanism of its initiation. Neurosci. Res. 70 (1), 2–8.
Orlov S.N., Mongin A.A. 2007. Salt-sensing mechanisms in blood pressure regulation and hypertension. Am. J. Physiol. Circ. Physiol. 293 (4), H2039–H2053.
Sobolevsky A.I., Rosconi M.P., Gouaux E. 2009. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 462 (7274), 745–756.
Linden D.J., Smeyne M., Connor J.A. 1993. Induction of cerebellar long-term depression in culture requires postsynaptic action of Sodium Ions. Neuron. 11 (6), 1093–1100.
Dickenson A.H. 2006. Amino acids: Excitatory. In: Neurotransmitters, Drugs Brain Funct. Chichester, UK: John Wiley & Sons, Ltd, p. 211–223.
Henley J.M., Wilkinson K.A. 2016. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17 (6), 337–350.
Luscher C., Malenka R.C. 2012. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 4, a005710.
Lalanne T., Oyrer J., Farrant M., Sjöström P.J. 2018. Synapse type-dependent expression of calcium-permeable AMPA receptors. Front. Synaptic Neurosci. 10, 1–8.
Malinow R., Malenka R.C. 2002. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25 (1), 103–126.
Liu S.Q.J., Cull-Candy S.G. 2000. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 405 (6785), 454–458.
Gielen M., Siegler Retchless B., Mony L., Johnson J.W., Paoletti P. 2009. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 459 (7247), 703–7.
Yuan H., Hansen K.B., Vance K.M., Ogden K.K., Traynelis S.F. 2009. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 29 (39), 12045–12058.
Hansen K.B., Furukawa H., Traynelis S.F. 2010. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol. Pharmacol. 78 (4), 535–549.
Karakas E., Furukawa H. 2014. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 344 (6187), 992–7.
Uteshev V. V. 2012. α7 nicotinic ACh receptors as a ligand-gated source of Ca2+ ions: The search for a Ca2+ optimum. Adv. Exp. Med. Biol. 740, 603–638.
Bean B.P. 2007. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8 (6), 451–465.
Yu F.H., Catterall W.A. 2003. Overview of the voltage-gated sodium channel family. Genome Biol. 4 (3), 207.
Catterall W.A. 2000. From ionic currents to molecular mechanisms. Neuron. 26 (1), 13–25.
George A.L. 2005. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115 (8), 1990–1999.
Savio-Galimberti E., Gollob M.H., Darbar D. 2012. Voltage-gated sodium channels: Biophysics, pharmacology, and related channelopathies. Front. Pharmacol. 3, 1–19.
Caldwell J.H., Schaller K.L., Lasher R.S., Peles E., Levinson S.R. 2000. Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA. 97 (10), 5616–5620.
Hu W., Tian C., Li T., Yang M., Hou H., Shu Y. 2009. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat. Neurosci. 12 (8), 996–1002.
Duflocq A., Le Bras B., Bullier E., Couraud F., Davenne M. 2008. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol. Cell. Neurosci. 39 (2), 180–192.
Solé L., Tamkun M.M. 2020. Trafficking mechanisms underlying Nav channel subcellular localization in neurons. Channels. 14 (1), 1–17.
Kim Y., Hsu C.L., Cembrowski M.S., Mensh B.D., Spruston N. 2015. Dendritic sodium spikes are required for long-term potentiation at distal synapses on hippocampal pyramidal neurons. Elife. 4, 1–30.
Miller C. 2000. An overview of the potassium channel family. Genome Biol. 1 (4), 1–5.
Zemel B.M., Ritter D.M., Covarrubias M., Muqeem T. 2018. A-Type KV channels in dorsal root ganglion neurons: Diversity, function, and dysfunction. Front. Mol. Neurosci. 11, 1–17.
Giese K.P., Storm J.F., Reuter D., Fedorov N.B., Shao L.R., Leicher T., Pongs O., Silva A.J. 1998. Reduced K+ channel inactivation, spike broadening, and after- hyperpolarization in Kvβ1.1-deficient mice with impaired learning. Learn. Mem. 5 (4–5), 257–273.
Hofmann F., Lacinová L., Klugbauer N. 1999. Voltage-dependent calcium channels: From structure to function. Rev. Physiol. Biochem. Pharmacol. 139, 33–87.
Brehm P., Eckert R. 1978. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 202 (4373), 1203–1206.
Striessnig J., Pinggera A., Kaur G., Bock G., Tuluc P. 2014. L-type Ca2+ channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 3 (2), 15–38.
Ma H., Cohen S., Li B., Tsien R.W. 2013. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci. Rep. 33 (1), 97–101.
Di Biase V., Obermair G.J., Szabo Z., Altier C., Sanguesa J., Bourinet E., Flucher B.E. 2008. Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J. Neurosci. 28 (51), 13845–13855.
Jenkins M.A., Christel C.J., Jiao Y., Abiria S., Kim K.Y., Usachev Y.M., Obermair G.J., Colbran R.J., Lee A. 2010. Ca2+-Dependent facilitation of cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30 (15), 5125–5135.
Striessnig J., Koschak A. 2008. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene Knockout models. Channels 2 (4), 233–251.
Moosmang S., Haider N., Klugbauer N., Adelsberger H., Langwieser N., Müller J., Stiess M., Marais E., Schulla V., Lacinova L., Goebbels S., Nave K.A., Storm D.R., Hofmann F., Kleppisch T. 2005. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 25 (43), 9883–9892.
White J.A., McKinney B.C., John M.C., Powers P.A., Kamp T.J., Murphy G.G. 2008. Conditional forebrain deletion of the L-type calcium channel Ca V1.2 disrupts remote spatial memories in mice. Learn. Mem. 15 (1), 1–5.
Barad M. 2006. Divide and conquer: An L-type voltage-gated calcium channel subtype finds a role in conditioned fear: Commentary. Learn. Mem. 13 (5), 560–561.
Horrigan F.T., Aldrich R.W. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120 (3), 267–305.
Rothberg B.S., Magleby K.L. 1999. Gating kinetics of single large-conductance Ca2+-activated K+ channels in high Ca2+ suggest a two-tiered allosteric gating mechanism. J. Gen. Physiol. 114 (1), 93–124.
Berkefeld H., Sailer C.A., Bildl W., Rohde V., Thumfart J.O., Eble S., Klugbauer N., Reisinger E., Bischofberger J., Oliver D., Knaus H.G., Schultes U., Fakler B. 2006. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 314 (5799), 615–620.
Berkefeld H., Fakler B. 2008. Repolarizing responses of BKCa-cav complexes are distinctly shaped by their cav subunits. J. Neurosci. 28 (33), 8238–8245.
Raffaelli G., Saviane C., Mohajerani M.H., Pedarzani P., Cherubini E. 2004. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J. Physiol. 557 (1), 147–157.
Zhang J., Guan X., Li Q., Meredith A.L., Pan H.L., Yan J. 2018. Glutamate-activated BK channel complexes formed with NMDA receptors. Proc. Natl. Acad. Sci. USA. 115 (38), E9006–E9014.
Li Q., Yan J. 2016. Modulation of BK channel function by auxiliary beta and gamma subunits. Physiol. Behav. 128, 51–90.
Li B., Gao T.M. 2016. Functional role of mitochondrial and nuclear BK channels. Int. Rev. Neurobiol. 128, 163–191.
Springer S.J., Burkett B.J., Schrader L.A. 2015. Modulation of BK channels contributes to activity-dependent increase of excitability through MTORC1 activity in CA1 pyramidal cells of mouse hippocampus. Front. Cell. Neurosci. 8, 1–12.
Xia X.M., Fakler B., Rivard A., Wayman G., Johnson-Pais T., Keen J.E., Ishii T., Hirschberg B., Bond C.T., Lutsenko S., Maylie J., Adelman J.P. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 395 (6701), 503–507.
Kohler M., Hirschberg B., Bond C.T., Kinzie J.M., Marrion N. V., Maylie J., Adelman J.P. 1996. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 273 (5282), 1709–1714.
Hirschberg B., Maylie J., Adelman J.P., Marrion N. V. 1998. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 111 (4), 565–581.
Logsdon N.J., Kang J., Togo J.A., Christian E.P., Aiyar J. 1997. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 272 (52), 32723–32726.
Ishii T.M., Silvia C., Hirschberg B., Bond C.T., Adelman J.P., Maylie J. 1997. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA. 94 (21), 11651–11656.
Sforna L., Megaro A., Pessia M., Franciolini F., Catacuzzeno L. 2018. Structure, gating and basic functions of the Ca2+-activated K channel of intermediate conductance. Curr. Neuropharmacol. 16 (5), 608–617.
King B., Rizwan A.P., Asmara H., Heath N.C., Engbers J.D.T., Dykstra S., Bartoletti T.M., Hameed S., Zamponi G.W., Turner R.W. 2015. IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep. 11 (2), 175–182.
Faber E.S.L., Delaney A.J., Sah P. 2005. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat. Neurosci. 8 (5), 635–641.
Marrion N. V., Tavalin S.J. 1998. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 395 (6705), 900–905.
Ngo-Anh T.J., Bloodgood B.L., Lin M., Sabatini B.L., Maylie J., Adelman J.P. 2005. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8 (5), 642–649.
Hammond B., Lemen J., Dudek R., Ward D., Jiang C., Nemeth M., Burns J. 2006. Results of a 90-day safety assurance study with rats fed grain from corn rootworm-protected corn. Food Chem. Toxicol. 44 (2), 147–160.
Dryer S.E. 1994. Na+-activated K+ channels: A new family of large-conductance ion channels. Trends Neurosci. 17 (4), 155–160.
Kameyama M., Kakei M., Sato R., Shibasaki T., Matsuda H., Irisawa H. 1984. Intracellular Na+ activates a K+ channel in mammalian cardiac cells. Nature. 309 (5966), 354–356.
Budelli G., Hage T.A., Wei A., Rojas P., Ivy Jong Y.J., O’Malley K., Salkoff L. 2009. Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat. Neurosci. 12 (6), 745–750.
Lu S., Das P., Fadool D.A., Kaczmarek L.K. 2010. The slack sodium-activated potassium channel provides a major outward current in olfactory neurons of Kv1.3-/- super-smeller mice. J. Neurophysiol. 103 (6), 3311–3319.
Nuwer M.O., Picchione K.E., Bhattacharjee A. 2010. PKA-induced internalization of Slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J. Neurosci. 30 (42), 14165–14172.
Hage T.A., Salkoff L. 2012. Sodium-activated potassium channels are functionally coupled to persistent sodium currents. J. Neurosci. 32 (8), 2714–2721.
Kaczmarek L.K. 2013. Slack, slick, and sodium-activated potassium channels. ISRN Neurosci. 2013, 1–14.
Pfeiffer B.E., Huber K.M. 2009. The state of synapses in fragile X syndrome. Neurosci. 15 (5), 549–567.
Noma A. 1983. ATP-regulated K+ channels in cardiac muscle. Nature. 305 (5930), 147–148.
Seino S., Miki T. 2003. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 81 (2), 133–176.
Ashcroft F.M., Harrison D.E., Ashcroft S.J.H. 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 312 (5993), 446–448.
Haller M., Mironov S.L., Karschin A., Richter D.W. 2001. Dynamic activation of K ATP channels in rhythmically active neurons. J. Physiol. 537 (1), 69–81.
Zawar C., Plant T.D., Schirra C., Konnerth A., Neumcke B. 1999. Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. J. Physiol. 514 (2), 327–341.
Choeiri C., Staines W.A., Miki T., Seino S., Renaud J.M., Teutenberg K., Messier C. 2006. Cerebral glucose transporters expression and spatial learning in the K-ATP Kir6.2-/- knockout mice. Behav. Brain Res. 172 (2), 233–239.
Moriguchi S., Ishizuka T., Yabuki Y., Shioda N., Sasaki Y., Tagashira H., Yawo H., Yeh J.Z., Sakagami H., Narahashi T., Fukunaga K. 2018. Blockade of the K ATP channel Kir6.2 by memantine represents a novel mechanism relevant to Alzheimer’s disease therapy. Mol. Psychiatry. 23 (2), 211–221.
Brini M., Carafoli E. 2011. The Plasma Membrane Ca2+ ATPase and the plasma membrane Sodium Calcium exchanger cooperate in the regulation of cell Calcium. Cold Spring Harb. Perspect. Biol. 3 (2), 1–15.
Jaffe D.B., Johnston D., Lasser-Ross N., Lisman J.E., Miyakawa H., Ross W.N. 1992. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 357 (6375), 244–246.
Bennay M., Langer J., Meier S.D., Kafitz K.W., Rose C.R. 2008. Sodium signals in cerebellar Purkinje neurons and Bergmann glial cells evoked by glutamatergic synaptic transmission. Glia 56 (10), 1138–1149.
Verkhratsky A. 2005. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85 (1), 201–279.
Block B.A., Imagawa T., Campbell K.P., Franzini-Armstrong C. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107 (6), 2587–2600.
Samsó M., Feng W., Pessah I.N., Allen P.D. 2009. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7 (4), e1000085.
Gao L., Tripathy A., Lu X., Meissner G. 1997. Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 412 (1), 223–226.
Kunerth S., Langhorst M.F., Schwarzmann N., Gu X., Huang L., Yang Z., Zhang L., Mills S.J., Zhang L.H., Potter B.V.L., Guse A.H. 2004. Amplification and propagation of pacemaker Ca2+ signals by cyclic ADP-ribose and the type 3 ryanodine receptor in T cells. J. Cell Sci. 117 (10), 2141–2149.
Gerasimenko J. V., Flowerdew S.E., Voronina S.G., Sukhomlin T.K., Tepikin A. V., Petersen O.H., Gerasimenko O. V. 2006. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J. Biol. Chem. 281 (52), 40154–40163.
Sukhareva M., Smith S. V., Maric D., Barker J.L. 2002. Functional properties of ryanodine receptors in hippocampal neurons change during early differentiation in culture. J. Neurophysiol. 88 (3), 1077–1087.
Korkotian E., Segal M. 2011. Synaptopodin regulates release of calcium from stores in dendritic spines of cultured hippocampal neurons. J. Physiol. 589 (24), 5987–5995.
Raymond C.R., Redman S.J. 2002. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J. Neurophysiol. 88 (1), 249–255.
Raymond C.R., Redman S.J. 2006. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J. Physiol. 570 (1), 97–111.
Balschun D., Wolfer D.P., Bertocchini F., Barone V., Conti A., Zuschratter W., Missiaen L., Lipp H.P., Frey J.U., Sorrentino V. 1999. Deletion of the ryanodine receptor type 3 (RyR3) impairs forms of synaptic plasticity and spatial learning. EMBO J. 18 (19), 5264–5273.
Santulli G., Nakashima R., Yuan Q., Marks A.R. 2017. Intracellular calcium release channels: An update. J. Physiol. 595 (10), 3041–3051.
Bezprozvanny I., Ehrlich B.E. 1994. Inositol (1,4,5)-trisphosphate (InsP3)-gated Ca channels from cerebellum: Conduction properties for divalent cations and regulation by intraluminal calcium. J. Gen. Physiol. 104 (5), 821–856.
Boehning D., Mak D.O.D., Foskett J.K., Joseph S.K. 2001. Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J. Biol. Chem. 276 (17), 13509–13512.
Boehning D., Joseph S.K., Mak D.O.D., Foskett J.K. 2001. Single-channel recordings of recombinant inositol trisphosphate receptors in mammalian nuclear envelope. Biophys. J. 81 (1), 117–124.
Marchenko S.M., Yarotskyy V.V., Kovalenko T.N., Kostyuk P.G., Thomas R.C. 2005. Spontaneously active and InsP3-activated ion channels in cell nuclei from rat cerebellar Purkinje and granule neurones. J. Physiol. 565 (3), 897–910.
Fujii S., Matsumoto M., Igarashi K., Kato H., Mikoshiba K. 2000. Synaptic plasticity in hippocampal CA1 neurons of mice lacking type 1 inositol-1,4,5-trisphosphate receptors. Learn. Mem. 7 (5), 312–320.
Qiu Z., Nicoll D.A., Philipson K.D. 2001. Helix packing of functionally important regions of the cardiac Na+-Ca2+ exchanger. J. Biol. Chem. 276 (1), 194–199.
Nicoll D.A., Ottolia M., Lu L., Lu Y., Philipson K.D. 1999. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J. Biol. Chem. 274 (2), 910–917.
Hilge M., Aelen J., Vuister G.W. 2006. Ca2+ Regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 22 (1), 15–25.
Lytton J. 2007. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem. J. 406 (3), 365–382.
Quednau B.D., Nicoll D.A., Philipson K.D. 2004. The sodium/calcium exchanger family – SLC8. Pflügers Arch. Eur. J. Physiol. 447 (5), 543–548.
Quednau B.D., Nicoll D.A., Philipson K.D. 1997. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am. J. Physiol. Physiol. 272 (4), C1250–C1261.
DiPolo R., Beaugé L. 2006. Sodium/calcium exchanger: Influence of metabolic regulation on ion carrier interactions. Physiol. Rev. 86 (1), 155–203.
Blaustein M.P., Juhaszova M., Golovina V.A., Church P.J., Stanley E.F. 2006. Na/Ca exchanger and PMCA localization in neurons and astrocytes. Ann. N. Y. Acad. Sci. 976 (1), 356–366.
Jeon D., Yang Y.M., Jeong M.J., Philipson K.D., Rhim H., Shin H.S. 2003. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron 38 (6), 965–976.
Molinaro P., Viggiano D., Nisticò R., Sirabella R., Secondo A., Boscia F., Pannaccione A., Scorziello A., Mehdawy B., Sokolow S., Herchuelz A., di Renzo G.F., Annunziato L. 2011. Na+-Ca2+ exchanger (NCX3) knock-out mice display an impairment in hippocampal long-term potentiation and spatial learning and memory. J. Neurosci. 31 (20), 7312–7321.
Scheiner-Bobis G. 2002. The sodium pump. Eur. J. Biochem. 269 (10), 2424–2433.
Blanco G. 2005. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 25 (5), 292–303.
Hieber V., Siegel G.J., Fink D.J., Beaty M.W., Mata M. 1991. Differential distribution of (Na, K)-ATPase? isoforms in the central nervous system. Cell. Mol. Neurobiol. 11 (2), 253–262.
McGrail K., Phillips J., Sweadner K. 1991. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 11 (2), 381–391.
Peng L., Martin-Vasallo P., Sweadner K.J. 1997. Isoforms of Na,K-ATPase α and β subunits in the rat cerebellum and in granule cell cultures. J. Neurosci. 17 (10), 3488–3502.
Akkuratov E.E., Lopacheva O.M., Kruusmägi M., Lopachev A. V., Shah Z.A., Boldyrev A.A., Liu L. 2015. Functional interaction between Na/K-ATPase and NMDA receptor in cerebellar neurons. Mol. Neurobiol. 52 (3), 1726–1734.
Munzer J.S., Daly S.E., Jewell-Motz E.A., Lingrel J.B., Blostein R. 1994. Tissue- and isoform-specific kinetic behavior of the Na,K-ATPase. J. Biol. Chem. 269 (24), 16668–16676.
Jewell E.A., Lingrel J.B. 1991. Comparison of the substrate dependence properties of the rat Na,K-ATPase α1, α2, and α3 isoforms expressed in HeLa cells. J. Biol. Chem. 266 (25), 16925–16930.
Zahler R., Zhang Z.-T., Manor M., Boron W.F. 1997. Sodium kinetics of Na,K-ATPase α isoforms in intact transfected HeLa cells. J. Gen. Physiol. 110 (2), 201–213.
Dobretsov M. 2005. Neuronal function and alpha3 isoform of the Na/K-ATPase. Front. Biosci. 10, 2373–2396.
Azarias G., Kruusmägi M., Connor S., Akkuratov E.E., Liu X.-L., Lyons D., Brismar H., Broberger C., Aperia A. 2013. A Specific and essential role for Na,K-ATPase α3 in neurons co-expressing α1 and α3. J. Biol. Chem. 288 (4), 2734–2743.
Kim J.H., Sizov I., Dobretsov M., Von Gersdorff H. 2007. Presynaptic Ca2+ buffers control the strength of a fast post-tetanic hyperpolarization mediated by the α3 Na+/K+-ATPase. Nat. Neurosci. 10 (2), 196–205.
Glushchenko T.S., Izvarina N.L. 1997. Na+,K+-ATPase activity in neurons and glial cells of the olfactory cortex of the rat brain during the development of long-term potentiation. Neurosci. Behav. Physiol. 27 (1), 49–52.
Reich C.G., Mason S.E., Alger B.E. 2004. Novel form of LTD induced by transient, partial inhibition of the Na, K-pump in rat hippocampal CA1 cells. J. Neurophysiol. 91 (1), 239–247.
Niggli V., Sigel E., Carafoli E. 1982. The purified Ca2+ pump of human erythrocyte membranes catalyzes an electroneutral Ca2+-H+ exchange in reconstituted liposomal systems. J. Biol. Chem. 257 (5), 2350–2356.
Smallwood J.I., Waisman D.M., Lafreniere D., Rasmussen H. 1983. Evidence that the erythrocyte calcium pump catalyzes a Ca2+:nH+ exchange. J. Biol. Chem. 258 (18), 11092–11097.
Di Leva F., Domi T., Fedrizzi L., Lim D., Carafoli E. 2008. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 476 (1), 65–74.
Greeb J., Shull G.E. 1989. Molecular cloning of a third isoform of the calmodulin-sensitive plasma membrane Ca2+-transporting ATPase that is expressed predominantly in brain and skeletal muscle. J. Biol. Chem. 264 (31), 18569–18576.
Stauffer T.P., Guerini D., Carafoli E. 1995. Tissue distribution of the four gene products of the plasma membrane Ca pump. J. Biol. Chem. 270 (20), 12 184–12 190.
Mata A.M. 2010. Plasma membrane Ca2+-ATPases in the nervous system during development and ageing. World J. Biol. Chem. 1 (7), 229.
Burette A.C., Strehler E.E., Weinberg R.J. 2009. ‘Fast’ plasma membrane calcium pump PMCA2a concentrates in GABAergic terminals in the adult rat brain. J. Comp. Neurol. 512 (4), 500–513.
Strehler E.E., Filoteo A.G., Penniston J.T., Caride A.J. 2007. Plasma-membrane Ca2+ pumps: Structural diversity as the basis for functional versatility. Biochem. Soc. Trans. 35 (5), 919–922.
Marques-da-Silva D., Gutierrez-Merino C. 2014. Caveolin-rich lipid rafts of the plasma membrane of mature cerebellar granule neurons are microcompartments for calcium/reactive oxygen and nitrogen species cross-talk signaling. Cell Calcium. 56 (2), 108–123.
Garside M.L., Turner P.R., Austen B., Strehler E.E., Beesley P.W., Empson R.M. 2009. Molecular interactions of the plasma membrane calcium ATPase 2 at pre- and post-synaptic sites in rat cerebellum. Neuroscience. 162 (2), 383–395.
Zanni G., Calì T., Kalscheuer V.M., Ottolini D., Barresi S., Lebrun N., Montecchi-Palazzi L., Hu H., Chelly J., Bertini E., Brini M., Carafoli E. 2012. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA. 109 (36), 14514–14519.
Kip S.N., Gray N.W., Burette A., Canbay A., Weinberg R.J., Strehler E.E. 2006. Changes in the expression of plasma membrane calcium extrusion systems during the maturation of hippocampal neurons. Hippocampus. 16 (1), 20–34.
Misquitta C.M., Mack D.P., Grover A.K. 1999. Sarco/endoplasmic reticulum Ca2+(SERCA)-pumps: Link to heart beats and calcium waves. Cell Calcium. 25 (4), 277–290.
Verboomen H., Wuytack F., Van den Bosch L., Mertens L., Casteels R. 1994. The functional importance of the extreme C-terminal tail in the gene 2 organellar Ca2+-transport ATPase (SERCA2a/b). Biochem. J. 303 (3), 979–984.
Majewska A., Brown E., Ross J., Yuste R. 2000. Mechanisms of calcium decay kinetics in hippocampal spines: Role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J. Neurosci. 20 (5), 1722–1734.
Emptage N.J., Reid C.A., Fine A. 2001. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 29 (1), 197–208.
Collingridge B.Y.G.L., Kehl S.J., Mclennan H. 1983. Excitatory amino acids in synaptic transmission in the schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 334, 33–46.
Bliss T. V., Collingridge G.L. 1993. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 361 (6407), 31–39.
Aniksztejn L., Ben-Ari Y. 1995. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. J. Neurophysiol. 74 (6), 2349–2357.
Berridge M.J. 1998. Neuronal calcium signaling. Neuron. 21 (1), 13–26.
Malenka R.C., Lancaster B., Zucker R.S. 1992. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron. 9 (1), 121–128.
Santana L.F. 2008. Editorial: NFAT-dependent excitation-transcription coupling in heart. Circ. Res. 103 (7), 681–683.
Gundersen K. 2011. Excitation-transcription coupling in skeletal muscle: The molecular pathways of exercise. Biol. Rev. 86 (3), 564–600.
Morgan J.I., Curran T. 1989. Stimulus-transcription coupling in neurons: Role of cellular immediate-early genes. Trends Neurosci. 12 (11), 459–462.
Sheng M., Greenberg M.E. 1990. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 4 (4), 477–485.
Cole A.J., Saffen D.W., Baraban J.M., Worley P.F. 1989. Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature. 340 (6233), 474–476.
Fleischmann A., Hvalby O., Jensen V., Strekalova T., Zacher C., Layer L.E., Kvello A., Reschke M., Spanagel R., Sprengel R., Wagner E.F., Gass P. 2003. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-fos in the CNS. J. Neurosci. 23 (27), 9116–9122.
Minatohara K., Akiyoshi M., Okuno H. 2016. Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front. Mol. Neurosci. 8, 1–11.
Greenberg M.E., Ziff E.B., Greene L.A. 1986. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science. 234 (4772), 80–83.
Orlov S.N., Aksentsev S.L., Kotelevtsev S. V. 2005. Extracellular calcium is required for the maintenance of plasma membrane integrity in nucleated cells. Cell Calcium. 38 (1), 53–57.
Koltsova S. V., Tremblay J., Hamet P., Orlov S.N. 2015. Transcriptomic changes in Ca2+-depleted cells: Role of elevated intracellular [Na+]/[K+] ratio. Cell Calcium. 58 (3), 317–324.
Deisseroth K., Heist E.K., Tsien R.W. 1998. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 392 (6672), 198–202.
Ma H., Groth R.D., Cohen S.M., Emery J.F., Li B., Hoedt E., Zhang G., Neubert T.A., Tsien R.W. 2014. γcaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 159 (2), 281–294.
Malenka R.C., Kauer J.A., Perkel D.J., Mauk M.D., Kelly P.T., Nicoll R.A., Waxham M.N. 1989. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 340 (6234), 554–557.
Mcdonald T.F., Pelzer S., Trautwein W., Pelzer D.J. 1994. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 74 (2), 365–507.
Greer P.L., Greenberg M.E. 2008. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron. 59 (6), 846–860.
Clapham D.E. 2007. Calcium signaling. Cell. 131 (6), 1047–1058.
Derkach V., Barria A., Soderling T.R. 1999. Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA. 96 (6), 3269–3274.
Kristensen A.S., Jenkins M. a., Banke T.G., Schousboe A., Johnson R.C., Huganir R., Traynelis S.F. 2011. Mechanism of CaMKII regulation of AMPA receptor gating. Nat. Neurosci. 14 (6), 727–735.
Mitsushima D., Ishihara K., Sano A., Kessels H.W., Takahashi T. 2011. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc. Natl. Acad. Sci. USA. 108 (30), 12503–12508.
Banke T.G., Bowie D., Lee H., Huganir R.L., Schousboe A., Traynelis S.F. 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20 (1), 89–102.
Chetkovich D.M., Gray R., Johnston D., Sweatt J.D. 1991. N-Methyl-D-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc. Natl. Acad. Sci. USA. 88 (15), 6467–6471.
Lau C.G., Takeuchi K., Rodenas-Ruano A., Takayasu Y., Murphy J., Bennett M.V.I., Zukin R.S. 2009. Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem. Soc. Trans. 37 (6), 1369–1374.
Aman T.K., Maki B.A., Ruffino T.J., Kasperek E.M., Popescu G.K. 2014. Separate intramolecular targets for protein kinase A control N-Methyl-D-aspartate receptor gating and Ca2+ permeability. J. Biol. Chem. 289 (27), 18805–18817.
Matthews R.P., Guthrie C.R., Wailes L.M., Zhao X., Means A.R., McKnight G.S. 1994. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol. Cell. Biol. 14 (9), 6107–6116.
Reymann K.G., Frey U., Jork R., Matthies H. 1988. Polymyxin B, an inhibitor of protein kinase C, prevents the maintenance of synaptic long-term potentiation in hippocampal CA1 neurons. Brain Res. 440 (2), 305–314.
Kauer J.A., Malenka R.C., Nicoll R.A. 1988. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1 (10), 911–917.
Lovinger D.M., Wong K.L., Murakami K., Routtenberg A. 1987. Protein kinase C inhibitors eliminate hippocampal long-term potentiation. Brain Res. 436 (1), 177–183.
Finkbeiner S., Greenberg M.E. 1996. Ca2+-dependent routes to Ras: Mechanisms for neuronal survival, differentiation, and plasticity? Neuron. 16 (2), 233–236.
Herring B.E., Nicoll R.A. 2016. Long-term potentiation: From CaMKII to AMPA receptor trafficking. Annu. Rev. Physiol. 78 (1), 351–365.
Murakoshi H., Wang H., Yasuda R. 2011. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 472 (7341), 100–104.
Lynch G., Rex C.S., Gall C.M. 2007. LTP consolidation: Substrates, explanatory power, and functional significance. Neuropharmacology. 52 (1), 12–23.
Amini M., Ma C. -l., Farazifard R., Zhu G., Zhang Y., Vanderluit J., Zoltewicz J.S., Hage F., Savitt J.M., Lagace D.C., Slack R.S., Beique J.-C., Baudry M., Greer P.A., Bergeron R., Park D.S. 2013. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J. Neurosci. 33 (13), 5773–5784.
Baeza-Lehnert F., Saab A.S., Gutiérrez R., Larenas V., Díaz E., Horn M., Vargas M., Hösli L., Stobart J., Hirrlinger J., Weber B., Barros L.F. 2019. Non-canonical control of neuronal energy status by the Na+ pump. Cell Metab. 29 (3), 668–680.e4.
Kiedrowski L., Wroblewski J.T., Costa E. 1994. Intracellular sodium concentration in cultured cerebellar granule cells challenged with glutamate. Mol. Pharmacol. 45 (5), 1050–1054.
Rose C.R., Konnerth A. 2001. NMDA receptor-mediated Na+ signals in spines and dendrites. J. Neurosci. 21 (12), 4207–4214.
Somjen G.G. 2002. Ion regulation in the brain: Implications for pathophysiology. Neuroscientist. 8 (3), 254–267.
Deitmer J.W., Rose C.R. 2010. Ion changes and signalling in perisynaptic glia. Brain Res. Rev. 63 (1–2), 113–129.
Verkhratsky A., Noda M., Parpura V., Kirischuk S. 2013. Sodium fluxes and astroglial function. Adv. Exp. Med. Biol. 961, 295–305.
Callaway J.C., Ross W.N. 1997. Spatial distribution of synaptically activated sodium concentration changes in cerebellar Purkinje neurons. J. Neurophysiol. 77 (1), 145–152.
Miyazaki K., Ross W.N. 2017. Sodium dynamics in pyramidal neuron dendritic spines: synaptically evoked entry predominantly through AMPA receptors and removal by diffusion. J. Neurosci. 37 (41), 9964–9976.
Behnisch T., Reymann K.G. 1995. Thapsigargin blocks long-term potentiation induced by weak, but not strong tetanisation in rat hippocampal CA1 neurons. Neurosci. Lett. 192 (3), 185–188.
Balschun D., Wolfer D.P., Gass P., Mantamadiotis T., Welzl H., Schütz G., Frey J.U., Lipp H.P. 2003. Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J. Neurosci. 23 (15), 6304–6314.
Koltsova S. V., Trushina Y., Haloui M., Akimova O.A., Tremblay J., Hamet P., Orlov S.N. 2012. Ubiquitous [Na+]i/[K+]i-sensitive transcriptome in mammalian cells: Evidence for Ca2+i-independent excitation-transcription coupling. PLoS One 7 (5), e38032.
Kasahara J., Fukunaga K., Miyamoto E. 2001. Activation of calcium/calmodulin-dependent protein kinase IV in long term potentiation in the rat hippocampal CA1 region. J. Biol. Chem. 276 (26), 24044–24050.
Taurin S., Dulin N.O., Pchejetski D., Grygorczyk R., Tremblay J., Hamet P., Orlov S.N. 2002. c-Fos expression in ouabain-treated vascular smooth muscle cells from rat aorta: Evidence for an intracellular-sodium-mediated, calcium-independent mechanism. J. Physiol. 543 (3), 835–847.
Haloui M., Taurin S., Akimova O.A., Guo D.F., Tremblay J., Dulin N.O., Hamet P., Orlov S.N. 2007. [Na+]i-induced c-Fos expression is not mediated by activation of the 5′-promoter containing known transcriptional elements. FEBS J. 274 (14), 3557–3567.
Orlov S.N., Taurin S., Tremblay J., Hamet P. 2001. Inhibition of Na+,K+ pump affects nucleic acid synthesis and smooth muscle cell proliferation via elevation of the [Na+]i/ [K+]i ratio: Possible implication in vascular remodelling. J. Hypertens. 19 (9), 1559–1565.
Smolyaninova L.V., Shiyan A.A., Kapilevich L.V., Lopachev A.V., Fedorova T.N., Klementieva T.S., Moskovtsev A.A., Kubatiev A.A., Orlov S.N. 2019. Transcriptomic changes triggered by ouabain in rat cerebellum granule cells: Role of α3- And α1-Na+,K+-ATPase-mediated signaling. PLoS One 14 (9), 1–23.
Yang H., Chen C. 2008. Cyclooxygenase-2 in synaptic signaling. Curr. Pharm. Des. 14 (14), 1443–1451.
Yamagata K., Andreasson K.I., Kaufmann W.E., Barnes C.A., Worley P.F. 1993. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron. 11 (2), 371–386.
Chen C., Magee J.C., Bazan N.G. 2002. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J. Neurophysiol. 87 (6), 2851–2857.
Sang N., Chen C. 2006. Lipid signaling and synaptic plasticity. Neuroscientist. 12 (5), 425–434.
Yagami T., Koma H., Yamamoto Y. 2016. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol. Neurobiol. 53 (7), 4754–4771.
Koelle M.R. 1997. A new family of G-protein regulators – the RGS proteins. Curr. Opin. Cell Biol. 9 (2), 143–147.
Ingi T., Krumins A.M., Chidiac P., Brothers G.M., Chung S., Snow B.E., Barnes C.A., Lanahan A.A., Siderovski D.P., Ross E.M., Gilman A.G., Worley P.F. 1998. Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J. Neurosci. 18 (18), 7178–7188.
Leslie J.H., Nedivi E. 2011. Activity-regulated genes as mediators of neural circuit plasticity. Prog. Neurobiol. 94 (3), 223–237.
Rudy J.W. 2014. The neurobiology of learning and memory. Sunderland, MA: Sinauer Associates, Inc. Publishers, p. 48–55.
Xu X., Shrager P. 2005. Dependence of axon initial segment formation on Na+ channel expression. J. Neurosci. Res. 79 (4), 428–441.
Oda T., Makino K., Yamashita I., Namba K., Maéda Y. 2001. Distinct structural changes detected by X-ray fiber diffraction in stabilization of F-actin by lowering pH and increasing ionic strength. Biophys. J. 80 (2), 841–851.
Rajamanickam G.D., Kroetsch T., Kastelic J.P., Thundathil J.C. 2017. Testis-specific isoform of Na/K-ATPase (ATP1A4) regulates sperm function and fertility in dairy bulls through potential mechanisms involving reactive oxygen species, calcium and actin polymerization. Andrology. 5 (4), 814–823.
Rajasekaran A.K., Rajasekaran S.A. 2003. Role of Na-K-ATPase in the assembly of tight junctions. Am. J. Physiol. – Ren. Physiol. 285, 388–396.
La J., Reed E.B., Koltsova S., Akimova O., Hamanaka R.B., Mutlu G.M., Orlov S.N., Dulin N.O. 2016. Regulation of myofibroblast differentiation by cardiac glycosides. Am. J. Physiol. – Lung Cell. Mol. Physiol. 310 (9), L815–L823.
Alder J., Thakker-Varia S., Bangasser D.A., Kuroiwa M., Plummer M.R., Shors T.J., Black I.B. 2003. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J. Neurosci. 23 (34), 10 800–10 808.
Altar C.A., Laeng P., Jurata L.W., Brockman J.A., Lemire A., Bullard J., Bukhman Y. V., Young T.A., Charles V., Palfreyman M.G. 2004. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J. Neurosci. 24 (11), 2667–2677.
Sidorenko S., Klimanova E., Milovanova K., Lopina O.D., Kapilevich L.V., Chibalin A.V., Orlov S.N. 2018. Transcriptomic changes in C2C12 myotubes triggered by electrical stimulation: Role of ${\text{Ca}}_{{\text{i}}}^{{2 + }}$ -mediated and ${\text{Ca}}_{{\text{i}}}^{{2 + }}$ -independent signaling and elevated [Na+]i /[K+]i ratio. Cell Calcium. 76, 72–86.
Ng A.N., Toresson H. 2011. Endoplasmic reticulum dynamics in hippocampal dendritic spines induced by agonists of type I metabotropic glutamate but not by muscarinic acetylcholine receptors. Synapse. 65 (4), 351–355.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии