

Биологические мембраны: Журнал мембранной и клеточной биологии, 2020, T. 37, № 6, стр. 442-452
Активация тромбоцитов через рецептор GPVI: вариабельность ответа
М. Г. Степанян a, b, А. А. Филькова a, b, c, А. К. Гарсон Дасгупта b, c, А. А. Мартьянов a, b, c, d, А. Н. Свешникова a, b, c, *
a Центр теоретических проблем физико-химической фармакологии РАН
109029 Москва, Россия
b Московский государственный университет им. М.В. Ломоносова,
физический факультет
119991 Москва, Россия
c Национальный медицинский исследовательский центр детской гематологии,
онкологии и иммунологии им. Д. Рогачева
117997 Москва, Россия
d Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, Россия
* E-mail: a.sveshnikova@physics.msu.ru
Поступила в редакцию 24.04.2020
После доработки 21.05.2020
Принята к публикации 22.05.2020
Аннотация
Тромбоциты – безъядерные клетки, ответственные за предотвращение кровопотери в случае нарушения целостности сосудистых стенок. При контакте с обнажившимся при повреждении сосуда коллагеном тромбоциты активируются через рецептор-гликопротеин, GPVI, что приводит к изменению формы, дегрануляции, проагрегантному и прокоагулянтному ответу. Целью данной работы было наблюдение изменения концентрации Ca2+ в цитозоле и функциональных ответов тромбоцитов при стимуляции через рецептор GPVI. В исследовании приняли участие здоровые взрослые добровольцы. Также были использованы домовые мыши Mus musculus линии C57Bl6 (дикий тип). Внутриклеточная концентрация Ca2+ оценивалась по изменению флуоресценции Fura-Red на проточном цитофлуориметре. Активация интегринов наблюдалась на проточном цитофлуориметре по связыванию фибриногена человека, коньюгированного с флуоресцентной меткой, или на оптическом агрегометре. Тромбоциты активировались при помощи пептида, сходного по аминокислотной последовательности с коллагеном (collagen-related peptide, CRP) при работе на цитометре и с помощью фибриллярного коллагена при работе на агрегометре. У тромбоцитов человека была обнаружена существенная междонорская вариабельность в относительном изменении уровня Ca2+ в цитозоле в ответ на стимуляцию; значительно меньшей была междонорская вариабельность в активации тромбоцитарных интегринов, изменении формы и в агрегационном ответе. У мышей такой вариабельности не наблюдалось. Наблюдаемая вариабельность кальциевого ответа тромбоцитов могла быть вызвана различиями в экспрессии GPVI у здоровых доноров и полиморфизмами в гене рецептора GPVI у изученных доноров. Обнаруженное сходство в функциональных ответах тромбоцитов, при различных сигнальных ответах, позволяет предположить вариабельный вклад фосфоинозитидной ветви внутриклеточной сигнализации в ответ на активацию GPVI у тромбоцитов.
ВВЕДЕНИЕ
Внутренней оболочкой кровеносного сосуда является интима. Она состоит из эндотелия и межклеточного матрикса, представляющего собой коллагеновые и ретикулиновые волокна, а также фибробласты и продольно ориентированные гладкомышечные клетки [1]. При повреждении интимы происходит обнажение фибриллярных коллагенов: фибронектина и ламинина [2]. Обнажившийся коллаген связывается с фактором Виллебранда (vWF) – мультимерным белком плазмы крови, разворачивающимся при связывании с коллагеном под действием гемодинамических сил сопротивления [3]. В развернутой конформации vWF привлекает тромбоциты крови через их механорецептор GPIb [4]. Тромбоциты – безъядерные клетки, входящие в состав крови, главной задачей которых является образование гемостатических пробок в области нарушения целостности кровеносных сосудов [5]. Переходя в активированное состояние, тромбоциты изменяют форму и выпускают наружу содержимое своих гранул, что привлекает другие тромбоциты и приводит к росту тромба [6]. Адгезия тромбоцитов к коллагену за счет связи GPIb и vWF не является стабильной, поэтому после привлечения тромбоцита к коллагену через vWF-GPIb, необходима его дальнейшая активация [7]. Коллаген запускает активацию тромбоцита через гликопротеин-VI (GPVI) и интегрин α2β1 [8]. В то время как последний является адгезионным, именно GPVI отвечает за генерацию внутриклеточных сигналов, запускающих активацию и функциональные ответы тромбоцитов – изменение формы, дегрануляцию, проагрегантный (активацию тромбоцитарных интегринов αIIbβ3) и прокоагулянтный ответы [8, 9]. Активированные интегрины αIIbβ3 связываются с плазменным белком фибриногеном, который служит своеобразным “мостиком” для связывания двух тромбоцитов, что приводит к формированию тромбоцитарных агрегатов [10].
Рецептор GPVI экспрессируется совместно с FcRγ-цепью, их комплекс формируется посредством нековалентного трансмембранного контакта [11]. GPVI присутствует только на поверхности тромбоцитов и производящих их мегакариоцитов [11]. GPVI мыши имеет идентичность 67.3 и 64.4% с GPVI человека на уровне нуклеотидной и аминокислотной последовательностей белка соответственно [12]. Внеклеточные и внутриклеточные домены рецептора GPVI человека и мыши не идентичны. Например, большинство блокирующих антител, направленных против GPVI человека, не связываются с GPVI мыши, что осложняет доклинические испытания анти-GPVI агентов [13]. Уровень гомологии между GPVI человека и мыши несколько выше в Ig-доменах (78%), содержащих сайты связывания для активаторов (коллагена, коллаген-подобного пептида (CRP) и конвульксина) [14]. Недавно было продемонстрировано, что фибрин также является активатором тромбоцитарного GPVI рецептора человека и мыши [15]. Наконец, иммобилизованный фибриноген способен активировать димеризованные рецепторы GPVI [16]. Данные исследования были проведены in vivo на человеческих тромбоцитах, а также на тромбоцитах мышей, в которых была экспрессирована человеческая форма рецептора GPVI [17].
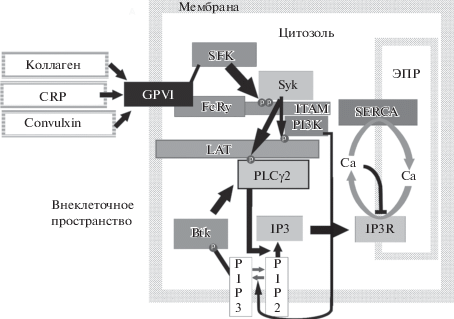
Каждая цепь FcRγ содержит мотив активации иммунорецептора на основе тирозина (ITAM), состоящий из двух последовательностей YxxL, разделенных 6–12 аминокислотами [18]. При активации GPVI связанные с ним тирозинкиназы семейтсва Src (SFK) фосфорилируют ITAM [19]. Это приводит к связыванию с ITAM и активации тирозин-киназ Syk. Syk-зависимый сигнальный каскад ведет к формированию LAT-сигналосомы, в комплексе которой активируется фосфоинозитид-3 киназа (PI3K), фосфорилирующая фосфоинозитол-4,5-бисфосфат (PIP2) до фосфоинозитол-3,4,5-трифосфата (PIP3). К PIP3 присоединяется тирозинкиназа Брутона (Btk), которая запускает активацию фосфолипазы Cγ2 (PLCγ2). Она гидролизует фосфоинозитол-4,5-бисфосфат (PIP2), в результате чего образуется инозитол-1,4,5-трифосфат (IP3). IP3 активирует находящийся на ЭПР IP3-рецептор, что приводит к выходу Ca2+ в цитозоль [20]. Схема сигнализации, опосредованной рецептором GPVI, представлена на рис. 1.
Рис. 1.
Сигнальный каскад при активации тромбоцита через рецептор GPVI. Связывание коллагена/CRP/конвульксина и GPVI запускает активацию SFK, фосфорилирующих ITAM [21]. Это приводит к связыванию киназы Syk, которая фосфорилирует адаптерный белок LAT. К фосфорилированным LAT присоединяются PLCγ2 и PI3K. Ассоциированные с LAT PI3K активируются и фосфорилируют PIP2, производя PIP3. Тирозинкиназы Брутона (Btk) связываются своим плекстрин-гомологичным доменом с PIP3, активируются и запускают активацию соединенных с LAT фосфолипаз PLCγ2. Активные PLCγ2 гидролизуют комплекс PIP2, производя IP3. IP3 активирует рецептор к инозитолтрифосфату (IP3R), находящийся на мембране ЭПР. Происходит выход кальция в цитозоль клетки, корректируемый также самим IP3R и сарко/эндоплазматическоретикулярной Ca2+ ATP-азой SERCA.
Мутации в гене рецептора GPVI (GP6) вызывают возникновение петехий, меноррагий и носовых кровотечений легкой степени тяжести [22–24]. Однако при этом у пациентов с мутациями в гене GP6 не наблюдалось тромбоцитопений [25]. Функциональное тестирование тромбоцитов показало ослабленную агрегацию богатой тромбоцитами плазмы пациентов с мутацией в гене GP6 в ответ на CRP и конвульксин (индукторы активации тромбоцитарного GPVI), но при этом нормальную агрегацию в ответ на ADP (активатор P2Y1 и P2Y12 рецепторов), арахидоновую кислоту, U46617 (активаторы TP рецептора), TRAP1 (активатор рецептора PAR1), ристоцетин (агент, вызывающий агглютинацию тромбоцитов через GPIbα-vWF) [26]. Также было показано, что мутация в гене рецептора GPVI приводит к нарушению свертываемости крови и формированию тромбов ex vivo как у людей, так и у мышей [17, 25–28].
В настоящее время рецептор GPVI рассматривается в качестве мишени для антитромботической и противовоспалительной терапии [29]. Показано, что введение растворимого GPVI (sGPVI) здоровым донорам не вызывает кровоточивости, однако ингибирует агрегацию тромбоцитов в ответ на коллаген in vitro [30]. Также рассматривались ингибиторы сигнального пути GPVI, Syk-киназы (пицеатаннол, малеат, десантиниб), однако отсутствие специфичности в связи с большим количеством различных биологических ролей у целевых киназ является основным недостатком данного подхода [31]. Использование антител к GPVI также было эффективным для терапии тромбозов [32–34].
Изучение сигнальных путей и описание доноров по их индивидуальным показателям открывают новые возможности для разработки более безопасных антитромботических лекарств, основанных на ингибировании рецептора GPVI. Цель данной работы – описание кальциевой сигнализации и активации тромбоцитарных интегринов при активации тромбоцитарного рецептора GPVI в человеческих и мышиных тромбоцитах.
МАТЕРИАЛЫ И МЕТОДЫ
Объекты исследования. В исследование были включены 14 здоровых добровольцев, как мужчин, так и женщин в возрасте от 18 до 58 лет, отрицавших наличие сердечно-сосудистых заболеваний в семейной истории и давших письменное информированное согласие. Кровь здоровых доноров забирали из локтевой вены в вакуумные пробирки, содержащие гирудин (SARSTEDT Monovette, для использования в проточной цитометрии) и цитрат натрия (Improvacuter, для турбидиметрической агрегометрии). Также были использованы 12 домовых мышей (Mus musculus) линии C57Bl6 возрастом от 4 до 5 месяцев, весом 25–35 г, обоих полов. Кровь мышей была взята на ACD (кислотно-цитратную декстрозу). Эксперименты проводились в соответствии с Хельсинкской декларацией и директивой Европейского парламента 63/2010/EU и были одобрены этическим комитетом ЦТП ФХФ РАН.
Реагенты. Были использованы следующие материалы: флуоресцентный зонд Fura-Red-AM (Molecular Probes, Eugene, Орегон, США); FITC, фибриноген человека, EGTA, HEPES, бычий сывороточный альбумин, апираза (степень очистки VII), D(+)глюкоза (Sigma, США); NaCl; Na2HPO4; KCl; NaHCO3; MgCl2; CaCl2; C6H5Na3O7 · 2(H2O); C6H8O7 · H2O (Агат-Мед, Москва, Россия), коллаген (Технология-Стандарт, Россия). Цистеинсодержащая версия сшитого подобного коллагену пептида (CRP) была любезно предоставлена профессором R.W. Farndale (Университет Кембриджа, Кембридж, Великобритания) [35].
Протокол выделения клеток для проточной цитометрии. Цельная кровь, антикоагулированная гирудином, была инкубирована с кальций-чувствительным зондом Fura-Red-AM в концентрации 2 мкМ в присутствии 0.2 ед/мл апиразы в течение 35 мин при 37°С. Апираза была добавлена для снижения негативного влияния секретируемого осажденными эритроцитами ADP на активацию тромбоцитов. По истечении 35 мин кровь была разведена буфером Тирода (134 мМ NaCl; 0.34 мМ Na2HPO4; 2.9 мМ KCl; 12 мМ NaHCO3; 20 мМ HEPES; 5 мМ глюкозы; 1 мМ MgCl2; 2 мМ CaCl2; BSA 2% по массе; pH 7.3) до концентрации тромбоцитов 1 × 103 в 1 мкл.
Кровь мышей была взята из брюшной артерии в шприц, содержащий антикоагулянт ACD (85 мМ дигидрата тринайтрийцитрата, 66.6 мМ моногидрата лимонной кислоты и 111 мМ D(+)глюкозы в растворе дистиллированной воды (осмолярность 450 мОсм/л, pH 4.5) в соотношении объемов 1 : 6 (антикоагулянт к крови). Тромбоциты были выделены согласно протоколу [36]. Клетки были окрашены Fura-Red в концентрации 2 мкМ в присутствии 0.2 ед/мл апиразы в течение 35 мин при 37°С.
Проточная цитометрия. Методика измерения концентрации кальция в цитозоле тромбоцита и активации интегринов аналогична описанной ранее [37–41]. В исследовании был использован проточный цитометр BD FACS Canto II. В начальный момент времени к пробам добавляли флуоресцентно меченный фибриноген в концентрации 100 мкг/мл. Через 60 с от начала исследования к пробам добавляли агонист рецептора GPVI CRP (0.25, 1 и 5 мкг/мл). При обработке результатов по значениям прямого и бокового светорассеяния клетками выбиралась область, соответствующая тромбоцитам. Затем оценивалась флуоресценция Fura-Red в свободном состоянии (возбуждение 488 нм, излучение 640 ± 30 нм) и в связанном с ионами кальция состоянии (возбуждение 405 нм, излучение 670 ± 30 нм). Усреднение проводилось по 1000 клеток. В качестве меры концентрации кальция в цитозоле тромбоцита рассматривалось отношение средней интенсивности флуоресценции (mean fluorescence intensity, MFI), R = (MFI405/MFI488). [Ca2+] измерялась в относительных единицах R/R0, где R0 – наблюдаемое для данного образца значение R в покое (до активации). Связывание фибриногена (возбуждение 488 нм, излучение 520 ± 20 нм) считалось максимальным после инкубации пробы с 1 мкМ иономицина в течение 10 мин и использовалось для расчета степени связывания фибриногена при активации CRP. Для оценки изменения формы клеток использовалось боковое светорассеяние (SSC, параметр проточной цитометрии, растущий при увеличении неоднородности поверхности клетки), где за 0% принималось усредненное значение бокового светорассеяния за первые 60 с измерений (без активации), а за 100% – отсутствие детектированных лучей.
Турбидиметрическая агрегометрия. Для исследования агрегации тромбоцитов методом турбидиметрической агрегометрии кровь доноров забирали из локтевой вены в пробирки, содержащие цитрат натрия (финальная концентрация Na3C6H5O7 в полученной крови 0.129 моль/л). Затем кровь центрифугировали (8 мин, 150 g) и отбирали богатую тромбоцитами плазму (platelet rich plasma, PRP). Оставшуюся часть крови центрифугировали 15 мин с ускорением 2000 g для получения бедной тромбоцитами плазмы с целью калибровки прибора. К богатой тромбоцитами плазме добавляли гирудин в концентрации 10 ед/мл и CaCl2 до концентрации 20 мМ (расчетная концентрация свободных ионов кальция в плазме составляет 2.5 мМ), и 3 мин инкубировали при температуре 37°С, затем проводили измерение на агрегометре Biola LA-230 при скорости перемешивания 800 оборотов в минуту. Активация проводилась коллагеном (1 и 2.5 мкг/мл).
Анализ данных. Обработка данных проточной цитометрии и агрегометрии проводилась посредством Python 3.7. Для оценки значимости различий был использован U-критерий Манна–Уитни, статистически значимым отличием считалось значение p < 0.05.
РЕЗУЛЬТАТЫ
Ответы тромбоцитов на активацию GPVI в проточной цитофлуориметрии. При исследовании внутриклеточной сигнализации в тромбоцитах здоровых доноров и мышей в качестве маркера сигнальной активности использовалось относительное изменение концентрации свободных ионов кальция в цитозоле в ответ на активацию (рис. 2а, 2г, 2ж). Функциональные характеристики тромбоцитов оценивались по связыванию фибриногена (проагрегантный тромбоцитарный ответ, рис. 2б, 2д, 2з) и изменению формы (первичный ответ тромбоцитов на активацию, рис. 2в, 2е, 2и). По сигнальным и функциональным ответам тромбоцитов в ходе исследования были выделены две отдельные группы (рис. 2а–2в и рис. 2г–2е соответственно). Доноры, максимальное изменение внутриклеточного кальция которых в ответ на активацию 5 мкг/мл CRP превышало 1.4 отн. ед. (рис. 2а, 2г), были отнесены к типу 1 (рис. 2а–2в), в обратном случае к типу 2 (рис. 2г–2е). Мышиные тромбоциты были выделены в отдельную группу (рис. 2ж–2и). Выбор границы был сделан на основании визуального разделения доноров на две группы при наблюдении кривых зависимости относительной концентрации внутриклеточного Ca2+ от времени.
Рис. 2.
Типичные кривые для трех видов ответов, полученные в ходе экспериментов. Стрелка указывает на время добавления активатора. Концентрация CRP в образце: 0.25 (1), 1 (2) и 5 мкг/мл (3). Относительное изменение концентрации кальция в цитозоле, R/R0 (а), связывание фибриногена (б) и изменение формы (в) тромбоцитов в течение 140 с после добавления CRP для получения трех различных концентраций активатора в образце для доноров типа 1. Относительное изменение концентрации кальция в цитозоле, R/R0 (г), связывание фибриногена (д) и изменение формы (е) тромбоцитов в течение 140 с после добавления CRP для получения трех различных концентраций активатора в образце для доноров типа 2. Относительное изменение концентрации кальция в цитозоле, R/R0 (ж), связывание фибриногена (з) и изменение формы (и) тромбоцитов в течение 140 с после добавления CRP для получения трех различных концентраций активатора в образце у мышей.
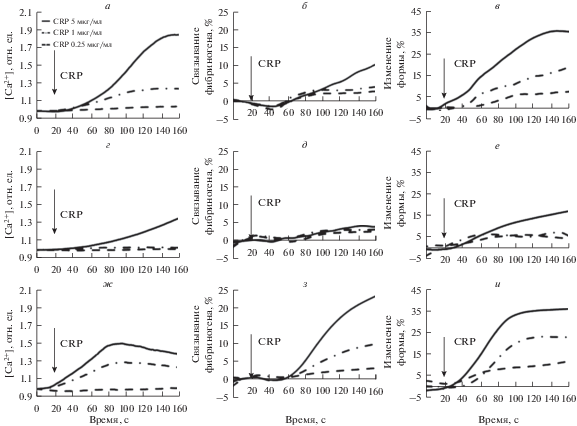
Дальнейший анализ полученных данных был проведен для выявления закономерностей в измеренных параметрах в зависимости от типа объекта исследования. Так, степень связывания фибриногена и процент изменения бокового светорассеяния (далее изменение формы) также различался в зависимости от группы: мышиные тромбоциты демонстрировали существенно большее связывание фибриногена по сравнению с человеческими (рис. 2б, 2д, 2з), а изменение формы у доноров типа 2 было снижено по сравнению с типом 1 и мышами (рис. 2в, 2е, 2и).
В ответ на высокую концентрацию CRP наблюдается вариабельность ответа по относительному изменению концентрации внутриклеточного кальция в тромбоцитах человека (2.02 ± ± 0.45 отн. ед. и 1.22 ± 0.13 отн. ед. у типа 1 и типа 2, соответственно). Тромбоциты мышей были выделены в группу 3 (1.49 ± 0.06 отн. ед.), статистически значимо отличающуюся от типа 2 (рис. 3а). Связывание фибриногена напрямую не коррелирует со значением внутриклеточной концентрации кальция (рис. 3а, 3б). Максимальный процент связывания фибриногена был наивысшим у мышей (17.6 ± 5.8), средним для типа 1 (9.1 ± 1.7) и низким для типа 2 (4.6 ± 2.3). Также была продемонстрирована двукратная вариабельность по изменению формы для типа 1 и типа 2 (рис. 3в).
Рис. 3.
Количественное сравнение ответов тромбоцитов на стимуляцию рецептора GPVI. а – Средние значения максимума R/R0 для трех типов ответов при трех различных концентрациях CRP. б – Средние значения максимального процента связывания фибриногена для трех типов ответов при трех различных концентрациях CRP. в – Средние значения максимального процента изменения формы для трех типов ответов при трех различных концентрациях CRP. Двумя звездочками обозначено статистически значимое различие при p < 0.01, одной – при p < 0.05.
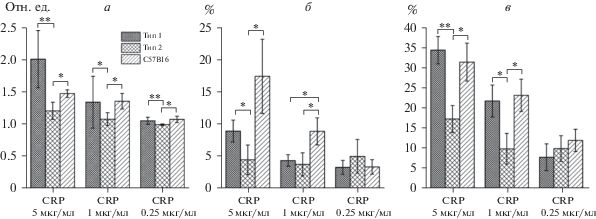
При помощи критерия Манна–Уитни были исследованы численные характеристики ответов доноров на стимуляцию CRP для поиска статистически значимых различий. При концентрациях активатора 5, 1 и 0.25 мкг/мл изменение внутриклеточной концентрации кальция различалось между типом 1 и типом 2 и между типом 2 и мышами (рис. 3а). Процент связывания фибриногена тромбоцитами типа 2 при концентрации CRP 5 мкг/мл был значимо ниже, чем у двух оставшихся групп (рис. 3б). При уменьшении количества активатора до 1 мкг/мл человеческие тромбоциты демонстрировали схожие ответы по фибриногену (4.5 ± 0.9 для типа 1 и 3.9 ± 1.8 для типа 2), меньшие, чем ответ мышиных клеток (9.0 ± 2.1) (рис. 3б). Оба типа ответа человеческих тромбоцитов при концентрациях агониста 5 и 1 мкг/мл различались по изменению формы, и сходным образом наблюдалось отличие между типом 2 и мышиными тромбоцитами (рис. 3в).
Снижение концентрации CRP приводило к уменьшению вариабельности функциональных видов ответов, так, различия по фибриногену и изменению формы при концентрации CRP 0.25 мкг/мл были незначимы (рис. 3б–3в).
Ответы тромбоцитов на активацию GPVI в агрегометрии. Плазма 12 доноров была исследована методом турбидиметрической агрегометрии. Используя данные, полученные экспериментальным путем, мы получили значения максимального изменения оптической плотности раствора при активации коллагеном I типа (концентрации 2.5 и 1 мкг/мл), а также начальную скорость агрегации. Значения данных параметров для различных концентраций активатора сходятся в пределах погрешности (рис. 4в).
Рис. 4.
Результаты агрегометрии в ответ на коллаген. Агрегационные кривые тромбоцитов 12 доноров в ответ на концентрации коллагена 1 (а) и 2.5 мкг/мл (б). в – Значения средних максимумов агрегации и начальной скорости агрегации для доноров двух типов при концентрациях коллагена 1 и 2.5 мкг/мл.
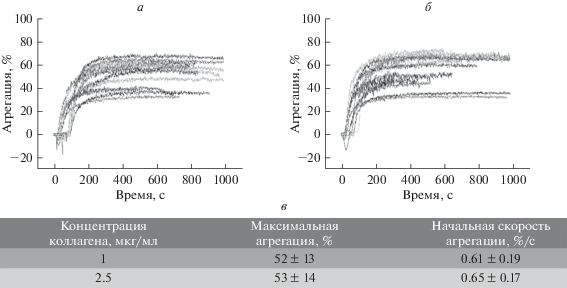
Агрегационные ответы различных доноров также оказались вариабельны, как при стимуляции 1 мкг/мл коллагена (рис. 4а), так и при стимуляции 2.5 мкг/мл коллагена (рис. 4б). Однако, в отличие от данных проточной цитометрии (рис. 2, 3), выделить две различные подгруппы доноров по данным агрегометрии оказалось невозможно (рис. 4а, 4б). Важно отметить, что группировка доноров по результатам проточной цитометрии проводилась по максимально достижимым концентрациям свободных ионов кальция в ответ на активацию, так как провести четкое разделение по степени связывания фибриногена данных групп было затруднительно (рис. 2б, 2д).
ОБСУЖДЕНИЕ
В данной работе были проанализированы различные показатели ответа тромбоцитов при их активации коллагеном или подобным коллагену пептидом через рецептор GPVI и показана вариабельность по уровню внутриклеточного кальция, связыванию фибриногена и изменению формы (рис. 2, 3) для разных доноров. В то время как кальциевые ответы позволяли выделить несколько групп здоровых доноров, различия по фибриногену между группами были менее выраженны или вовсе отсутствовали (рис. 3а, 3б). Результаты турбидиметрической агрегометрии не выявили разделения доноров на группы. Мышиные тромбоциты по концентрации свободных ионов кальция и изменению формы были близки к ответам типа 1, а также демонстрировали существенно большее связывание фибриногена (рис. 3). Согласно литературным данным, существует множество потенциальных причин наблюдаемого нами феномена.
Авторами [42] было показано, что экспрессия GPVI у здоровых доноров может иметь пятикратные различия, а также была продемонстрирована значительная разница в индуцированной конвульксином активности протромбиназы на тромбоцитах, которая коррелировала с уровнем GPVI-рецепторов. В той же работе прямой связи между агрегацией тромбоцитов в ответ на конвульксин и CRP и экспрессией GPVI не наблюдалось, и корреляции между адгезией тромбоцитов к лигандам GPVI и содержанию данного рецептора также не было продемонстрировано. Однако существуют исследования, в которых экспрессия рецептора имеет меньшую вариабельность, так, среди 102 доноров она различалась только в 1.5 раза [43]. Для проверки гипотезы о том, что изменение уровня внутриклеточного кальция в ответ на активацию CRP (рис. 2, 3) зависит от уровня экспрессии GPVI, необходимо дальнейшее изучение феномена, включающее в себя подсчет числа интересующих нас рецепторов на мембране. Вопрос о причинах столь значительных отличий экспрессии тромбоцитарного рецептора GPVI также остается открытым на сегодняшний день.
Другой причиной вариабельных ответов у здоровых доноров могли быть различные полиморфизмы в аминокислотной последовательности рецептора GPVI. Пять однонуклеотидных полиморфизмов (SNP) были найдены в гене GP6. Были выделены два общих гаплотипа, каждый из которых определяется пятью связанными полиморфизмами, три из которых находятся во внеклеточном домене и два в цитоплазматическом (аллель “а” – SKTQH и аллель “b” – PEALN с аллельными частотами 0.85 и 0.13) [44, 45]. Аллель “b” ассоциирована с низкой плотностью GPVI и уменьшает функциональные ответы на коллаген и CRP [43]. С другой стороны, Трифиро и коллеги показали отсутствие связи между генотипом и экспрессией GPVI [46]. Также было показано, что гаплотипы SKTQH и PEALN отвечали только за 35% вариабельности [47]. Была найдена связь между гаплотипом “bb” и коронарным тромбозом [48]. Гомозиготность по “b” аллелю была ассоциирована с развитием инфаркта миокарда у японцев [49]. При этом была найдена корреляция между уменьшением повторов сердечно-сосудистых событий и снижением смертности у пациентов с аллелем “b”, но не первым инфарктом миокарда [50]. Интерес также представляют различия в отдельных SNP. N322 ослабляет связывание с Fyn/Lyn и, следовательно, фосфорилирование Syk [46]. Для проверки предположения, что гаплотип доноров связан с наблюдаемой вариабельностью кальциевого ответа (рис. 3а), необходимо дальнейшее расширение выборки и скрининг доноров на предмет наличия полиморфизмов.
Механизмы взаимодействия GPVI индуцированной кальциевой сигнализации в тромбоцитах и функциональных ответов тромбоцита не являются до конца определенными. Отдельный интерес представляет фосфоинозитидная сигнализация в тромбоцитах. Тромбоцитарные интегрины αIIbβ3, ответственные за проагрегантный ответ, запускаются GTP-азой Rap1, ключевым регулятором которой является Ca2+-регулируемый фактор обмена нуклеотидов CalDAG-GEFI [51, 52]. Было продемонстрировано, что активация Rap1 по GPVI-зависимому пути зависит от активности PI3K [53]. Таким образом, при разной концентрации внутриклеточного кальция (рис. 3а) может быть достигнута схожесть в активации комплекса αIIbβ3 (рис. 3б) и агрегационного ответа (рис. 4) у здоровых доноров.
Кроме того, при активации GPVI происходит активация металлопротеиназы ADAM10, которая отделяет эктодомен GPVI рецептора [54]. В результате данного процесса в плазму выделяется 55 кДа белок sGPVI [55]. Ингибиторы кальциевой сигнализации и компонентов сигнального пути рецептора GPVI блокируют данный процесс [54, 56]. Растворимый GPVI связывается с коллагеном, а также уменьшает адгезию и агрегацию тромбоцитов in vivo [57]. Участие sGPVI в агрегации (рис. 4) может быть причиной меньших функциональных различий по сравнению с проточной цитометрией (рис. 2, 3) в связи с большей концентрацией клеток в растворе.
Оптическая агрегометрия является одним из широко используемых методов выявления нарушений функционирования тромбоцитов, однако существует большое количество ограничений при использовании данного метода и численной оценке данных. Так, результат одного и того же пациента зависит от большого числа условий: типа антикоагулянта, количества липидов в плазме, гемолиза, количества тромбоцитов, свежести образца. Отсутствие стандартизации и контроля качества существенно усложняет наблюдение малых различий [58]. Таким образом, не выявленное в ходе этого исследования разделение доноров на подгруппы тестом турбидиметрической агрегометрии (рис. 4) может также быть следствием низкой точности теста.
В настоящей работе нами было продемонстрировано существование различных типов активации тромбоцитов по GPVI-пути и не связанных между собой напрямую кальциевого и фибриногенового ответа тромбоцитов и выдвинуто несколько гипотез о природе данного явления. Полученные нами данные позволяют предположить, что в тромбоците существует больше одного уровня регуляции внутриклеточных процессов, отвечающих за активацию и агрегацию. Дальнейшее изучение вопроса необходимо для уточнения механизмов и потенциального использования знаний о группе пациента (рис. 2, 3) для персонализированной терапии.
Авторы благодарны М.А. Пантелееву (ЦТП ФХФ РАН, Москва, РФ) за обсуждения в ходе работы и В.В. Попову (факультет фундаментальной медицины МГУ, Москва, РФ) за консультации по работе с животными. Разработка предложенной методики наблюдения активации тромбоцитов на проточном цитометре поддержана грантом Российского научного фонда № 17-74-20 045, анализ полученных данных поддержан стипендией Президента РФ СП-2675.2019.4, работа со здоровыми донорами поддержана грантом фонда “Врачи, инновации, наука – детям”.
Список литературы
Weinberg C.B., Bell E. 1986. A blood vessel model constructed from collagen and cultured vascular cells. Science. 231 (4736), 397–400.
Monroe D.M., Hoffman M. 2006. What does it take to make the perfect clot? Arterioscler. Thromb. Vasc. Biol. 26 (1), 41–48.
Jackson S.P., Nesbitt W.S., Kulkarni S. 2003. Signaling events underlying thrombus formation. J. Thromb. Haemost. 1 (7), 1602–1612.
Siedlecki C.A., Lestini B.J., Kottke-Marchant K., Eppell S.J., Wilson D.L., Marchant R.E. 1996. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 88 (8), 2939–2950.
Koupenova M., Kehrel B.E., Corkrey H.A., Freedman J.E. 2016. Thrombosis and platelets: an update. Eur. Heart J. 38 (11), 785–791.
Yun S.H., Sim E.H., Goh R.Y., Park J.I., Han J.Y. 2016. Platelet activation: The mechanisms and potential biomarkers. Biomed Res. Int. 2016, 9060143.
Jackson S.P. 2011. Arterial thrombosis-insidious, unpredictable and deadly. Nature Medicine. 17 (11), 1423–1436.
Snell D.C., Schulte V., Jarvis G.E., Arase K., Sakurai D., Saito T., Watson S.P., Nieswandt B. 2002. Differential effects of reduced glycoprotein VI levels on activation of murine platelets by glycoprotein VI ligands. Biochem. J. 368 (1), 293–300.
Versteeg H.H., Heemskerk J.W.M., Levi M., Reitsma P.H. 2013. New fundamentals in hemostasis. Physiol. Rev. 93 (1), 327–358.
Bennett J.S. 2015. Regulation of integrins in platelets. Biopolymers. 104 (4), 323–333.
Lagrue-Lak-Hal A.H., Debili N., Kingbury G., Lecut C., Le Couedic J.P., Villeval J.L., Jandrot-Perrus M., Vainchenker W. 2001. Expression and function of the collagen receptor GPVI during megakaryocyte maturation. J. Biol. Chem. 276 (18), 15 316–15 325.
Nieswandt B., Watson S.P. 2003. Platelet-collagen interaction: Is GPVI the central receptor? Blood. 102 (2), 449–461.
Boylan B., Berndt M.C., Kahn M.L., Newman P.J. 2006. Activation-independent, antibody-mediated removal of GPVI from circulating human platelets: development of a novel NOD/SCID mouse model to evaluate the in vivo effectiveness of anti-human platelet agents. Blood. 108 (3), 908–914.
Polgár J., Clemetson J.M., Kehrel B.E., Wiedemann M., Magnenat E.M., Wells T.N.C., Clemetson K.J. 1997. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (Tropical rattlesnake) venom via the p62/GPVI collagen receptor. J. Biol. Chem. 272 (21), 13 576–13 583.
Alshehri O.M., Hughes C.E., Montague S., Watson S.K., Frampton J., Bender M., Watson S.P. 2015. Fibrin activates GPVI in human and mouse platelets. Blood. 126 (13), 1601–1608.
Induruwa I., Moroi M., Bonna A., Malcor J.D., Howes J.M., Warburton E.A., Farndale R.W., Jung S.M. 2018. Platelet collagen receptor glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J. Thromb. Haemost. 16 (2), 389–404.
Mangin P.H., Onselaer M.B., Receveur N., Le Lay N., Hardy A.T., Wilson C, Sanchez X., Loyau S., Dupuis A., Babar A.K., Miller J.L.C., Philippou H., Hughes C.E., Herr A.B., Ariëns R.A.S., Mezzano D., Jandrot-Perrus M., Gachet C., Watson S.P. 2018. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 103 (5), 898–907.
Getahun A., Cambier J.C. 2015. Of ITIMs, ITAMs, and ITAMis: Revisiting immunoglobulin Fc receptor signaling. Immunol. Rev. 268 (1), 66–73.
Andrews R.K., Suzuki-Inoue K., Shen Y., Tulasne D., Watson S.P., Berndt M.C. 2002. Interaction of calmodulin with the cytoplasmic domain of platelet glycoprotein VI. Blood. 99 (11), 4219–4221.
Moroi M., Jung S.M. 2004. Platelet glycoprotein VI: Its structure and function. Thromb. Res. 114 (4), 221–233.
Quek L., Pasquet J., Hers I., Cornall R., Knight G., Barnes M., Hibbs M., Dunn A., Lowell C., Watson S. 2001. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood. 96, 4246–4253.
Dumont B., Lasne D., Rothschild C., Bouabdelli M., Ollivier V., Oudin C., Ajzenberg N., Grandchamp B., Jandrot-Perrus M. 2009. Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood. 114 (9), 1900–1903.
Hermans C., Wittevrongel C., Thys C., Smethurst P.A., Van Geet C., Freson K. 2009. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J. Thromb. Haemost. 7 (8), 1356–1363.
Matus V., Valenzuela G., Sáez C.G., Hidalgo P., Lagos M., Aranda E., Panes O., Pereira J., Pillois X., Nurden A.T., Mezzano D. 2013. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J. Thromb. Haemost. 11 (9), 1751–1759.
Arthur J.F., Dunkley S., Andrews R.K. 2007. Platelet glycoprotein VI-related clinical defects. British J. Haematology. 139 (3), 363–372.
Moroi M., Shinmyozut K., Hospital K.C. 1989. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J. Clin. Investig. 84, 1440–1445.
Nieswandt B., Bergmeier W., Schulte V., Rackebrandt K., Gessner J.E., Zirngibl H. (2000) Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRγ chain. J. Biol. Chem. 275 (31), 23998–24002.
Nieswandt B., Schulte V., Bergmeier W., Mokhtari-Nejad R., Rackebrandt K., Cazenave J.P., Ohlmann P., Gachet C., Zirngibl H. 2001. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 193 (4), 459–469.
Dütting S., Bender M., Nieswandt B. 2012. Platelet GPVI: A target for antithrombotic therapy?! Trends Pharmacol. Sci. 33, 583–590.
Ungerer M., Rosport K., Bültmann A., Piechatzek R., Uhland K., Schlieper P., Gawaz M., Münch G. 2011. Novel antiplatelet drug revacept (dimeric glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 123 (17), 1891–1899.
Geahlen R.L. (2014) Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 35 (8), 414–422.
Mangin P.H., Tang C.J., Bourdon C., Loyau S., Freund M., Hechler B., Gachet C., Jandrot-Perrus M. 2012. A humanized glycoprotein VI (GPVI) mouse model to assess the antithrombotic efficacies of anti-GPVI agents. J. Pharmacol. Exp. Ther. 341 (1), 156–163.
Li H., Lockyer S., Concepcion A., Gong X., Takizawa H., Guertin M., Matsumoto Y., Kambayashi J., Tandon N.N., Liu Y. 2007. The fab fragment of a novel anti-GPVI monoclonal antibody, OM4, reduces in vivo thrombosis without bleeding risk in rats. Arterioscler. Thromb. Vasc. Biol. 27 (5), 1199–1205.
Takayama H., Hosaka Y., Nakayama K., Shirakawa K., Naitoh K., Matsusue T., Shinozaki M., Honda M., Yatagai Y., Kawahara T., Hirose J., Yokoyama T., Kurihara M., Furusako S. 2008. A novel antiplatelet antibody therapy that induces cAMP-dependent endocytosis of the GPVI/Fc receptor γ-chain complex. J. Clin. Investig. 118 (5), 1785–1795.
Morton L.F., Hargreaves P.G., Farndale R.W., Young R.D., Barnes M.J. 1995. Integrin α2β1-independent activation of platelets by simple collagen-like peptides: Collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for α2β1-independent platelet reactivity. Biochem. J. 306 (2), 337–344.
Cazenave J.P., Ohlmann P., Cassel D., Eckly A., Hechler B., Gachet C. 2004. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol. Biol. 272 (5), 13–28.
Alberio L., Ravanat C., Hechler B., Mangin P.H., Lanza F., Gachet C. 2017. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb. Haemost. 117 (06), 1101–1114.
Martyanov A.A., Morozova D.S., Sorokina M.A., Filkova A.A., Fedorova D.V., Uzueva S.S., Suntsova E.V., Novichkova G.A., Zharkov P.A., Panteleev M.A., Sveshnikova A.N. 2020. Heterogeneity of integrin αIIbβ3 function in pediatric immune thrombocytopenia revealed by continuous flow cytometry analysis. Int. J. Mol. Sci. 21 (9), 3035.
Martyanov A.A., Balabin F.A., Dunster J.L., Panteleev M.A., Gibbins J.M., Sveshnikova A.N. 2020. Control of platelet CLEC-2-mediated activation by receptor clustering and tyrosine kinase signaling. Biophys. J. 118 (11), 2641–2655. https://doi.org/10.1016/j.bpj.2020.04.023
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A.V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Мартьянов А.А., Морозова Д.С., Хорева А.Л., Пантелеев М.А., Щербина А.Ю., Свешникова А.Н. 2020. Особенности внутриклеточной кальциевой сигнализации тромбоцитов при синдроме Вискотта-Олдрича. Вопросы онкологии/гематологии и иммунопатологии в педиатрии. 19 (1), 100–107.
Furihata K., Clemetson K.J., Deguchi H., Kunicki T.J. 2001. Variation in human platelet glycoprotein VI content modulates glycoprotein VI-specific prothrombinase activity. Arterioscler. Thromb. Vasc. Biol. 21 (11), 1857–1863.
Best D., Senis Y.A., Jarvis G.E., Eagleton H.J., Roberts D.J., Saito T., Jung S.M., Moroi M., Harrison P., Green F.R., Watson S.P. 2003. GPVI levels in platelets: relationship to platelet function at high shear. Blood. 102 (8), 2811–2818.
Joutsi-Korhonen L., Smethurst P.A., Rankin A., Gray E., IJsseldijk M., Onley C.M., Watkins N.A., Williamson L.M., Goodall A.H., de Groot P.G., Farndale R.W., Ouwehand W.H. 2003. The low-frequency allele of the platelet collagen signaling receptor glycoprotein VI is associated with reduced functional responses and expression. Blood. 101 (11), 4372–4379.
Croft S.A., Samani N.J., Teare M.D., Hampton K.K., Steeds K.S., Channer K.S., Daly M.E. 2001. Novel platelet membrane glycoprotein VI dimorphism is a risk factor for myocardial infarction. Circulation. 104 (13), 1459–1463.
Trifiro E., Williams S.A., Cheli Y., Furihata K., Pulcinelli F.M., Nugent D.J., Kunicki T.J. 2009. The low-frequency isoform of platelet glycoprotein VIb attenuates ligand-mediated signal transduction but not receptor expression or ligand binding. Blood. 114 (9), 1893–1899.
Jones C.I., Garner S.F., Angenent W., Bernard A., Berzuini C., Burns P., Farndale R.W., Hogwood J., Rankin A., Stephens J.C., Tom B.D., Walton J., Dudbridge F., Ouwehand W.H., Goodall A.H. 2007. Mapping the platelet profile for functional genomic studies and demonstration of the effect size of the GP6 locus. J. Thromb. Haemost. 5 (8), 1756–1765.
Ollikainen E., Mikkelsson J., Perola M., Penttilä A., Karhunen P.J. (2004) Platelet membrane collagen receptor glycoprotein VI polymorphism is associated with coronary thrombosis and fatal myocardial infarction in middle-aged men. Atherosclerosis. 176 (1), 95–99.
Takagi S., Iwai N., Baba S., Mannami T., Ono K., Tanaka C., Miyata T., Miyazaki S., Nonogi H., Goto Y. 2002. A GPVI polymorphism is a risk factor for myocardial infarction in Japanese. Atherosclerosis. 165 (2), 397–398.
Snoep J.D., Gaussem P., Eikenboom J.C.J., Emmerich J., Zwaginga J.J., Holmes C.E., Vos H.L., De Groot P.H.G., Herrington D.M., Bray P.F., Rosendaal F.R., Van der Bom J.G. 2010. The minor allele of GP6 T13254C is associated with decreased platelet activation and a reduced risk of recurrent cardiovascular events and mortality: Results from the SMILE–Platelets project. J. Thromb. Haemost. 8 (11), 2377–2384.
Bernardi B., Guidetti G.F., Campus F., Crittenden J.R., Graybiel A.M., Balduini C., Torti M. 2006. The small GTPase Rap1b regulates the cross talk between platelet integrin alpha2beta1 and integrin alphaIIbbeta3. Blood. 107 (7), 2728–2735.
Bertoni A., Tadokoro S., Eto K., Pampori N., Parise L.V., White G.C., Shattil S.J. 2002. Relationships between Rap1b, affinity modulation of integrin αIIbβ3, and the actin cytoskeleton . J. Biol. Chem. 277 (28), 25 715–25 721.
Gilio K., Munnix I.C.A., Mangin P., Cosemans J.M.E.M., Feijge M.A.H., van der Meijden P.E.J., Olieslagers S., Chrzanowska-Wodnicka M.B., Lillian R., Schoenwaelder S., Koyasu S., Sage S.O., Jackson S.P., Heemskerk J.W.M. 2009. Non-redundant roles of phosphoinositide 3-kinase isoforms alpha and beta in glycoprotein VI-induced platelet signaling and thrombus formation. J. Biol. Chem. 284 (49), 33750–33762.
Gardiner E.E., Karunakaran D., Shen Y., Arthur J.F., Andrews R.K., Berndt M.C. 2007. Controlled shedding of platelet glycoprotein (GP)VI and GPIb–IX–V by ADAM family metalloproteinases. J. Thromb. Haemost. 5 (7), 1530–1537.
Andrews R.K., Karunakaran D., Gardiner E.E., Berndt M.C. 2007. Platelet receptor proteolysis: A mechanism for downregulating platelet reactivity. Arterioscler. Thromb. Vasc. Biol. 27 (7), 1511–1520.
Stephens G., Yan Y., Jandrot-Perrus M., Villeval J.L., Clemetson K.J., Phillips D.R. 2005. Platelet activation induces metalloproteinase-dependent GP VI cleavage to down-regulate platelet reactivity to collagen. Blood. 105 (1), 186–191.
Massberg S., Konrad I., Bültmann A., Schulz C., Münch G., Peluso M., Lorenz M., Schneider S., Besta F., Müller I., Hu B.I.N., Langer H., Kremmer E., Rudelius M., Heinzmann U., Ungerer M., Gawaz M. 2003. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 18 (2), 397–399.
Alessi M.C., Sié P., Payrastre B. 2020. Strengths and weaknesses of light transmission aggregometry in diagnosing hereditary platelet function disorders. J. Clin. Med. 9 (3), 763.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии