

Биологические мембраны: Журнал мембранной и клеточной биологии, 2021, T. 38, № 4, стр. 237-256
Фактор Виллебранда в норме и при патологии
П. П. Авдонин a, Н. В. Цветаева b, Н. В. Гончаров c, d, Е. Ю. Рыбакова a, С. К. Труфанов a, А. А. Цитрина a, П. В. Авдонин a, *
a Институт биологии развития им. Н.К. Кольцова РАН
119334 Москва, Россия
b Национальный медицинский исследовательский центр гематологии Минздрава России
125167 Москва, Россия
c Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
194223 Санкт-Петербург, Россия
d Научно-исследовательский институт гигиены, профпатологии и экологии человека ФМБА России
188663 Ленинградская обл., ст. Капитолово, Россия
* E-mail: pvavdonin@yandex.ru
Поступила в редакцию 15.04.2021
После доработки 29.04.2021
Принята к публикации 30.04.2021
Аннотация
Фактор Виллебранда (фВ), ключевой компонент гемостаза, синтезируется в эндотелиальных клетках и мегакариоцитах и высвобождается в кровь в виде высокомолекулярных мультимерных гликопротеинов с массой до 20 миллионов дальтон. Металлопротеаза плазмы крови ADAMTS13 расщепляет сверхкрупные мультимеры фВ до более мелких мультимерных и олигомерных молекул. Молекулы фВ присоединяются к местам повреждения поверхности артериол и капилляров и разворачиваются в условиях быстрого кровотока. На развернутой молекуле фВ экспонируются участки, взаимодействующие с рецепторами на мембране тромбоцитов. После связывания с нитями фВ тромбоциты активируются, к ним дополнительно присоединяются циркулирующие в сосудах тромбоциты, и в итоге происходит формирование тромбов, закупорка микрососудов и остановка кровотечения. В обзоре будет описана история открытия фВ, представлены данные о механизмах секреции фВ и его структуре, охарактеризованы процессы обмена фВ в организме в норме и при патологических состояниях.
ИСТОРИЯ ОТКРЫТИЯ ФАКТОРА ВИЛЛЕБРАНДА И МЕТАЛЛОПРОТЕАЗЫ ADAMTS13
Ключевой компонент системы гемостаза, фактор Виллебранда (фВ), назван по имени врача Эрика Адольфа фон Виллебранда (1.02.1870– 12.12.1949), получившего образование в Императорском Александровском университете в Гельсингфорсе (Хельсинки) и работавшего в Финляндии после распада Российской империи. В 1924 году фон Виллебранда попросили осмотреть пятилетнюю девочку из деревни на Аландских островах, страдавшую от кровотечений. Родственники девочки со стороны и отца, и матери имели склонность к сильным кровотечениям при малейшем повреждении кожи и слизистых. Из 11 братьев и сестер девочки четверо умерли по этой причине в раннем возрасте. В 1926 г. по итогам своих исследований Эрик фон Виллебранд опубликовал на шведском языке статью о ранее неизвестной форме наследственной гемофилии, для которой было характерно нормальное свертывание крови, но увеличенное время кровотечения [1]. Он назвал это заболевание псевдогемофилией. Позднее в его честь оно получило наименование болезнь Виллебранда (см. обзор [2]). Девочка, которую обследовал Эрик фон Виллебранд, умерла в возрасте 13 лет во время четвертого менструального периода. Эрик фон Виллебранд описал одну из наиболее тяжелых форм данной болезни. К настоящему времени показано, что у болезни Виллебранда имеется порядка 20 разновидностей от почти незаметных до крайне тяжелых. Помимо наследственных встречаются приобретенные формы болезни Виллебранда.
Кровотечение из мелких сосудов, которое является главным признаком болезни Виллебранда, в норме останавливается в результате прикрепления тромбоцитов к поврежденной поверхности и образования микротромбов. Переливание цельной крови или плазмы здорового человека позволяет остановить кровотечение при данной болезни [3]. В конце 50-х годов путем криопреципитации была получена уменьшающая время кровотечения фракция плазмы крови. Выделенная фракция содержала фактор VIII свертывания крови, однако свойством сокращать время кровотечения обладал также криопреципитат, который получали из плазмы больных гемофилией А [4]. Неизвестный активный компонент плазмы получил название фактор фон Виллебранда (фВ). Для того, чтобы исследовать его природу, необходимо было иметь модельную систему, имитирующую кровотечение из мелких сосудов и процесс его остановки. Первоначально для этого использовали пластиковую трубку, заполненную стеклянными шариками диаметром 0.5 мм, через которую пропускали кровь [5, 6]. Адгезивные свойства тромбоцитов оценивали по времени их задержки при протекании крови. Задержка тромбоцитов при протекании крови от пациентов с болезнью Виллебранда была короче, но ее можно было скорректировать, если предварительно через трубку с шариками пропускали плазму здоровых доноров или больных гемофилией А, криопреципитаты или частично очищенные фракции плазмы (см. обзор [7]). С помощью этой тест-системы проводилось определение активности фВ в белковых фракциях при гель-фильтрации, и таким образом удалось получить достаточно хорошо очищенный препарат фВ, к которому были выработаны антитела [8, 9]. С их помощью вскоре была показана мультимерная структура фВ [10]. Методом иммунопреципитации было доказано, что фактор VIII и фВ – это разные субстанции [11]. Для разделения мультимеров фВ используется агарозный гель [12, 13]. Мультимерные белки образованы в результате сшивания дисульфидными связями димерных молекул. В самом димере мономерные белки также соединены дисульфидной связью [14]. В плазме крови фВ представляет собой смесь таких мультимеров с разбросом молекулярной массы от примерно 600 тысяч (одиночный димер) до 20 миллионов дальтон и более. Более детально вопрос о структуре фВ будет рассмотрен ниже.
Изучению фВ в большой степени способствовало открытие индуцированной антибиотиком ристоцетином агрегации тромбоцитов в плазме крови [15]. В присутствии ристоцетина при перемешивании обогащенной тромбоцитами плазмы от здоровых доноров происходит агрегация тромбоцитов, тогда как в плазме пациентов с болезнью Виллебранда агрегации нет. С помощью ристоцетина в тесте агрегации проводится количественное определение активности фВ в плазме крови у пациентов с подозрением на наличие болезни Виллебранда. При этом можно использовать отмытые и фиксированные формальдегидом тромбоциты, которые сохраняют способность к агглютинации в присутствии ристоцетина и нормальной плазмы. Как в дальнейшем выяснилось, ристоцетин вызывает частичное раскручивание высокомолекулярных мультимеров, что придает фактору Виллебранда способность связываться с тромбоцитами. В 1972 были впервые выявлены пациенты с болезнью Виллебранда 2-типа с нормальным уровнем антигена фВ, но с пониженной активностью [16]. Такое же действие, как ристоцетин, оказывает белок из змеиного яда ботроцитин [17].
Немногим ранее открытия Эриком фон Виллебрандом болезни, связанной с дефицитом фВ, в США была опубликована статья Э. Мошковица, посвященная описанию болезни с прямо противоположным механизмом патогенеза [18]. Был впервые описан случай тромботической тромбоцитопенической пурпуры (ТТП) или синдрома Мошковица – болезни, вызванной гиперактивностью фВ в плазме. У 16-летней девочки наблюдались многочисленные кровоизлияния на поверхности тела (петехии), гемолиз, с последующими параличом, потерей сознания и смертью. При вскрытии были видны многочисленные мелкие гиалиновые (стекловидные) тромбы в артериолах и капиллярах разных органов. Связь этой патологии с фВ стала проясняться после того, как в 1982 г. было показано, что у больных с ТТП во время ремиссии в плазме присутствуют в избытке тромбогенные сверхкрупные мультимеры фВ [19]. При рецидиве болезни, напротив, уменьшается доля сверхкрупных мультимеров фВ и возрастает содержание мультимеров малых размеров. На основе данных о повышенном содержании высокомолекулярных мультимеров при ТТП было предположено, что в плазме больных не происходит или снижено их расщепление. Расщепление гигантских мультимеров фВ до более мелких олигомеров осуществляет металлопротеаза ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 motifs). Этот фермент был обнаружен в двух лабораториях – в Швейцарии в 1997 г. [20] и спустя год в США [21]. ADAMTS13 разлагает мультимеры путем разрыва пептидных связей фВ между 1605 и 1606 остатками тирозина и метионина в мономерных субъединицах. Основной причиной развития ТТП является образование в организме антител IgG, блокирующих активность этого фермента [22]. При другой форме тромботической микроангиопатии (ТМА), гемолитико-уремическом синдроме (ГУС), подъем активности фВ вызван увеличением его производства эндотелием, а не подавлением распада. Во время рецидивов ГУС содержание антигена фВ возрастает в несколько раз, причем также преобладают небольшие олигомеры фВ, но в отличие от ТТП, при ремиссии ГУС не происходит сдвиг мультимерного состава в сторону высокомолекулярных мультимеров [23]. Помимо ТТП и ГУС известны другие формы ТМА, при которых происходит повышение активности фВ. Этиология ТМА не всегда установлена.
СИНТЕЗ ФАКТОРА ВИЛЛЕБРАНДА
Ключевой компонент системы гемостаза, фактор фон Виллебранда (фВ), синтезируется в эндотелиальных клетках и в мегакариоцитах, дающих начало тромбоцитам. фВ секретируется в кровь и обеспечивает присоединение тромбоцитов к поврежденной стенке сосуда, связываясь с коллагеном, а также выполняет функцию носителя фактора VIII свертывания крови [24]. Исходный белок-мономер, из которого образуется мультимерная цепь фВ, кодируется геном, расположенным на хромосоме 12p2.1 [25]. Ген VWF имеет длину примерно 178 тысяч пар оснований (п.о.) и содержит 52 экзона [26], из которых 17 экзонов кодируют сигнальный пептид и пропептид фВ (так называемый антиген II фактора Виллебранда). Размер экзонов варьирует от 40 до 1379 п.о., размер интронов – от 97 до 19.9 тысяч п.о. Зрелая субъединица фактора Виллебранда и 3'-нетранслируемая область кодируются частью гена, включающей приблизительно 80 тысяч п.о. В гаплоидном геноме человека имеется один экземпляр гена фВ [27]. Экспрессия контролируется транскрипционным фактором GATA2 [28].
Первичный генный продукт фВ образован 2813 аминокислотами (а.к.), включающий в себя сигнальный пептид из 22 а.к. и пропептид 741 а.к. [29–31]. Доменная структура мономера фВ с многочисленными участками, по которым происходит гликозилирование, показана на рис. 1. Процесс созревания мультимерной молекулы фВ и его секреция в эндотелиальных клетках происходит следующим образом. В шероховатом эндоплазматическом ретикулуме начинается процесс гликозилирования молекулы фВ путем присоединения крупных маннозных остатков. Зрелый гликозилированный мономер массой 260 кДа образует димер в эндоплазматическом ретикулуме через С-концевые дисульфидные связи и перемещается по антероградному пути в аппарат Гольджи. За счет дополнительного роста гликозильных остатков масса мономера возрастает до 275 кДа. Далее происходит мультимеризация фВ, отщепление пропептида у большей части мономеров и начало спирализации. Масса мономера снижается до 220 кДа, однако в некоторых секретируемых мультимерах фВ сохраняются мономеры с массой 275 кДа. Формирующиеся мультимеры фВ поступают в транс-цистерны аппарата Гольджи (trans-Golgi network), в которых уже можно видеть под электронным микроскопом начинающие формироваться тубулы мультимеров фВ [32]. Отшнуровывающиеся от аппарата Гольджи везикулы преобразуются в характерные только для эндотелиальных клеток структуры – тельца Вейбеля–Паладе, сигарообразная форма которых формируется под влиянием спиралевидных трубок, образованных растущими мультимерами фВ [33, 34]. В результате спирализации занимаемый мультимерами объем уменьшается на 2 порядка [35, 36]. Диаметр телец Вейбеля–Паладе составляет 100–200 нм, а длина от 1 до 5 мкм. Молекулярная масса мультимеров фВ в зрелых тельцах может превышать 20 миллионов дальтон [36]. Секретируемые эндотелиальными клетками сверхкрупные мультимеры фВ расщепляются присутствующей в плазме протеазой ADAMTS13 до более мелких фрагментов [37]. В опытах in vivo показано, что в брыжеечных артериях мышей с нокаутом гена ADAMTS13 в ответ на активацию эндотелиальных клеток мультимеры фВ, выстраиваясь в ряд, соединяются друг с другом и формируют прикрепленные к клеткам нити длиной от 20 до 100 мкм [38–40]. При окрашивании тромбоцитов родамином можно видеть, как они прикрепляются к этим нитям подобно бусинам. В культивируемых эндотелиальных клетках из пупочной вены человека длина образованных пучками молекул фВ нитей достигает миллиметра и более [39]. Из этих нитей может формироваться сетчатая структура, которая не образуется in vivo. В прикреплении нитей фВ к мембране культивируемых эндотелиальных клеток участвуют Р-селектин и αVβ3 интегрин [38].
Рис. 1.
Доменная структура мономера зрелого фактора Виллебранда. Показаны участки взаимодействия с фактором VIII, Р-селектином, рецепторами фВ на мембране тромбоцитов GP1bα, GPIIbIIIa, коллагена типов I, III, IV, VI, участки присоединения ADAMTS13 и расщепления фВ, участки, по которым происходит димеризация и мультимеризация фВ. TIL' – trypsin inhibitor-like domain. Гликозильные остатки представлены в виде черных (N-гликаны) и белых (О-гликаны) шариков на ножках. Гликозилирование происходит по остаткам аминокислот аргинина (N), треонина (Т) и серина (S). Адаптировано из [30] и [90].
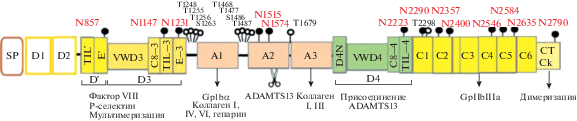
Помимо фВ в тельцах Вейбеля–Паладе присутствуют такие белки, как Р-селектин, интерлейкин-8, остеопротегерин, ангиопоэтин-2, эндотелин-1 [41, 42]. Р-селектин и ангиопоэтин-2 хранятся в разных фракциях телец Вейбеля–Паладе [43]. Кроме того, есть свидетельства, что высвобождение фВ и Р-селектина из телец Вейбеля–Паладе по-разному регулируется [34]. Показано, что только в части культивируемых эндотелиальных клеток из аорты человека одновременно с фВ экспрессируется Р-селектин [44]. Фактор VIII также секретируется не всеми эндотелиальными клетками, производящими фВ [45]. Физиологические эффекты секретируемых из телец Вейбеля–Паладе белков и пептидов рассмотрены в обзоре [42].
В тромбоцитах фВ находится в альфа-гранулах, которые имеют округлую форму. фВ в них также упакован в виде трубочек диаметром 200–250 ангстрем, расположенных эксцентрично [46].
РЕГУЛЯЦИЯ ЭКЗОЦИТОЗА ФАКТОРА ВИЛЛЕБРАНДА
Секреция фВ в эндотелиальных клетках происходит в результате конститутивного и регулируемого экзоцитоза телец Вейбеля–Паладе [45, 47, 48]. Помимо этих двух описан третий механизм экзоцитоза – с участием аутофагосом [49]. Активация экзоцитоза фВ происходит при различных условиях, включая воспаление, повреждение сосудов, гипоксию, напряжение сдвига, активацию мембранных рецепторов [42, 50, 51]. Регулируемый экзоцитоз происходит преимущественно в апикальном направлении, конститутивный экзоцитоз идет и в апикальном, и в базальном направлениях [40, 52].
Предполагается, что конститутивный экзоцитоз происходит при высвобождении содержимого одиночных телец Вейбеля–Паладе. Высвобождаемый таким путем фВ в комплексе с фактором VIII необходим для поддержания нормального гемоcтаза. Показано, что при активации экзоцитоза гистамином и тромбином сразу несколько телец Вейбеля–Паладе высвобождают содержащийся в них фВ в особые секреторные везикулы (secretory pods). Молекулы фВ, присутствующие в секреторных везикулах в большом количестве, объединяются в пучки, из которых на поверхности эндотелия формируются нити, цепляющие на себе тромбоциты [38].
В передаче действия агонистов на секрецию фВ участвуют вторичные посредники cAMP и Са2+ [40, 53]. Тромбин и гистамин, активирующие Са2+-сигнальную систему, оказывают более выраженную активацию экзоцитоза фВ [54–56] по сравнению с адреналином и вазопрессином, действующими через cAMP [57, 58]. Кроме этого, адреналин и вазопрессин, в отличие от гистамина и тромбина, не вызывают или слабо активируют экзоцитоз телец Вейбеля–Паладе, содержащих, помимо фВ, Р-селектин [59]. Механизм экзоцитоза телец Вейбеля–Паладе до конца не изучен, но известны некоторые базовые сигнальные механизмы, запускающие данный процесс. При действии как ионов Са2+, так и cAMP, происходит активация обмена GDP на GTP в белке RalA под влиянием RalGDS [60, 61]. RalA, действуя через фосфолипазу D (PLD), стимулирует опосредованное SNARE слияние мембран на плазматической мембране [62]. В этом процессе альфа-синуклеин регулирует активность RalA и может предотвратить слияние телец Вейбеля–Паладе с мембраной [63]. В передаче активирующего сигнала от cAMP помимо протеинкиназы А принимает участие Epac через сигнальный путь Rap1 –> PREX1 –> Rac1 –> PLD [40].
Регуляция экзоцитоза происходит также на более ранней стадии – во время транспорта телец Вейбеля–Паладе вдоль микротрубочек. После присоединения Rab27A к тельцам Вейбеля–Паладе посредством белков MyRIP и MyoVa происходит их “заякоривание” на актиновых филаментах и тем самым предотвращается экзоцитоз незрелых везикул [64, 65]. Помимо привязки к актиновому цитоскелету, Rab27A регулирует экзоцитоз телец Вейбеля–Паладе посредством присоединения к ним эффекторных белков Slp-4a и Munc14-3 [66–69]. Slp-4a и Munc14-3 необходимы для слияния телец Вейбеля–Паладе с плазматической мембраной. В регуляции экзоцитоза участвуют также Rab15, Rab3B, Rab3D [69] и синтаксин-3 [70, 71]. Разрушение микротрубочек ингибирует Са2+-зависимый экзоцитоз, а дестабилизация актина, наоборот, его усиливает [55, 67, 72]. В случае cAMP-индуцируемого экзоцитоза данные эффекты не наблюдаются. Ионы кальция активируют транспорт телец Вейбеля–Паладе в сторону мембраны, при повышении концентрации Са2+ в цитоплазме эндотелиальных клеток весь фВ высвобождается из клеток. Напротив, при повышении уровня cAMP секретируется только часть фВ [53, 57, 73]. Интересно, что агонисты, активирующие аденилатциклазу, вызывают концентрирование телец Вейбеля–Паладе в околоядерной области клетки (см. обзор [42]).
В последние годы накапливается все больше свидетельств связи воспаления и тромбозов [74, 75]. В качестве индукторов тромбообразования могут выступать активные формы кислорода, генерируемые при воспалительных процессах. Ранее было показано, что секрецию фВ стимулирует супероксид анион [76]. В отношении влияния на секрецию фВ продукта дисмутации супероксид аниона – пероксида водорода, сведения противоречивы. В исследованиях, представленных в цитируемой статье [76], такой эффект Н2О2 не был выявлен. С другой стороны, в работе Yang и соавт. [77] приведены данные об увеличении секреции фВ в 1.5–2 раза под действием экзогенного Н2О2 в достаточно высокой концентрации – 0.5 мМ. В наших исследованиях мы исходили из идеи, что тромбообразование при воспалении определяется не столько циркулирующим фВ, сколько прикрепленными к поверхности активированного эндотелия мультимерами фВ. Поэтому было проведено исследования действия пероксида водорода на экспрессию мультимеров фВ на поверхности эндотелиальных клеток из пупочной вены человека. Мы использовали два метода окрашивания фВ на мембране – с помощью антител [78] и с помощью флуоресцентно-меченного аптамера, связывающего с доменом А1 в молекуле фВ [79]. Было установлено, что пероксид водорода в физиологически релевантной концентрации (100 мкМ) в 2–4 раза увеличивает экспонирование фВ на мембране эндотелиальных клеток. Показано, что при этом формируются нити фВ длиной в десятки микрометров. Таким образом, эти данные свидетельствуют о роли Н2О2 в тромбообразовании при воспалительных процессах. Помимо поступления в эндотелиальные клетки экзогенного пероксида водорода происходит его образование внутри клеток при воздействии VEGF [80], TNFα [81]. Оба эти агониста активируют экзоцитоз фВ [82, 83]. Исходя из этого можно предполагать, что Н2О2 участвует в вызываемом этими агонистами экзоцитозе фВ в качестве вторичного посредника.
СТРУКТУРА ФАКТОРА ВИЛЛЕБРАНДА
Исходный мономер фВ образован доменами D1-D2-D'-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK. D-домены образованы более мелкими модулями, которые видны на электронных фотографиях [45, 84]. Ключевой элемент мономера фВ образован доменами А: домен A1 обеспечивает связывание фВ с рецептором GPIbα на мембране тромбоцитов, в A2 расположена расщепляемая ADAMTS13 пептидная связь, гидролиз которой регулирует размер мультимера фВ в кровотоке, домен А3 осуществляет присоединение к коллагену в месте повреждения сосудистого эндотелия. Дисульфидный мостик между N- и C-концами A1- и A3-доменов фиксирует каждый из этих доменов в относительно жесткой конфигурации. Напротив, А2-домен не имеет жесткой структуры и растягивается в условиях быстрого кровотока при высоком уровне напряжения сдвига, что делает его доступным для протеолиза [85]. Связывание домена А3 с коллагенами I и III, находящимися в субэндотелиальном пространстве, происходит за счет электростатического взаимодействия отрицательно заряженных аминокислотных остатков в пептидной цепи А3 и положительно заряженных остатков в коллагенах. Ван-дер-Ваальсово взаимодействие слабо выражено [86]. Этим объясняется невысокое сродство А3 к коллагену и необходимость одновременного присоединения фВ к коллагену несколькими участками для прочной адгезии. По-видимому, отчасти по этой причине сверхкрупные мультимеры обладают большей тромбогенностью. Домены D1, D2, D3 включают модули VWD (von Willebrand D domain), C8, TIL и E-модуль. Домен D' не содержит VWD и C8, а в D4 нет Е-модуля и присутствует субдомен D4N. В последовательности фВ необычно высокое содержание остатков цистеина (8.3%). Это в 4 раза больше, чем в среднем для белков. Предполагается, что большая часть цистеиновых остатков формирует дисульфидные связи, причем эти связи перестраиваются во время созревания и спирализации мультимера фВ [87]. Функции доменов подробно описаны в обзорах [88, 89].
Благодаря образованию дисульфидных связей происходит сначала сшивание мономеров фВ в димеры, а затем образование из димеров мультимерных молекул фВ. Протодимеры образуются в результате сшивки двух первичных мономеров тремя дисульфидными связями между цистеиновыми остатками 2771, 2773 и 2811, находящимися в С-концевой части пептидной цепи. Эти дисульфидные мостики структурно защищены от восстановления, что обеспечивает прочность димеров фВ. Находясь в везикулах аппарата Гольджи при кислых значениях рН, протодимеры структурно напоминают букет с двумя переплетенными “стеблями”, образованными доменами A2-A3-D4-C1-C2-C3-C4-C5-C6, и “цветками” из доменов D1-D2-D'-D3-A1 [84]. Следующим этапом является ковалентная сшивка протодимеров двумя дисульфидными связями между цистеинами 889 и 898 в D3-доменах на N-концах полипептидных цепей. После образования мультимеров протеаза фурин отщепляет N-концевые хвосты мономеров.
Мультимерная структура фВ выявляется при электрофорезе в агарозном геле с концентрацией агарозы от 1 до 2–3% [13]. Сверхкрупные мультимеры можно увидеть при электрофорезе в геле с наименьшей концентрацией агарозы. По результатам электрофореза, максимальная масса мультимеров, образующихся в эндотелиальных клетках, составляет 20 и более миллионов дальтон, что соответствует массе 35–40 димеров [45]. Для улучшения разделения малых компонентов можно дополнительно вводить в состав геля акриламид. Благодаря этому методу у людей, страдающих болезнью Виллебранда типа IIA, выявлены низкомолекулярные белковые молекулы [13]. Сверхкрупные мультимеры фВ способны спонтанно связываться с тромбоцитами в отличие от мультимеров среднего и небольшого размера, которые присоединяются к тромбоцитам только в присутствии ристоцетина или ботроцитина [91]. Согласно модели, предложенной Гуриа и соавт. [92, 93], более высокая тромбогенная активность сверхкрупных мультимеров фВ в сосудах объясняется меньшим пороговым значением напряжения сдвига, при котором происходит их разворачивание.
В нормальной плазме мультимерный состав фВ регулирует металлопротеаза ADAMTS13, расщепляющая связь Tyr1605-Met1606 в домене А2 [94]. Недавние исследования свидетельствуют, что некоторые другие протеазы в плазме, включая плазмин, могут расщеплять мультимеры фВ [95]. Плазмин разрезает связь между аминокислотными остатками K1491-R1492 в участке пептидной цепи, соединяющем домены А1 и А2.
ГЛИКОЗИЛИРОВАНИЕ И СИАЛИРОВАНИЕ ФАКТОРА ВИЛЛЕБРАНДА
В течение всего времени с момента образования мономера и до секреции гигантских мультимеров происходит посттрансляционная модификация фВ, заключающаяся в гликозилировании [96]. После окончания этого процесса масса N- и O-гликанов достигает 20% от конечной массы фВ [90]. Гликаны присоединяются по остаткам аргинина (N-гликаны) и серина с треонином (О-гликаны). Анализ структуры мономера фВ первоначально выявил 13 потенциальных мест N-гликозилирования, последующие масс-спектрометрические исследования показали наличие 12 N-гликанов [97]. Присоединение первичного олигосахарида (Glc3Man9GlcNAc2) к аргинину происходит в ретикулуме, после чего сначала в ретикулуме, а далее в аппарате Гольджи идет ремоделирование и построение разветвленной структуры гликозильных N-гликанов [96]. После отщепления двух крайних остатков глюкозы гликозидазами мономер связывается с лектинами ретикулума калнексином и калретикулином и происходит его сворачивание (фолдинг). По завершению фолдинга белок далее транспортируется в аппарат Гольджи, где продолжается модификация N-гликанов. В итоге образуются сложные гетерогенные углеводные структуры, в их формировании принимают участие многочисленные гликозилтрансферазы. Образуется три группы N-гликанов – наиболее близкие к исходной структуре богатые маннозой гликаны, гибридные гликаны и комплексные гликаны (complex-type) c двух-, трех- и четырехантенными цепями. Доминирующими формами являются моно- и дисиалированные двухантенные комплексные N-гликаны. У 15% N-гликанов имеются детерминанты групп крови АВ0(Н). 80% сиаловых кислот в молекуле фВ присоединены к N-гликанам и оставшиеся 20% к О-гликанам.
Связанные с серином или треонином олигосахариды (О-гликаны) синтезируются путем последовательного гликозилирования уже после выхода фВ из комплекса Гольджи [96]. По структуре О-гликаны гораздо проще N-гликанов. Из них только 1% несут АВ0-детерминанты групп крови.
Эндотелиальные клетки являются главным источником циркулирующего в крови фВ [98]. Секретируемый тромбоцитами фВ, судя по экспериментам in vitro, остается связанным с рецепторами на тромбоцитарной мембране [99, 100]. В плазме крови происходит постепенное отщепление остатков сиаловых кислот от гликозильных остатков на молекуле фВ нейраминидазами. Этот процесс происходит при старении фВ. Десиалированный фВ имеет большую функциональную активность по сравнению с исходным – он вызывает спонтанную агрегацию тромбоцитов и сильнее связывает тромбоциты с коллагеном при напряжении сдвига [90].
Интересно, что фВ в тромбоцитах по характеру гликозилирования существенно отличается от этого гликопротеина в эндотелиальных клетках и от циркулирующего в крови фВ [98]. Тромбоцитарный фВ значительно беднее сиалированными N-гликанами, и у него отсутствуют детерминанты групп крови [96]. фВ тромбоцитов хуже связывается с GPIbα и сильнее с интегрином GPIIb/IIIa и гепарином.
СВЯЗЫВАНИЕ ТРОМБОЦИТОВ С ФАКТОРОМ ВИЛЛЕБРАНДА
Тромбоциты содержат 2 рецептора, которые связывают VWF: комплекс гликопротеина Ib-IX-V (GPIb-IX-V) и интегрин αIIbβ3 (GPIIb-IIIa). В местах повреждения сосудов экспонируется субэндотелиальная поверхность, к которой прикрепляются нити фВ. В микрососудах или в местах сужения артерий в условиях высокого напряжения сдвига прикрепленные к поверхности мультимерные молекулы фВ разворачиваются и на них раскрываются участки, соответствующие пептидной цепи от Leu-480/Val-481 до Gly-718 в первичной молекуле мономера [101]. Комплекс GPIb-IX-V на мембране тромбоцитов взаимодействует с этими последовательностями на молекуле фВ. Узнавание комплекса GPIb-IX-V обеспечивает участок Asp-514-Glu-542 внутри этих сайтов [17]. Вторым рецептором фВ на поверхности тромбоцитов является интегрин αIIbβ3 (GPIIb-IIIa). C GPIIb-IIIa связываются фВ и фибриноген, формируя молекулярные мостики между агрегирующими тромбоцитами. Количество GPIIb-IIIa на поверхности тромбоцитов коррелирует со степенью ADP-индуцированной агрегации тромбоцитов здоровых доноров и больных с острым коронарным синдромом [102]. При низких значениях напряжения сдвига в отсутствие фибриногена оба рецептора обеспечивают присоединение фВ при агрегации тромбоцитов [103]. При высоком значении напряжения сдвига важным фактором агрегации становиться взаимодействие фВ с GPIb-IX-V [104]. Каневой и соавт. [105] предложена математическая модель, описывающая роль рецепторов фВ в адгезии и последующем тромбообразовании.
Рецептор фВ GPIb-IX-V экспрессируется только в тромбоцитах в количестве примерно 25000 рецепторных комплексов на клетку. В состав GPIb-IX-V входят по 2 субъединицы GPIbα, GP1bβ, GPIX и 1 субъединица GPV [35]. фВ связывается с участком 1-282 внеклеточной части GPIbα. В молекуле фВ сайт связывания с GPIbα находится в домене А1. Помимо фВ лигандами GPIbα являются также тромбоспондин, фактор XII, фактор XI, тромбин, высокомолекулярный кининоген, P-селектин и Mac-1. Взаимодействие фВ и гликопротеина Ib-IX-V не ограничивается только пассивным присоединением тромбоцитов к этому белковому комплексу. Через свой цитоплазматический хвостовой домен (Phe568-Trp570), GPIbα взаимодействует с PI3-киназой, актинсвязывающим белком фламином, адапторным белком 14-3-3ζ. Связывание фВ с GPIb-IX-V вызывает активацию тромбоцитов и переход интегрина αIIbβ3 из низкоаффинного в высокоаффинное состояние, в котором он способен взаимодействовать с доменом C4 в мономере фВ. Это способствует устойчивому прикреплению тромбоцитов к фВ и их распластыванию. Вопрос о том, как взаимодействие VWF/GPIb-IX-V способствует активации тромбоцитов, все еще остается спорным. Разные сигнальные пути активируются в зависимости от величины напряжения сдвига. В активации тромбоцитов, вызванной GPIb-IX-V, показано участие нескольких внутриклеточных молекул – киназ семейства Src, Rac1, PI3-киназы/Akt, cGMP-киназы и MAP-киназы [35].
ОБМЕН ФАКТОРА ВИЛЛЕБРАНДА В ПЛАЗМЕ КРОВИ
Как отмечено выше, эндотелиальные клетки постоянно секретируют фВ в апикальном направлении в плазму крови и в базальном направлении в межклеточное пространство. При активации клеток агонистами секреция фВ в кровь увеличивается. В норме в условиях кровотока нити фВ, выходящие из секреторных пузырьков, в которых аккумулируется фВ из нескольких телец Вейбеля–Паладе, расщепляются протеазой ADAMTS13 [39]. Сверхкрупные мультимеры частично расщепляются и в плазме при разворачивании их в быстром потоке. Благодаря этому поддерживается баланс между крупными мультимерными, средними и мелкими олигомерными формами фВ. В обзоре Садлера [106] приводится среднее значение для уровня антигена фВ в плазме, которое составляет около 100 МЕ/дл. По данным других исследователей [107], концентрация антигена фВ у здоровых доноров составляет 61 МЕ/дл (95%CI:51-91 МЕ/дл). Одна международная единица (МЕ) соответствует 10 мкг фВ. Если выражать содержание фВ в других единицах (мкг в мл), среднее значение концентрации составляет около 10 мкг/мл, причем у людей с группой крови 0(I) оно на 20–25% ниже [108, 109]. Десмопрессин, аналог вазопрессина с заменой L-аргинина на D-аргинин, действующий через V2-рецепторы [110], вызывает практически немедленный 2-кратный подъем уровня антигена фВ в плазме крови человека, сохраняющийся в течение не менее 2 ч и возвращающийся к норме через 24 ч. Введение здоровым донорам гистамина или эндотелина-1 повышало уровень фВ в плазме на 11% [111] и 19% [112], соответственно. Сильно выраженные изменения наблюдаются при системном воспалении, вызванном внутривенным введением 2 нг/кг эндотоксина здоровым добровольцам. Через 4 ч уровень антигена фВ возрастал на 259%, через 24 ч на 192% и через 7 дней возвращался к норме [113]. Было исследовано, как агонисты, активирующие эндотелиальные клетки, влияют на секрецию других белков, находящихся в тельцах Вейбеля–Паладе. Поступление в кровь протегерина не изменяется при введении десмопрессина и возрастает в 2 раза в ответ на липополисахарид [114]. Это говорит о том, что десмопрессин и липополисахарид in vivo вызывают экзоцитоз из разных популяций телец Вейбеля–Паладе. Десмопрессин стимулирует секрецию фВ в кровь, но при этом не вызывает поступления в кровь Р-селектина [107], что также подтверждает действие этого агониста на отдельную популяцию телец Вейбеля–Паладе.
Продолжительность жизни фВ варьирует от 4.2 до 26 ч (меньше у группы 0(I), чем у АВ(IV)) [35]. Предполагалось, что выведение фВ из плазмы происходит после его расщепления металлопротеазой ADAMTS13. Однако разными способами было показано, что это не так. У пациентов с болезнью Виллебранда 1-типа или с гемофилией А скорость снижения уровня фВ после введения десмопрессина не зависит от активности ADAMTS13 [115]. Опыты на мышах ADAMTS-13+/+ и ADAMTS-13–/– также показали, что скорость выведения фВ не зависит от присутствия этого фермента в крови [116].
Согласно имеющимся данным, клиренс фВ происходит в результате его захвата макрофагами, эндотелиальными клетками синусов печени и гепатоцитами (см. обзор [117]), причем основную роль играют макрофаги. При введении мышам в кровь радиоактивно меченного фВ он накапливается большей частью в печени и в значительно меньшем количестве в селезенке и почках [118, 119]. Выведение крупных и мелких мультимеров проходило с одинаковой скоростью.
При захвате макрофагами, эндотелиоцитами и гепатоцитами фВ он сначала связывается рецепторами на плазматической мембране. Присоединение фВ осуществляют два типа рецепторов – лектины и скэвенджер-рецепторы (scavenger receptors) [117]. Связывание фВ лектинами и последующий клиренс зависят от наличия сиаловой кислоты в гликозильных остатках. Сиаловая кислота защищает фВ от захвата отдельными видами лектиновых рецепторов [120–122]. В макрофагах фВ с десиалированными гликанами связывается с рецептором MGL (macrophage galactose-type lectin) [123], в гепатоцитах – с рецептором AMR (Ashwell-Morell receptor) [124, 125]. Значение MGL для клиренса фВ является преобладающим. Рецепторы MGL и AMR связываются с β-D-галактозой и N-ацетил-D-галактозамином. Эти углеводные остатки демаскируются после отщепления сиаловой кислоты при старении фВ. Рецептор MGL присоединяет также гипосиалированный фВ [123, 126]. Помимо MGL и AMR в макрофагах экспрессируется рецептор лектинового типа, связывающий сиалированный фВ. Это лектин Siglec-5 (sialic-acid binding immunoglobulin-like lectin) [127, 128]. Он присутствует также в нейтрофилах и Т-лимфоцитах [129]. Рецептор фВ CLEC4M (C-type lectin domain family 4 member M), специфически связывающий маннозу [130], экспрессируется в эндотелиальных клетках синусоидов печени и в лимфатических узлах [131].
К скэвенджер-рецепторам, осуществляющим захват фВ, относятся рецепторы LRP1 (lipoprotein receptor-related protein-1) [132] и SR-A1 (scavenger receptor class A member I) [133] в макрофагах и стабилин-2 в эндотелиальных клетках синусоидов печени [134]. фВ связывается с LRP1 участками, расположенными в доменах А1А2А3 [135]. Наличие N-гликанов защищает фВ от фагоцитоза макрофагами печени и селезенки, опосредованного рецепторами LRP1. В регуляции связывания фВ с SR-A1 участвуют участок пептидной цепи доменов D'D3, домен A1 и домен D4 [136].
Вторым хранилищем фВ в крови являются тромбоциты. Содержание фВ в тромбоцитах составляет 15–20% от общего количества фВ в крови или в пересчете на количество клеток 2.8 мкг в 109 тромбоцитов [99]. фВ секретируется из альфа-гранул при активации тромбоцитов под действием ADP, коллагена, тромбина [137]. В экспериментах со свиньями показано, что секреция фВ из тромбоцитов не оказывает заметного влияния на общий уровень фВ в плазме крови [98]. Секретируемый из альфа-гранул фВ при наличии в среде ионов кальция связывается с тромбоцитами [99, 100]. Количество секретируемого тромбоцитами фВ можно определить, если в среду добавлять хелатор двухвалентных катионов. В бескальциевой среде в условиях in vitro концентрация свободного фВ при активации тромбоцитов в суспензии может подниматься до 50 МЕ/дл или 5 мкг/мл [138]. Тромбоцитарный фВ играет важную роль в адгезии тромбоцитов к коллагену в условиях напряжения сдвига [98]. В экспериментах со свиньями показано, что тромбоцитарный фВ, в отличие от фВ, циркулирующего в плазме, не принимает непосредственного участия в образовании артериальных тромбов [139].
НАРУШЕНИЯ ОБМЕНА ФАКТОРА ВИЛЛЕБРАНДА КАК ПРИЧИНА ПАТОЛОГИЧЕСКОГО КРОВОТЕЧЕНИЯ И ТРОМБОЗОВ
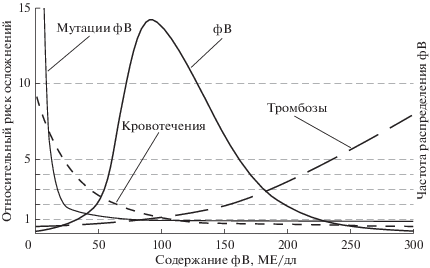
Кривая частоты распределения фВ по содержанию в плазме крови людей (рис. 2) имеет несимметричную колоколообразную форму с расположением 95% всех значений в приблизительном интервале от 50 до 200 МЕ/дл [104]. Популяционный анализ показывает, что при крайне высоких и крайне низких значениях содержания фВ повышен риск тромбозов и кровотечений соответственно. Как отмечено выше, средний уровень фВ в плазме для людей с разными группами крови различается, и это не позволяет строго определить те границы, в которые укладываются относительно нормальные значения этого показателя. Выявлены мутации в гене фВ, которые приводят к снижению продукции фВ. Примерная зависимость между наличием мутаций и содержанием антигена фВ в плазме показана тонкой линией (рис. 2). Как будет показано ниже, более объективным критерием для определения патологического состояния является определение мультимерного состава фВ и его активности в тесте агглютинации с ристоцетином [140].
Рис. 2.
Связь между содержанием фВ в плазме и риском развития кровотечений и тромбозов и мутациями в гене фВ. Прерывистые линии отражают связь между содержанием антигена фВ в плазме и относительным риском кровотечений или тромбозов. Тонкой сплошной линией показана зависимость между наличием мутаций в гене фВ и продукцией фВ. Относительный риск кровотечений или тромбозов принят за единицу при содержании фВ 100 МЕ/дл. Адаптировано из [106].
БОЛЕЗНЬ ВИЛЛЕБРАНДА
Причинами болезни Виллебранда являются сниженное содержание фВ в плазме крови или структурно-функциональные нарушения в молекуле фВ [29, 140]. Болезнь Виллебранда – это наиболее распространенное наследственное нарушение свертывания крови. Для него характерны частые гематомы (подкожные кровоизлияния), аномально длительные кровотечения после незначительных травм и кровоточивость поверхностей слизистых оболочек, включая слизистую желудочно-кишечного тракта. Известно по меньшей мере 20 разновидностей болезни Виллебранда [29]. Они подразделяются на 3 типа. Признаком первого типа является снижение содержания фВ в плазме крови – менее 30 мкг/мл [141]. Соотношение мультимеров фВ при первом типе болезни Виллебранда соответствует норме (рис. 3а). Причиной сниженного содержания фВ может быть уменьшение синтеза/секреции фВ [142] или более быстрый клиренс фВ [141]. Для 1-го типа болезни Виллебранда в основном характерно аутосомно-доминантное наследование. Выявлено более 10 мутаций, вызывающих развитие этой формы заболевания [143]. Большинство этих мутаций вызывают снижение секреции фВ эндотелием.
Рис. 3.
Изменения мультимерного состава фактора Виллебранда, характерные для болезни Виллебранда I (а), II (б) и III (в) типа. К – мультимеры фВ в плазме здоровых доноров. Электрофорез проводили в 1.5% агарозе, переносили белки на нитроцеллюлозную мембрану, инкубировали с антителами против фВ и вторыми антителами, конъюгированными с пероксидазой хрена. Проявление проводили с помощью дитионитробензоата.
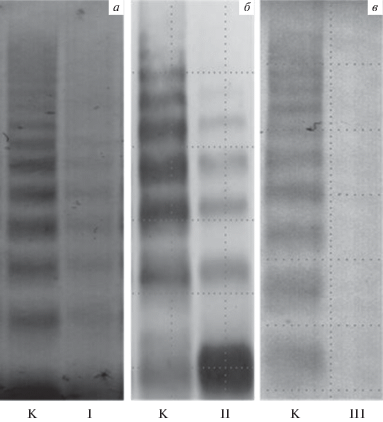
При третьем типе болезни Виллебранда, который наследуется аутосомно-рецессивно, фВ в плазме практически полностью отсутствует (рис. 3в). Концентрация фВ в плазме меньше 3 мкг/мл рассматривается как диагностический признак 3-го типа болезни Виллебранда. При 3-м типе болезни Виллебранда резко снижено содержание фактора VIII свертывание крови. Известно более 80 мутаций разного характера, приводящих к этому типу данной патологии [144].
Болезнь Виллебранда 2-го типа вызвана мутациями, приводящими к структурным и функциональным изменениям фВ. Мультимерный состав фВ при разных видах болезни Виллебранда 2-го типа представлен в ряде публикаций [145–147]. Есть следующие подтипы 2-го типа болезни Виллебранда – 2A (аутосомно-доминантный и аутосомно-рецессивный), 2B (аутосомно-доминантный), 2M (аутосомно-доминантный) и 2N (аутосомно-рецессивный) [148]. При 2А, 2В и 2N подтипах в плазме снижено относительное содержание сверхкрупных и крупных мультимеров фВ. Пример мультимерного состава фВ при 2-м типе болезни Виллебранда представлен на рис. 3б. Классификация наследственных форм болезни Виллебранда рассмотрена в сводке данных международных исследований [149].
Тип 2А характеризуется сниженным сродством фВ к тромбоцитам вследствие падения уровня высокомолекулярных мультимеров фВ в плазме. Эта патология ассоциирована с более чем 50 различными миссенс-мутациями, которые приводят к двум типам патогенетических механизмов: нарушению биосинтеза димера или мультимера фВ (мутации группы 1), либо к биосинтезу белка с повышенной чувствительностью к протеолизу металлопротеазой ADAMTS13 (мутации группы 2) [144]. Мутации 1 группы приводят к аминокислотным заменам в пропептиде фВ, в доменах D3 и A2 и на C-конце зрелой субъединицы фВ. Мутации, приводящие к ускоренному протеолизу фВ, локализованы в участке экзона 28, кодирующем А2-домен.
Тип 2В болезни Виллебранда включает патологические формы, при которых повышено сродство фВ к гликопротеину 1В тромбоцитов. Мутации вызывают аминокислотные замены в домене А1 и также локализованы в домене 28 [144]. При 2В-типе происходит спонтанное связывание фВ с тромбоцитами и выведение крупных мультимеров из кровотока.
Тип 2M включает варианты фВ со сниженным сродством к тромбоцитам при нормальном соотношении мультимеров. Это обусловлено аминокислотной заменой, нарушающей фолдинг петли домена А1 и тем самым приводящей к снижению сродства к гликопротеину 1b тромбоцитов [150].
Тип 2N (Normandy) вызван мутациями в сайте связывания фактора VIII, расположенном в доменах D' и D3 на N-конце молекулы фВ [151]. Наиболее частая причина этого фенотипа – мутация R854Q. При 2N-типе отсутствуют минорные полосы в триплетах мультимеров фВ, снижена секреция и содержание крупных мультимеров.
Количественное соотношение больных разными типами болезни Виллебранда было проанализировано во Франции (общее число установленных случаев 1167 из 670 семей). Показано, что 25% составляют больные с типом 1, 8% типом 3, 66% типом 2 (2A: 18%, 2B: 17%, 2M: 19%, 2N: 12%) и 1% больных с неопределенным типом [152].
Приобретенная форма болезни Виллебранда в ряде случаев вызвана расщеплением сверхкрупных мультимеров фВ в условиях высокого напряжения сдвига [153]. По мультимерному составу фВ данная форма похожа на болезнь Виллебранда 2А-типа. Эта разновидность приобретенной болезни Виллебранда наблюдается при стенозе аорты и сопровождается кровотечением в желудочно-кишечном тракте [154]. Снижение содержания фВ может быть вызвано образованием аутоантител, причем в части случаев они избирательно направлены на фВ в активированном состоянии [155]. Приобретенная форма болезни Виллебранда бывает ассоциирована с лимфопролиферативными и миелопролиферативными заболеваниями.
ОБМЕН ФАКТОРА ВИЛЛЕБРАНДА ПРИ ТРОМБОТИЧЕСКИХ МИКРОАНГИОПАТИЯХ
Повышенная активность фВ в плазме крови, вызванная увеличением доли тромбогенных высокомолекулярных мультимеров фВ при нормальном уровне его секреции или обусловленная общим ростом поступления фВ в плазму из эндотелия, является причиной тромбозов при тромботических микроангиопатиях [106, 156]. Тромботические микроангиопатии (ТМА) включают наследственную (синдром Апшоу–Шульмана) или аутоиммунную формы тромботической тромбоцитопенической пурпуры (ТТП), ТМА, вызванные раком, инфекциями, трансплантацией, химиотерапией и некоторыми лекарствами, а также гемолитико-уремический синдром (ГУС), ассоциированный с инфекцией кишечной палочкой, производящей Shiga-токсин, и атипичный ГУС, вызванный наследственными или приобретенными нарушениями в системе комплемента [157]. При беременности и в послеродовой период встречаются случаи ТМА, ассоциированной с HELLP-синдромом и преэклампсией [158]. При аутоиммуной ТТП активность ADAMTS13 падает ниже 10% от нормального уровня. Снижение вызвано выработкой ингибиторных аутоантител против ADAMTS13. При других формах ТМА уровень активности ADAMTS13 не падает ниже 20% [159]. Мы исследовали мультимерный состав фВ у больной с ТМА неясной этиологии и у больной с аутоиммунной ТТП, развившейся на поздних сроках беременности и после родоразрешения, а также у онкологического больного в терминальной стадии (рис. 4). Активности ADAMTS13 у больных с ТМА и аутоиммунной ТТП составляли соответственно 53% и менее 5% от нормы (рис. 5а). Эти показатели были определены в остром периоде болезни. У больной с ТМА в плазме крови наблюдалось заметное увеличение содержания фВ, что видно по яркости полос мультимеров фВ (рис. 4а). Нарастание уровня фВ в плазме данной больной, по всей вероятности, есть результат его повышенного производства, вызванного воспалительной реакцией в эндотелии. Но при этом отчетливо видно, что относительное содержание сверхкрупных мультимеров резко снижено. Причиной уменьшения доли сверхкрупных мультимеров на фоне общего повышенного уровня антигена фВ является потребление их во время тромбообразования в микрососудах. Аналогичная картина может наблюдаться при терминальной стадии онкологического заболевания (рис. 4в). В плазме больного было существенно повышено содержание антигена фВ, но при этом доля тромбогенных сверхкрупных мультимеров фВ резко снижена (рис. 4в).
Рис. 4.
Мультимерный состав фВ в плазме крови больной с ТМА неясной природы (а), больной с аутоиммунной ТТП (б), онкологического больного в терминальной стадии (в). Б – плазмы больных, К – контрольные плазмы здоровых доноров. Б', Б'', Б''' – плазма больной до и через 10 и 20 дней после начала плазмообменов. Условия электрофореза и анализа мультимерного состава фВ приведены в подписи к рис. 3.

Рис. 5.
а – Активность ADAMTS13 в плазме крови больной с ТМА неясной этиологии и больной с ТТП. б – Содержание антигена фВ и активность фВ в плазме больной с ТТП до начала (0) и через 10, 20 и 75 дней после начала плазмообменов. Активности ADAMTS13 и фВ выражены в % от среднего значения этих показателей у здоровых доноров. Активность фВ определяли в тесте агрегации лиофилизированных тромбоцитов с ристоцетином с использованием агрегометра НПФ “Биола” и диагностического набора НПО “Ренам”.
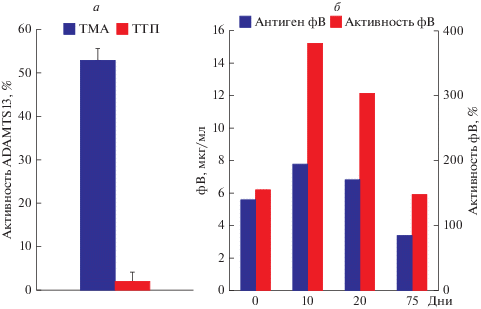
На рис. 4б приведены электрофореграммы мультимеров фВ в плазме больной с аутоимунной ТТП на высоте клинических проявлений и через 10 и 20 дней после проведения интенсивных плазмообменов. Клинические картина ТТП проявилась в конце беременности и нарастала после родоразрешения: гемолитический криз с активностью лактатдегидрогеназы (ЛДГ), превышающей 20 норм, тромбоцитопения, шистоцитоз в мазке крови, неврологические нарушения завершились комой. В этот момент анализ мультимерного состава фВ показал снижение относительного содержания сверхкрупных и крупных мультимеров (рис. 4б). Однако при этом не было повышения уровня антигена фВ по сравнению с контролем, как у больной с ТМА (рис. 4а). Наоборот, содержание антигена фВ в острой фазе не превышало средний уровень в норме, который составляет, по разным данным, от 6.1 [107] до 10 мкг/мл [106]. Через 10 дней после эффективных плазмообменов стали заметны изменения мультимерного состава фВ. Появились полосы сверхкрупных мультимеров, выросло их относительное содержание, что было расценено как торможение тромбообразования. Об этом же говорило прекращение гемолиза, увеличение числа тромбоцитов в крови больной от 20–30 × 109 до 120 × 109/л, нормализация активности ЛДГ. Через 20 дней после начала плазмообменов мультимерный состав фВ еще более приблизился к норме. Яркость полос уменьшилась, что свидетельствовало о понижении содержания антигена фВ, но сохранялся повышенный уровень сверхкрупных мультимеров. Изменения мультимерного состава фВ коррелировали с данными количественного определения антигена и активности фВ (рис. 5б). Как уже отмечено выше, в момент пика болезни содержание в плазме антигена фВ и активность фВ, которую определяли в тесте агглютинации с ристоцетином, практически не превышали норму. Небольшой рост концентрации антигена фВ происходил через 10 дней после начала плазмообменов. Но в это же время происходило резкое увеличение активности фВ, что отражает появление в плазме тромбогенных сверхкрупных мультимеров. Через 20 дней отмечалось постепенное снижение активности фВ, спустя 2.5 месяца активность фВ вернулась к исходной практически нормальной величине, а содержание антигена опустилось ниже первоначального уровня.
Общим признаком всех трех описанных патологических состояний является снижение в момент обострения болезни доли сверхкрупных мультимеров фВ, которые потребляются в процессах тромбообразования. Причины исходного повышения уровня сверхкрупных тромбогенных мультимеров фВ различны. При аутоиммунной ТТП количество секретируемого фВ, судя по приведенным данным, не превышает норму, но на фоне нулевой активности ADAMTS13 система менее устойчива, так как исходно соотношение мультимеров сдвинуто в сторону сверхкрупных форм, и тромбозы инициируются легче. При ТМА неясной этиологии и при онкологическом заболевании в финальной стадии, по-видимому, происходит гиперпродукция фВ. Остается не ясным, что является триггером, запускающим тромбообразование, поскольку для этого недостаточно только наличия сверхкрупных мультимеров. Есть данные о связи тромбозов и воспалительных процессов в сосудах [74]. В частности, известно, что фактор комплемента С5а вызывает экзоцитоз Р-селектина и фВ [160]. Однако обсуждение данной проблемы выходит за рамки настоящего обзора.
ЗАКЛЮЧЕНИЕ
Прошло без малого 100 лет с открытия заболеваний, связанных с нарушением обмена фактора Виллебранда. С тех пор показано, что этот уникальный гликопротеин, имеющий много мультимерных форм, различающийся у разных людей по характеру гликозилирования и полиморфизму, играет очень важную роль в реализации нормальных функций организма и в патогенезе многих болезней [161]. Исследования фактора Виллебранда интенсивно проводятся в течение последних десятков лет. Охарактеризованы генетические формы и пути превращения фВ в плазме крови, выявлено, как эти процессы связаны с патогенезом отдельных разновидностей болезни Виллебранда и аутоиммунной тромботической тромбоцитопенической пурпуры. Наименее исследованным представляется вопрос о том, чем определяется избыточная секреция фВ, которая является, по-видимому, одной из причин крайне тяжелых и не всегда поддающихся эффективному лечению форм тромботической микроангиопатии. И кроме того, требует изучения вопрос о том, какие дополнительные факторы инициируют тромбозы при повышенной активности фВ в плазме.
Источники финансирования. Авторы благодарны проф. А.В. Мазурову за ценные замечания при обсуждении данной статьи и Е.Е. Ефремову за помощь при определении содержания антигена фВ в плазме крови. Работа выполнена при поддержке Российского научного фонда (грант № 21-15-00441).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Соблюдение этических норм. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
von Willebrand A. 1926. Hereditar pseudohemofili. Finska Lakarsallskapetes Handl. 67, 7–112.
Favaloro E. J. 2014. Diagnosing von Willebrand disease: A short history of laboratory milestones and innovations, plus current status, challenges, and solutions. Semin Thromb Hemost. 40, 551–570. https://doi.org/10.1055/s-0034-1383546
Nilsson I.M., Blomback M., Jorpes E., Blomback B., Johansson S.A. 1957. Von Willebrand’s disease and its correction with human plasma fraction 1-0. Acta Med. Scand. 159, 179–188. https://doi.org/10.1111/j.0954-6820.1957.tb00123.x
Cornu P., Larrieu M.J., Caen J., Bernard J. 1963. Transfusion studies in von Willebrand’s disease: Effect on bleeding time and factor VIII. Br. J. Haematol. 9, 189–202. https://doi.org/10.1111/j.1365-2141.1963.tb05457.x
Hellem A.J. 1960. The adhesiveness of human blood platelets in vitro. Scand. J. Clin. Lab. Invest. 12 Suppl, 1–117.
Bowie E.J., Owen C.A., Jr., Thompson J.H., Didisheim P. 1969. Platelet adhesiveness in von Willebrand’s disease. Am. J. Clin. Pathol. 52, 69–77. https://doi.org/10.1093/ajcp/52.1.69
Bouma B.N., Van Mourik J.A. 2006. Unraveling the mystery of von Willebrand factor. J. Thromb. Haemost. 4, 489–495. https://doi.org/10.1111/j.1538-7836.2006.01813.x
Bouma B.N., Wiegerinck Y., Sixma J.J., Van Mourik J.A., Mochtar I.A. 1972. Immunological characterization of purified anti-haemophilic factor A (factor VIII) which corrects abnormal platelet retention in Von Willebrand’s disease. Nat. New Biol. 236, 104–106. https://doi.org/10.1038/newbio236104a0
Zimmerman T.S., Ratnoff O.D., Powell A.E. 1971. Immunologic differentiation of classic hemophilia (factor 8 deficiency) and von Willebrand’s dissase, with observations on combined deficiencies of antihemophilic factor and proaccelerin (factor V) and on an acquired circulating anticoagulant against antihemophilic factor. J. Clin. Invest. 50, 244–254. https://doi.org/10.1172/JCI106480
van Mourik J.A., Bouma B.N., LaBruyere W.T., de Graaf S., Mochtar I.A. 1974. Factor VIII, a series of homologous oligomers and a complex of two proteins. Thromb. Res. 4, 155–164. https://doi.org/10.1016/0049-3848(74)90211-4
Zimmerman T.S., Edgington T.S. 1973. Factor VIII coagulant activity and factor VIII-like antigen: Independent molecular entities. J. Exp. Med. 138, 1015–1020. https://doi.org/10.1084/jem.138.4.1015
Meyer D., Obert B., Pietu G., Lavergne J.M., Zimmerman T.S. 1980. Multimeric structure of factor VIII/von Willebrand factor in von Willebrand’s disease. J. Lab. Clin. Med. 95, 590–602.
Ruggeri Z.M., Zimmerman T.S. 1981. The complex multimeric composition of factor VIII/von Willebrand factor. Blood. 57, 1140–1143.
Counts R.B., Paskell S.L., Elgee S.K. 1978. Disulfide bonds and the quaternary structure of factor VIII/von Willebrand factor. J. Clin. Invest. 62, 702–709. https://doi.org/10.1172/JCI109178
Howard M.A., Firkin B.G. 1971. Ristocetin – a new tool in the investigation of platelet aggregation. Thromb. Diath. Haemorrh. 26, 362–369.
Holmberg L., Nilsson I.M. 1972. Genetic variants of von Willebrand’s disease. Br. Med. J. 3, 317–320. https://doi.org/10.1136/bmj.3.5822.317
Berndt M.C., Ward C.M., Booth W.J., Castaldi P.A., Mazurov A.V., Andrews R.K. 1992. Identification of aspartic acid 514 through glutamic acid 542 as a glycoprotein Ib-IX complex receptor recognition sequence in von Willebrand factor. Mechanism of modulation of von Willebrand factor by ristocetin and botrocetin. Biochemistry. 31, 11 144–11 151. https://doi.org/10.1021/bi00160a027
Moschcowitz E. 1925. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: an undescribed disease. Arch. Intern. Med. 36, 89–93.
Moake J.L., Rudy C.K., Troll J.H., Weinstein M.J., Colannino N.M., Azocar J., Seder R.H., Hong S.L., Deykin D. 1982. Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N. Engl. J. Med. 307, 1432–1435. https://doi.org/10.1056/NEJM198212023072306
Furlan M., Robles R., Solenthaler M., Wassmer M., Sandoz P., Lammle B. 1997. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 89, 3097–3103.
Tsai H.M., Lian E.C. 1998. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N. Engl. J. Med. 339, 1585–1594. https://doi.org/10.1056/NEJM199811263392203
Kwaan H.C., Bennett C.L. 2005. Thrombotic thrombocytopenic purpura-2005. Semin. Thromb. Hemost. 31, 611–614. https://doi.org/10.1055/s-2005-925466
Moake J.L., Byrnes J.J., Troll J.H., Rudy C.K., Weinstein M.J., Colannino N.M., Hong S.L. 1984. Abnormal VIII: von Willebrand factor patterns in the plasma of patients with the hemolytic-uremic syndrome. Blood. 64, 592–598.
Terraube V., O’Donnell J.S., Jenkins P.V. 2010. Factor VIII and von Willebrand factor interaction: Biological, clinical and therapeutic importance. Haemophilia. 16, 3–13. https://doi.org/10.1111/j.1365-2516.2009.02005.x
Verweij C.L., de Vries C.J., Distel B., van Zonneveld A.J., van Kessel A.G., van Mourik J.A., Pannekoek H. 1985. Construction of cDNA coding for human von Willebrand factor using antibody probes for colony-screening and mapping of the chromosomal gene. Nucleic Acids Res. 13, 4699–4717. https://doi.org/10.1093/nar/13.13.4699
Mancuso D.J., Tuley E.A., Westfield L.A., Worrall N.K., Shelton-Inloes B.B., Sorace J.M., Alevy Y.G., Sadler J.E. 1989. Structure of the gene for human von Willebrand factor. J. Biol. Chem. 264, 19514–19527.
Collins C.J., Underdahl J.P., Levene R.B., Ravera C.P., Morin M.J., Dombalagian M.J., Ricca G., Livingston D.M., Lynch D.C. 1987. Molecular cloning of the human gene for von Willebrand factor and identification of the transcription initiation site. Proc. Natl. Acad. Sci. USA. 84, 4393–4397. https://doi.org/10.1073/pnas.84.13.4393
Jahroudi N., Lynch D.C. 1994. Endothelial-cell-specific regulation of von Willebrand factor gene expression. Mol. Cell. Biol. 14, 999–1008.
Tuddenham E.G. 1989. von Willebrand factor and its disorders: An overview of recent molecular studies. Blood Rev. 3, 251–262.
Gogia S., Neelamegham S. 2015. Role of fluid shear stress in regulating VWF structure, function and related blood disorders. Biorheology. 52, 319–335. https://doi.org/10.3233/BIR-15061
Lenting P.J., Casari C., Christophe O.D., Denis C.V. 2012. von Willebrand factor: The old, the new and the unknown. J. Thromb. Haemost. 10, 2428–2437. https://doi.org/10.1111/jth.12008
Zenner H.L., Collinson L. M., Michaux G., Cutler D.F. 2007. High-pressure freezing provides insights into Weibel-Palade body biogenesis. J. Cell. Sci. 120, 2117–2125. https://doi.org/10.1242/jcs.007781
Wagner D.D., Marder V.J. 1984. Biosynthesis of von Willebrand protein by human endothelial cells: processing steps and their intracellular localization. J. Cell. Biol. 99, 2123–2130. https://doi.org/10.1083/jcb.99.6.2123
Nightingale T.D., McCormack J.J., Grimes W., Robinson C., Lopes da Silva M., White I.J., Vaughan A., Cramer L.P., Cutler D.F. 2018. Tuning the endothelial response: Differential release of exocytic cargos from Weibel-Palade bodies. J. Thromb. Haemost. 16, 1873–1886. https://doi.org/10.1111/jth.14218
Bryckaert M., Rosa J.P., Denis C.V., Lenting P.J. 2015. Of von Willebrand factor and platelets. Cell. Mol. Life Sci. 72, 307–326. https://doi.org/10.1007/s00018-014-1743-8
Huang R.H., Wang Y., Roth R., Yu X., Purvis A.R., Heuser J.E., Egelman E.H., Sadler J.E. 2008. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc. Natl. Acad. Sci. USA. 105, 482–487. https://doi.org/10.1073/pnas.0710079105
Dong J.F., Whitelock J., Bernardo A., Ball C., Cruz M.A. 2004. Variations among normal individuals in the cleavage of endothelial-derived ultra-large von Willebrand factor under flow. J. Thromb. Haemost. 2, 1460–1466. doi JTH830https://doi.org/10.1111/j.1538-7836.2004.00830.x
De Ceunynck K., De Meyer S.F., Vanhoorelbeke K. 2013. Unwinding the von Willebrand factor strings puzzle. Blood. 121, 270–277. https://doi.org/10.1182/blood-2012-07-442285
Dong J.F., Moake J.L., Nolasco L., Bernardo A., Arceneaux W., Shrimpton C.N., Schade A.J., McIntire L.V., Fujikawa K., Lopez J.A. 2002. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 100, 4033–4039.https://doi.org/10.1182/blood-2002-05-1401
Mourik M., Eikenboom J. 2017. Lifecycle of Weibel-Palade bodies. Hamostaseologie. 37, 13–24. https://doi.org/10.5482/HAMO-16-07-0021
van Breevoort D., van Agtmaal E.L., Dragt B.S., Gebbinck J.K., Dienava-Verdoold I., Kragt A., Bierings R., Horrevoets A.J., Valentijn K.M., Eikenboom J.C., Fernandez-Borja M., Meijer A.B., Voorberg J. 2012. Proteomic screen identifies IGFBP7 as a novel component of endothelial cell-specific Weibel-Palade bodies. J. Proteome Res. 11, 2925–2936. https://doi.org/10.1021/pr300010r
Rondaij M.G., Bierings R., Kragt A., van Mourik J.A., Voorberg J. 2006. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26, 1002–1007. https://doi.org/10.1161/01.ATV.0000209501.56852.6c
Fiedler U., Scharpfenecker M., Koidl S., Hegen A., Grunow V., Schmidt J.M., Kriz W., Thurston G., Augustin H.G. 2004. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 103, 4150–4156. https://doi.org/10.1182/blood-2003-10-3685
Byzova T.V., Romanov Iu.A., Blasik T.N., Mazurov A.V. 1995. Interaction of a monoclonal antibody to P-selectin with activated platelets and endothelial cells. Heterogeneity of expressing P-selectin in human aorta endothelial cells. Biokhimiia (Rus.). 60, 1292–1301.
Lenting P.J., Christophe O.D., Denis C.V. 2015. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 125, 2019–2028. https://doi.org/10.1182/blood-2014-06-528406
Cramer E.M., Meyer D., le Menn R., Breton-Gorius J. 1985. Eccentric localization of von Willebrand factor in an internal structure of platelet alpha-granule resembling that of Weibel-Palade bodies. Blood. 66, 710–713.
Valentijn K.M., van Driel L.F., Mourik M.J., Hendriks G.J., Arends T.J., Koster A.J., Valentijn J.A. 2010. Multigranular exocytosis of Weibel-Palade bodies in vascular endothelial cells. Blood. 116, 1807–1816. https://doi.org/10.1182/blood-2010-03-274209
Valentijn K.M., Eikenboom J. 2013. Weibel-Palade bodies: A window to von Willebrand disease. J. Thromb. Haemost. 11, 581–592. https://doi.org/10.1111/jth.12160
Torisu T., Torisu K., Lee I.H., Liu J., Malide D., Combs C.A., Wu X.S., Rovira, II, Fergusson M.M., Weigert R., Connelly P.S., Daniels M.P., Komatsu M., Cao L., Finkel T. 2013. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 19, 1281–1287. https://doi.org/10.1038/nm.3288
Antonova O.A., Loktionova S.A., Golubeva N.V., Romanov Y.A., Mazurov A.V. 2007. Damage and activation of endothelial cells during in vitro hypoxia. Bull. Exp. Biol. Med. 144, 504–506. https://doi.org/10.1007/s10517-007-0362-x
Okhota S., Melnikov I., Avtaeva Y., Kozlov S., Gabbasov Z. 2020. Shear stress-induced activation of von Willebrand factor and cardiovascular pathology. Int. J. Mol. Sci. 21. https://doi.org/10.3390/ijms21207804
Lopes da Silva M., Cutler D.F. 2016. von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood. 128, 277–285. https://doi.org/10.1182/blood-2015-10-677054
Rondaij M.G., Bierings R., Kragt A., van Mourik J.A., Voorberg J. 2006. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26, 1002–1007. https://doi.org/10.1161/01.ATV.0000209501.56852.6c
Sporn L.A., Marder V.J., Wagner D.D. 1986. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 46, 185–190. https://doi.org/10.1016/0092-8674(86)90735-x
Vischer U.M., Barth H., Wollheim C.B. 2000. Regulated von Willebrand factor secretion is associated with agonist-specific patterns of cytoskeletal remodeling in cultured endothelial cells. Arterioscler. Thromb. Vasc. Biol. 20, 883–891.
Birch K.A., Pober J.S., Zavoico G.B., Means A.R., Ewenstein B.M. 1992. Calcium/calmodulin transduces thrombin-stimulated secretion: studies in intact and minimally permeabilized human umbilical vein endothelial cells. J. Cell. Biol. 118, 1501–1510. https://doi.org/10.1083/jcb.118.6.1501
Vischer U.M., Wollheim C.B. 1997. Epinephrine induces von Willebrand factor release from cultured endothelial cells: Involvement of cyclic AMP-dependent signalling in exocytosis. Thromb. Haemost. 77, 1182–1188.
Kaufmann J.E., Oksche A., Wollheim C.B., Gunther G., Rosenthal W., Vischer U.M. 2000. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J. Clin. Invest. 106, 107–116. https://doi.org/10.1172/JCI9516
Cleator J.H., Zhu W.Q., Vaughan D.E., Hamm H.E. 2006. Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood. 107, 2736–2744. https://doi.org/10.1182/blood-2004-07-2698
Rondaij M.G., Bierings R., van Agtmaal E.L., Gijzen K.A., Sellink E., Kragt A., Ferguson S.S., Mertens K., Hannah M.J., van Mourik J.A., Fernandez-Borja M., Voorberg J. 2008. Guanine exchange factor RalGDS mediates exocytosis of Weibel-Palade bodies from endothelial cells. Blood. 112, 56–63. https://doi.org/10.1182/blood-2007-07-099309
de Leeuw H.P., Wijers-Koster P.M., van Mourik J.A., Voorberg J. 1999. Small GTP-binding protein RalA associates with Weibel-Palade bodies in endothelial cells. Thromb. Haemost. 82, 1177–1181.
Valentijn K.M., Sadler J.E., Valentijn J.A., Voorberg J., Eikenboom J. 2011. Functional architecture of Weibel-Palade bodies. Blood. 117, 5033–5043. https://doi.org/10.1182/blood-2010-09-267492
Kim K.S., Park J.Y., Jou I., Park S.M. 2010. Regulation of Weibel-Palade body exocytosis by alpha-synuclein in endothelial cells. J. Biol. Chem. 285, 21 416–21 425. https://doi.org/10.1074/jbc.M110.103499
Rojo Pulido I., Nightingale T.D., Darchen F., Seabra M.C., Cutler D.F., Gerke V. 2011. Myosin Va acts in concert with Rab27a and MyRIP to regulate acute von-Willebrand factor release from endothelial cells. Traffic. 12, 1371–1382. https://doi.org/10.1111/j.1600-0854.2011.01248.x
Conte I.L., Hellen N., Bierings R., Mashanov G.I., Manneville J.B., Kiskin N.I., Hannah M.J., Molloy J.E., Carter T. 2016. Interaction between MyRIP and the actin cytoskeleton regulates Weibel-Palade body trafficking and exocytosis. J. Cell. Sci. 129, 592–603. https://doi.org/10.1242/jcs.178285
Holthenrich A., Drexler H. C.A., Chehab T., Nass J., Gerke V. 2019. Proximity proteomics of endothelial Weibel-Palade bodies identifies novel regulator of von Willebrand factor secretion. Blood. 134, 979–982. https://doi.org/10.1182/blood.2019000786
Nightingale T.D., Pattni K., Hume A.N., Seabra M.C., Cutler D.F. 2009. Rab27a and MyRIP regulate the amount and multimeric state of VWF released from endothelial cells. Blood. 113, 5010–5018. https://doi.org/10.1182/blood-2008-09-181206
Bierings R., Hellen N., Kiskin N., Knipe L., Fonseca A.V., Patel B., Meli A., Rose M., Hannah M.J., Carter T. 2012. The interplay between the Rab27A effectors Slp4-a and MyRIP controls hormone-evoked Weibel-Palade body exocytosis. Blood. 120, 2757–2767. https://doi.org/10.1182/blood-2012-05-429936
Zografou S., Basagiannis D., Papafotika A., Shirakawa R., Horiuchi H., Auerbach D., Fukuda M., Christoforidis S. 2012. A complete Rab screening reveals novel insights in Weibel-Palade body exocytosis. J. Cell. Sci. 125, 4780–4790. https://doi.org/10.1242/jcs.104174
Schillemans M., Karampini E., van den Eshof B.L., Gangaev A., Hofman M., van Breevoort D., Meems H., Janssen H., Mulder A.A., Jost C.R., Escher J.C., Adam R., Carter T., Koster A.J., van den Biggelaar M., Voorberg J., Bierings R. 2018. Weibel-Palade body localized syntaxin-3 modulates von Willebrand factor secretion from endothelial cells. Arterioscler. Thromb. Vasc. Biol. 38, 1549–1561. https://doi.org/10.1161/ATVBAHA.117.310701
Schillemans M., Karampini E., Hoogendijk A.J., Wahedi M., van Alphen F.P.J., van den Biggelaar M., Voorberg J., Bierings R. 2019. Interaction networks of Weibel-Palade body regulators syntaxin-3 and syntaxin binding protein 5 in endothelial cells. J. Proteomics. 205, 103417. https://doi.org/10.1016/j.jprot.2019.103417
Manneville J.B., Etienne-Manneville S., Skehel P., Carter T., Ogden D., Ferenczi M. 2003. Interaction of the actin cytoskeleton with microtubules regulates secretory organelle movement near the plasma membrane in human endothelial cells. J. Cell. Sci. 116, 3927–3938. https://doi.org/10.1242/jcs.00672
Romani de Wit T., Rondaij M.G., Hordijk P.L., Voorberg J., van Mourik J.A. 2003. Real-time imaging of the dynamics and secretory behavior of Weibel-Palade bodies. Arterioscler. Thromb. Vasc. Biol. 23, 755–761. https://doi.org/10.1161/01.ATV.0000069847.72001.E8
Kawecki C., Lenting P.J., Denis C.V. 2017. von Willebrand factor and inflammation. J. Thromb. Haemost. 15, 1285–1294. https://doi.org/10.1111/jth.13696
Chen J., Chung D.W. 2018. Inflammation, von Willebrand factor, and ADAMTS13. Blood. 132, 141–147. https://doi.org/10.1182/blood-2018-02-769000
Vischer U.M., Jornot L., Wollheim C.B., Theler J.M. 1995. Reactive oxygen intermediates induce regulated secretion of von Willebrand factor from cultured human vascular endothelial cells. Blood. 85, 3164–3172.
Yang S., Zheng Y., Hou X. 2019. Lipoxin A4 restores oxidative stress-induced vascular endothelial cell injury and thrombosis-related factor expression by its receptor-mediated activation of Nrf2-HO-1 axis. Cell. Signal. 60, 146–153. https://doi.org/10.1016/j.cellsig.2019.05.002
Avdonin P.V., Tsitrina A.A., Mironova G.Y., Avdonin P.P., Zharkikh I.L., Nadeev A.D., Goncharov N.V. 2017. Hydrogen peroxide stimulates exocytosis of von Willebrand factor in human umbilical vein endothelial cells. Biol. Bull. 44, 531–537. https://doi.org/10.1134/s106235901705003x
Avdonin P.P., Trufanov S.K., Rybakova E.Y., Tsitrina A.A., Goncharov N.V., Avdonin P.V. 2021. The use of fluorescently labeled ARC1779 aptamer for assessimg the effect of H2O2 on von Willebrand factor exocytosis. Biochemistry (Moscow). 86, 123–131. https://doi.org/10.1134/S0006297921020012
Evangelista A.M., Thompson M.D., Bolotina V.M., Tong X., Cohen R.A. 2012. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Biol. Med. 53, 2327–2334. https://doi.org/10.1016/j.freeradbiomed.2012.10.546
Mo S.J., Son E.W., Rhee D.K., Pyo S. 2003. Modulation of TNF-alpha-induced ICAM-1 expression, NO and H2O2 production by alginate, allicin and ascorbic acid in human endothelial cells. Arch. Pharm. Res. 26, 244–251.
Bernardo A., Ball C., Nolasco L., Moake J.F., Dong J.F. 2004. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 104, 100–106.https://doi.org/10.1182/blood-2004-01-0107
Matsushita K., Yamakuchi M., Morrell C.N., Ozaki M., O’Rourke B., Irani K., Lowenstein C.J. 2005. Vascular endothelial growth factor regulation of Weibel-Palade-body exocytosis. Blood. 105, 207–214. https://doi.org/10.1182/blood-2004-04-1519
Zhou Y.F., Eng E.T., Zhu J., Lu C., Walz T., Springer T.A. 2012. Sequence and structure relationships within von Willebrand factor. Blood. 120, 449–458. https://doi.org/10.1182/blood-2012-01-405134
Zhang X., Halvorsen K., Zhang C.Z., Wong W.P., Springer T.A. 2009. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 324, 1330–1334. https://doi.org/10.1126/science.1170905
Huizinga E.G., Martijn van der Plas R., Kroon J., Sixma J.J., Gros P. 1997. Crystal structure of the A3 domain of human von Willebrand factor: Implications for collagen binding. Structure. 5, 1147–1156. https://doi.org/10.1016/s0969-2126(97)00266-9
Shapiro S.E., Nowak A.A., Wooding C., Birdsey G., Laffan M.A., McKinnon T.A. 2014. The von Willebrand factor predicted unpaired cysteines are essential for secretion. J. Thromb. Haemost. 12, 246–254. https://doi.org/10.1111/jth.12466
Dong X., Leksa N.C., Chhabra E.S., Arndt J.W., Lu Q., Knockenhauer K.E., Peters R.T., Springer T.A. 2019. The von Willebrand factor D'D3 assembly and structural principles for factor VIII binding and concatemer biogenesis. Blood. 133, 1523–1533. https://doi.org/10.1182/blood-2018-10-876300
Yee A., Kretz C.A. 2014. Von Willebrand factor: Form for function. Semin. Thromb. Hemost. 40, 17–27. https://doi.org/10.1055/s-0033-1363155
Ward S., O’Sullivan J.M., O’Donnell J.S. 2019. von Willebrand factor sialylation-A critical regulator of biological function. J. Thromb. Haemost. 17, 1018–1029. https://doi.org/10.1111/jth.14471
Arya M., Anvari B., Romo G.M., Cruz M.A., Dong J.F., McIntire L.V., Moake J.L., Lopez J.A. 2002. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: Studies using optical tweezers. Blood. 99, 3971–3977. https://doi.org/10.1182/blood-2001-11-0060
Pushin D.M., Salikhova T.Y., Zlobina K.E., Guria G.T. 2020. Platelet activation via dynamic conformational changes of von Willebrand factor under shear. PLoS One. 15, e0234501. https://doi.org/10.1371/journal.pone.0234501
Zlobina K.E., Guria G.T. 2016. Platelet activation risk index as a prognostic thrombosis indicator. Sci. Rep. 6, 30508. https://doi.org/10.1038/srep30508
Sadler J.E. 2008. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 112, 11–18. https://doi.org/10.1182/blood-2008-02-078170
Brophy T.M., Ward S.E., McGimsey T.R., Schneppenheim S., Drakeford C., O’Sullivan J.M., Chion A., Budde U., O’Donnell J.S. 2017. Plasmin cleaves von Willebrand factor at K1491-R1492 in the A1-A2 linker region in a shear- and glycan-dependent manner in vitro. Arterioscler. Thromb. Vasc. Biol. 37, 845–855. https://doi.org/10.1161/ATVBAHA.116.308524
Preston R.J., Rawley O., Gleeson E.M., O’Donnell J.S. 2013. Elucidating the role of carbohydrate determinants in regulating hemostasis: insights and opportunities. Blood. 121, 3801–3810. https://doi.org/10.1182/blood-2012-10-415000
Canis K., McKinnon T.A., Nowak A., Haslam S.M., Panico M., Morris H.R., Laffan M. A., Dell A. 2012. Mapping the N-glycome of human von Willebrand factor. Biochem. J. 447, 217–228. https://doi.org/10.1042/BJ20120810
McGrath R.T., McRae E., Smith O.P., O’Donnell J.S. 2010. Platelet von Willebrand factor – structure, function and biological importance. Br. J. Haematol. 148, 834–843. https://doi.org/10.1111/j.1365-2141.2009.08052.x
Schmugge M., Rand M.L., Freedman J. 2003. Platelets and von Willebrand factor. Transfus. Apher. Sci. 28, 269–277. https://doi.org/10.1016/S1473-0502(03)00046-6
Furby F. H., Berndt M. C., Castaldi P. A., Koutts J. 1984. Characterization of calcium-dependent binding of endogenous factor VIII/von Willebrand factor to surface activated platelets. Thromb. Res. 35, 501–511. https://doi.org/10.1016/0049-3848(84)90282-2
Andrews R.K., Gorman J.J., Booth W.J., Corino G.L., Castaldi P.A., Berndt M.C. 1989. Cross-linking of a monomeric 39/34-kDa dispase fragment of von Willebrand factor (Leu-480/Val-481-Gly-718) to the N-terminal region of the alpha-chain of membrane glycoprotein Ib on intact platelets with bis(sulfosuccinimidyl) suberate. Biochemistry. 28, 8326–8336. https://doi.org/10.1021/bi00447a010
Yakushkin V.V., Zyuryaev I.T., Khaspekova S.G., Sirotkina O.V., Ruda M.Y., Mazurov A.V. 2011. Glycoprotein IIb-IIIa content and platelet aggregation in healthy volunteers and patients with acute coronary syndrome. Platelets. 22, 243–251. https://doi.org/10.3109/09537104.2010.547959
Naimushin Y.A., Mazurov A.V. 2004. Von Willebrand factor can support platelet aggregation via interaction with activated GPIIb-IIIa and GPIb. Platelets. 15, 419–425. https://doi.org/10.1080/09537100410001721333
Wu Y.P., Vink T., Schiphorst M., van Zanten G.H., IJsseldijk M.J., de Groot P.G., Sixma J.J. 2000. Platelet thrombus formation on collagen at high shear rates is mediated by von Willebrand factor-glycoprotein Ib interaction and inhibited by von Willebrand factor-glycoprotein IIb/IIIa interaction. Arterioscler. Thromb. Vasc. Biol. 20, 1661–1667. https://doi.org/10.1161/01.atv.20.6.1661
Kaneva V.N., Dunster J.L., Volpert V., Ataullahanov F., Panteleev M.A., Nechipurenko D.Y. 2021. Modeling thrombus shell: Linking adhesion receptor properties and macroscopic dynamics. Biophys. J. 120, 334–351. https://doi.org/10.1016/j.bpj.2020.10.049
Sadler J.E. 2005. New concepts in von Willebrand disease. Annu. Rev. Med. 56, 173–191. https://doi.org/10.1146/annurev.med.56.082103.104713
Jilma B., Eichler H.G., Vondrovec B., Breiteneder H., Kyrle P. A., Kitzweger E., Kapiotis S., Speiser W. 1996. Effects of desmopressin on circulating P-selectin. Br. J. Haematol. 93, 432–436. https://doi.org/10.1046/j.1365-2141.1996.5031040.x
Murphy S.J.X., Lim S.T., Hickey F., Kinsella J.A., Smith D.R., Tierney S., Egan B., Feeley T.M., Murphy S.M., Collins D.R., Coughlan T., O’Neill D., Harbison J.A., Madhavan P., O’Neill S.M., Colgan M.P., O’Donnell J.S., O’Sullivan J.M., Hamilton G., McCabe D.J.H. 2021. von Willebrand factor antigen, von Willebrand factor propeptide, and ADAMTS13 in carotid stenosis and their relationship with cerebral microemboli. Thromb. Haemost. 121, 86–97. https://doi.org/10.1055/s-0040-1715440
Vischer U.M., Ingerslev J., Wollheim C.B., Mestries J.C., Tsakiris D.A., Haefeli W.E., Kruithof E.K. 1997. Acute von Willebrand factor secretion from the endothelium in vivo: Assessment through plasma propeptide (vWf:AgII) levels. Thromb. Haemost. 77, 387–393.
Kaufmann J.E., Vischer U.M. 2003. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J. Thromb. Haemost. 1, 682–689.
Jilma B., Pernerstorfer T., Dirnberger E., Stohlawetz P., Schmetterer L., Singer E.A., Grasseli U., Eichler H.G., Kapiotis S. 1998. Effects of histamine and nitric oxide synthase inhibition on plasma levels of von Willebrand factor antigen. J. Lab. Clin. Med. 131, 151–156. https://doi.org/10.1016/s0022-2143(98)90157-3
Leitner G.C., Schmetterer L., Kapiotis S., Jilma B. 2010. Effects of endothelin-1 and phenylephrine on plasma levels of von Willebrand factor and protein S. Thromb. Res. 125, e5–8. https://doi.org/10.1016/j.thromres.2009.08.004
Reiter R.A., Varadi K., Turecek P.L., Jilma B., Knobl P. 2005. Changes in ADAMTS13 (von-Willebrand-factor-cleaving protease) activity after induced release of von Willebrand factor during acute systemic inflammation. Thromb. Haemost. 93, 554–558. https://doi.org/10.1160/TH04-08-0467
Buchtele N., Kovacevic K.D., Brostjan C., Schwameis M., Hayden H., Derhaschnig U., Firbas C., Jilma B., Schoergenhofer C. 2020. Differential osteoprotegerin kinetics after stimulation with desmopressin and lipopolysaccharides in vivo. Thromb. Haemost. 120, 1108–1115. https://doi.org/10.1055/s-0040-1712448
Millar C.M., Riddell A.F., Brown S.A., Starke R., Mackie I., Bowen D.J., Jenkins P.V., van Mourik J.A. 2008. Survival of von Willebrand factor released following DDAVP in a type 1 von Willebrand disease cohort: Influence of glycosylation, proteolysis and gene mutations. Thromb. Haemost. 99, 916–924. https://doi.org/10.1160/TH07-09-0565
Badirou I., Kurdi M., Rayes J., Legendre P., Christophe O.D., Lenting P.J., Denis C.V. 2010. von Willebrand factor clearance does not involve proteolysis by ADAMTS-13. J. Thromb. Haemost. 8, 2338–2340. https://doi.org/10.1111/j.1538-7836.2010.04012.x
O'Sullivan J.M., Ward S., Lavin M., O’Donnell J.S. 2018. von Willebrand factor clearance – biological mechanisms and clinical significance. Br. J. Haematol. 183, 185–195. https://doi.org/10.1111/bjh.15565
Lenting P.J., Westein E., Terraube V., Ribba A.S., Huizinga E.G., Meyer D., de Groot P.G., Denis C.V. 2004. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J. Biol. Chem. 279, 12102–12109. https://doi.org/10.1074/jbc.M310436200
van Schooten C.J., Shahbazi S., Groot E., Oortwijn B.D., van den Berg H.M., Denis C.V., Lenting P.J. 2008. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 112, 1704–1712. https://doi.org/10.1182/blood-2008-01-133181
Chion A., O’Sullivan J.M., Drakeford C., Bergsson G., Dalton N., Aguila S., Ward S., Fallon P.G., Brophy T.M., Preston R.J., Brady L., Sheils O., Laffan M., McKinnon T.A., O’Donnell J.S. 2016. N-linked glycans within the A2 domain of von Willebrand factor modulate macrophage-mediated clearance. Blood. 128, 1959–1968. https://doi.org/10.1182/blood-2016-04-709436
van Schooten C.J., Denis C.V., Lisman T., Eikenboom J.C., Leebeek F.W., Goudemand J., Fressinaud E., van den Berg H.M., de Groot P.G., Lenting P.J. 2007. Variations in glycosylation of von Willebrand factor with O-linked sialylated T antigen are associated with its plasma levels. Blood. 109, 2430–2437. https://doi.org/10.1182/blood-2006-06-032706
Sodetz J.M., Pizzo S.V., McKee P.A. 1977. Relationship of sialic acid to function and in vivo survival of human factor VIII/von Willebrand factor protein. J. Biol. Chem. 252, 5538–5546.
Ward S.E., O’Sullivan J.M., Drakeford C., Aguila S., Jondle C.N., Sharma J., Fallon P.G., Brophy T.M., Preston R.J.S., Smyth P., Sheils O., Chion A., O’Donnell J.S. 2018. A novel role for the macrophage galactose-type lectin receptor in mediating von Willebrand factor clearance. Blood. 131, 911–916. https://doi.org/10.1182/blood-2017-06-787853
Grewal P.K. 2010. The Ashwell-Morell receptor. Methods Enzymol. 479, 223–241. https://doi.org/10.1016/S0076-6879(10)79013-3
Grewal P.K., Uchiyama S., Ditto D., Varki N., Le D.T., Nizet V., Marth J.D. 2008. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat. Med. 14, 648–655. https://doi.org/10.1038/nm1760
Denis C.V., Lenting P.J. 2018. VWF clearance: It’s glycomplicated. Blood. 131, 842–843. https://doi.org/10.1182/blood-2018-01-824904
Pegon J.N., Kurdi M., Casari C., Odouard S., Denis C.V., Christophe O.D., Lenting P.J. 2012. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica. 97, 1855–1863. https://doi.org/10.3324/haematol.2012.063297
Lock K., Zhang J., Lu J., Lee S.H., Crocker P.R. 2004. Expression of CD33-related siglecs on human mononuclear phagocytes, monocyte-derived dendritic cells and plasmacytoid dendritic cells. Immunobiology. 209, 199–207. https://doi.org/10.1016/j.imbio.2004.04.007
Jandus C., Simon H.U., von Gunten S. 2011. Targeting siglecs – a novel pharmacological strategy for immuno- and glycotherapy. Biochem. Pharmacol. 82, 323–332. https://doi.org/10.1016/j.bcp.2011.05.018
Rydz N., Swystun L.L., Notley C., Paterson A.D., Riches J.J., Sponagle K., Boonyawat B., Montgomery R.R., James P.D., Lillicrap D. 2013. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. 121, 5228–5237. https://doi.org/10.1182/blood-2012-10-457507
Khoo U.S., Chan K.Y., Chan V.S., Lin C.L. 2008. DC-SIGN and L-SIGN: The SIGNs for infection. J. Mol. Med. (Berl.). 86, 861–874. https://doi.org/10.1007/s00109-008-0350-2
Rastegarlari G., Pegon J.N., Casari C., Odouard S., Navarrete A.M., Saint-Lu N., van Vlijmen B.J., Legendre P., Christophe O.D., Denis C.V., Lenting P.J. 2012. Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood. 119, 2126–2134. https://doi.org/10.1182/blood-2011-08-373605
Zani I.A., Stephen S.L., Mughal N.A., Russell D., Homer-Vanniasinkam S., Wheatcroft S.B., Ponnambalam S. 2015. Scavenger receptor structure and function in health and disease. Cells. 4, 178–201. https://doi.org/10.3390/cells4020178
Swystun L.L., Lai J.D., Notley C., Georgescu I., Paine A.S., Mewburn J., Nesbitt K., Schledzewski K., Geraud C., Kzhyshkowska J., Goerdt S., Hopman W., Montgomery R.R., James P.D., Lillicrap D. 2018. The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J. Clin. Invest. 128, 4057–4073. https://doi.org/10.1172/JCI96400
Fazavana J., Brophy T.M., Chion A., Cooke N., Terraube V., Cohen J., Parng C., Pittman D., Cunningham O., Lambert M., O’Donnell J.S., O’Sullivan J.M. 2020. Investigating the clearance of VWF A-domains using site-directed PEGylation and novel N-linked glycosylation. J. Thromb. Haemost. 18, 1278–1290. https://doi.org/10.1111/jth.14785
Wohner N., Muczynski V., Mohamadi A., Legendre P., Proulle V., Ayme G., Christophe O.D., Lenting P.J., Denis C.V., Casari C. 2018. Macrophage scavenger receptor SR-AI contributes to the clearance of von Willebrand factor. Haematologica. 103, 728–737. https://doi.org/10.3324/haematol.2017.175216
Koutts J., Walsh P.N., Plow E.F., Fenton J.W., 2nd, Bouma B.N., Zimmerman T.S. 1978. Active release of human platelet factor VIII-related antigen by adenosine diphosphate, collagen, and thrombin. J. Clin. Invest. 62, 1255–1263. https://doi.org/10.1172/JCI109246
Fernandez M.F., Ginsberg M.H., Ruggeri Z.M., Batlle F.J., Zimmerman T.S. 1982. Multimeric structure of platelet factor VIII/von Willebrand factor: The presence of larger multimers and their reassociation with thrombin-stimulated platelets. Blood. 60, 1132–1138.
Nichols T.C., Bellinger D.A., Reddick R.L., Read M.S., Koch G.G., Brinkhous K.M., Griggs T.R. 1991. Role of von Willebrand factor in arterial thrombosis. Studies in normal and von Willebrand disease pigs. Circulation. 83, IV56–64.
Keesler D.A., Flood V.H. 2018. Current issues in diagnosis and treatment of von Willebrand disease. Res. Pract. Thromb. Haemost. 2, 34–41. https://doi.org/10.1002/rth2.12064
O'Donnell J.S. 2020. Low VWF: Insights into pathogenesis, diagnosis, and clinical management. Blood Adv. 4, 3191–3199. https://doi.org/10.1182/bloodadvances.2020002038
Eikenboom J.C., Matsushita T., Reitsma P.H., Tuley E.A., Castaman G., Briet E., Sadler J.E. 1996. Dominant type 1 von Willebrand disease caused by mutated cysteine residues in the D3 domain of von Willebrand factor. Blood. 88, 2433–2441.
Eikenboom J., Hilbert L., Ribba A.S., Hommais A., Habart D., Messenger S., Al-Buhairan A., Guilliatt A., Lester W., Mazurier C., Meyer D., Fressinaud E., Budde U., Will K., Schneppenheim R., Obser T., Marggraf O., Eckert E., Castaman G., Rodeghiero F., Federici A.B., Batlle J., Goudemand J., Ingerslev J., Lethagen S., Hill F., Peake I., Goodeve A. 2009. Expression of 14 von Willebrand factor mutations identified in patients with type 1 von Willebrand disease from the MCMDM-1VWD study. J. Thromb. Haemost. 7, 1304–1312. https://doi.org/10.1111/j.1538-7836.2009.03486.x
James P., Lillicrap D. 2008. The role of molecular genetics in diagnosing von Willebrand disease. Semin. Thromb. Hemost. 34, 502–508. https://doi.org/10.1055/s-0028-1103361
Zimmerman T.S., Dent J.A., Ruggeri Z.M., Nannini L.H. 1986. Subunit composition of plasma von Willebrand factor. Cleavage is present in normal individuals, increased in IIA and IIB von Willebrand disease, but minimal in variants with aberrant structure of individual oligomers (types IIC, IID, and IIE). J. Clin. Invest. 77, 947–951. https://doi.org/10.1172/JCI112394
Schneppenheim R., Budde U., Ruggeri Z.M. 2001. A molecular approach to the classification of von Willebrand disease. Best Pract. Res. Clin. Haematol. 14, 281–298. https://doi.org/10.1053/beha.2001.0134
Schneppenheim R., Budde U. 2005. Phenotypic and genotypic diagnosis of von Willebrand disease: A 2004 update. Semin. Hematol. 42, 15–28. https://doi.org/10.1053/j.seminhematol.2004.10.002
Fogarty H., Doherty D., O’Donnell J. S. 2020. New developments in von Willebrand disease. Br. J. Haematol. 191, 329–339. https://doi.org/10.1111/bjh.16681
Sadler J.E., Budde U., Eikenboom J.C., Favaloro E.J., Hill F.G., Holmberg L., Ingerslev J., Lee C.A., Lillicrap D., Mannucci P.M., Mazurier C., Meyer D., Nichols W.L., Nishino M., Peake I.R., Rodeghiero F., Schneppenheim R., Ruggeri Z.M., Srivastava A., Montgomery R.R., Federici A.B. 2006. Update on the pathophysiology and classification of von Willebrand disease: A report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 4, 2103–2114.
Stepanian A., Ribba A.S., Lavergne J.M., Fressinaud E., Juhan-Vague I., Mazurier C., Girma J.P., Meyer D. 2003. A new mutation, S1285F, within the A1 loop of von Willebrand factor induces a conformational change in A1 loop with abnormal binding to platelet GPIb and botrocetin causing type 2M von Willebrand disease. Br. J. Haematol. 120, 643–651. https://doi.org/10.1046/j.1365-2141.2003.04168.x
Castaman G., Giacomelli S.H., Jacobi P., Obser T., Budde U., Rodeghiero F., Haberichter S.L., Schneppenheim R. 2010. Homozygous type 2N R854W von Willebrand factor is poorly secreted and causes a severe von Willebrand disease phenotype. J. Thromb. Haemost. 8, 2011–2016. https://doi.org/10.1111/j.1538-7836.2010.03971.x
Veyradier A., Boisseau P., Fressinaud E., Caron C., Ternisien C., Giraud M., Zawadzki C., Trossaert M., Itzhar-Baikian N., Dreyfus M., d’Oiron R., Borel-Derlon A., Susen S., Bezieau S., Denis C. V., Goudemand J., French Reference Center for von Willebrand d. 2016. A laboratory phenotype/genotype correlation of 1167 french patients from 670 families with von Willebrand disease: A new epidemiologic picture. Medicine (Baltimore). 95, e3038. https://doi.org/10.1097/MD.0000000000003038
Mehta R., Athar M., Girgis S., Hassan A., Becker R.C. 2019. Acquired von Willebrand syndrome (AVWS) in cardiovascular disease: A state of the art review for clinicians. J. Thromb. Thrombolysis. 48, 14–26. https://doi.org/10.1007/s11239-019-01849-2
Vincentelli A., Susen S., Le Tourneau T., Six I., Fabre O., Juthier F., Bauters A., Decoene C., Goudemand J., Prat A., Jude B. 2003. Acquired von Willebrand syndrome in aortic stenosis. N. Engl. J. Med. 349, 343–349. https://doi.org/10.1056/NEJMoa022831
Hayakawa M., Matsumoto M. 2020. Acquired von Willebrand syndrom. Rinsho Ketsueki. 61, 809–817. https://doi.org/10.11406/rinketsu.61.809
Moake J.L., McPherson P.D. 1989. Abnormalities of von Willebrand factor multimers in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. Am. J. Med. 87, 9N–15N.
Sadler J.E. 2006. Thrombotic thrombocytopenic purpura: a moving target. Hematology Am. Soc. Hematol. Educ. Program. 415–420. https://doi.org/10.1182/asheducation-2006.1.415
Gupta M., Feinberg B.B., Burwick R.M. 2018. Thrombotic microangiopathies of pregnancy: Differential diagnosis. Pregnancy Hypertens. 12, 29–34. https://doi.org/10.1016/j.preghy.2018.02.007
Zheng X.L. 2015. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med. 66, 211–225. https://doi.org/10.1146/annurev-med-061813-013241
Foreman K.E., Vaporciyan A.A., Bonish B.K., Jones M.L., Johnson K.J., Glovsky M.M., Eddy S.M., Ward P.A. 1994. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Invest. 94, 1147–1155. https://doi.org/10.1172/JCI117430
Gragnano F., Golia E., Natale F., Bianchi R., Pariggiano I., Crisci M., Diana V., Fimiani F., Limongelli G., Russo M., Cirillo P., Calabro P. 2017. Von Willebrand factor and cardiovascular disease: From a biochemical marker to an attractive therapeutic target. Curr. Vasc. Pharmacol. 15, 404–415. https://doi.org/10.2174/1570161115666170201114835
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии