

Цитология, 2019, T. 61, № 10, стр. 769-786
Cемейство Polycomb: стволовые, опухолевые стволовые клетки и рак предстательной железы
Б. В. Попов *
Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: borisvp478@gmail.com
Поступила в редакцию 20.06.2019
После доработки 22.07.2019
Принята к публикации 22.07.2019
Аннотация
Семейство Polycomb (PcG) играет важную роль в эпигенетической регуляции клеточных фенотипов, контролируя в постнатальной жизни формирование и поддержание клеточной идентичности, дифференцировку соматических стволовых клеток. Конститутивное повышение продукции отдельных членов семейства PcG способствует формированию опухолевых стволовых клеток (ОСК) и возникновению опухолей различных органов, включая рак предстательной железы (ПЖ), который в настоящее время играет лидирующую роль в заболеваемости и смертности среди онкологических заболеваний. В обзоре рассматривается структурная организация и функциональная роль семейства PcG, которая с достаточной полнотой изучена на модели эмбриональных стволовых клеток, клеточная организация ПЖ, клетки-мишени и ОСК при раке ПЖ (РПЖ). Описаны функциональные последствия конститутивной гиперпродукции ключевых белков семейства PcG: Ezh2 и Bmi1, в ОСК и опухолях различной тканевой специфичности. Скрининг РПЖ основан в настоящем на определении в сыворотке крови уровня простат-специфического антигена (ПСА), что не позволяет эффективно диагностировать его агрессивные и быстро метастазирующие формы. Лечение РПЖ включает иcпользование специфических ингибиторов Ezh2 и Bmi1. Дальнейшая характеристика роли всего семейства PcG при РПЖ будет способствовать его эффективному скринингу, диагностике и терапии.
Эпигенетический уровень регуляции надстроен над генетическим кодом для формирования и поддержания клеточного фенотипа. Эпигенетические программы закодированы в химических метках ДНК, гистоновых белках, нуклеосомах, тканеспецифических взаимодействиях между белками, ДНК и РНК, которые регулируют структурную организацию и функции генома в клетках. Нарушения в геноме и его эпигенетической регуляции могут взаимодействовать в развитии опухолей. Крупномасштабное секвенирование генома опухолевых клеток показывает, что мутации в генах, кодирующих ферментную машину для формирования эпигенетического уровня регуляции, являются наиболее частыми при раковых заболеваниях человека, включая РПЖ (Yegnasubramanian, 2016).
Семейство белков Polycomb (PcG) занимает важное место в эпигенетической регуляции в антенатальном развитии, формировании и поддержании тканеспецифической клеточной идентичности и дифференцировки клеток в постнатальной жизни. Структурная организация и функциональная роль PcG в значительной степени охарактеризованы на модели эмбриональных стволовых клеток (ЭСК). Установлено, что отдельные члены семейства PcG играют ключевую роль в формировании соматических стволовых клеток (СК) и опухолевых стволовых клеток (ОСК), возникновении опухолей различной тканевой специфичности, включая рак РПЖ. Задачей будущего является моделирование роли всего семейства PcG в формировании соматических СК и ОСК в каждой ткани с целью выработки тканеспецифической противораковой терапии.
Настоящий обзор посвящен описанию данных литературы о структуре и функциях белков PcG и их роли в формировании ОСК предстательной железы. Хотя данные литературы по этому вопросу в настоящем фрагментарны, их представление послужит для создания полной картины участия PcG в опухолевой инвазии и метастазировании при РПЖ.
СТРУКТУРА И ФУНКЦИОНАЛЬНАЯ РОЛЬ СЕМЕЙСТВА PcG
Структурная организация семейства PcG. Семейство белков Polycomb (PcG) играет ключевую роль в формировании клеточной памяти благодаря своей способности поддерживать детерминированное состояние в различных тканеспецифических линиях в ходе долгосрочной пролиферации клеток (Hadorn, 1968). Члены семейства PcG были открыты как транскрипционные репрессоры генов HOX, контролирующих формирование плана и сегментацию тела при развитии Drosophila (Kennison, 1995). Белки семейства PcG формируют две группы репрессивных комплексов: Рolycomb-репрессивный комплекс 1 (PRC1), который опосредует моноубиквитилирование лизина 119 гистона Н2А (Wang et al., 2010; Gutierrez et al., 2012), и Рolycomb-репрессивный комплекс 2 (PRC2), который триметилирует лизин 27 гистона Н3 (Н3К27) (Cao et al., 2002; Muller et al., 2002). Стабильная репрессия генов-мишеней, опосредованная PcG, основана на взаимодействии комплексов PRC1 и PRC2. Каноническая модель предполагает взаимодействие PRC1 и PRC2 в ходе двух последовательных шагов: сначала PRC2 триметилирует H3K27 на промоторах генов-мишеней, создавая сайт связывания для PRC1, затем PRC1 способствует стабильному связыванию комплексов с хроматином путем моноубиквитилирования лизина 119 гистона H2A (рис. 1).
Рис. 1.
Структурная организация комплексов PRC1 и PRC2 семейства Polycom (PcG) и канонический механизм их взаимодействия. Метилтрансфераза Ezh2, входящая в состав комплекса PRC2, триметилирует лизин 27 гистона Н3 (Н3К27me3), который распознается белком Cbx комплекса PRC1. Убиквитиновая лигаза Ring1 в составе коровой субъединицы Ring1/Bmi1 моноубиквитинирует лизин 119 гистона Н2А (Н2А119ub), создавая стабильную связь PcG с хроматином. Ezh2, Suz12, Eed, Rbbp4/Rbbp7 – белки корового комплекса PRC2; Bmi1, Ring1, Cbx, Phc – белки корового комплекса PRC1. Здесь и на рис. 2, 3: RNAPII – РНК-полимераза II.
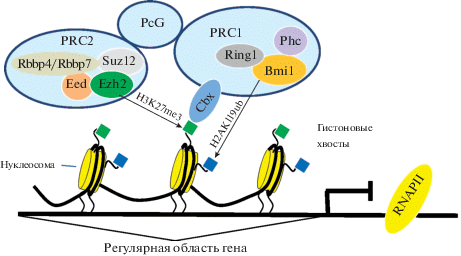
Коровый комплекс PRC2 формируется четырьмя белками: Ezh2/Ezh1 (set-domain containing histone methyltransferases enhancer of zeste), Eed (embryonic ectoderm development), Suz12 (suppressor of zeste), и Rbbp4/Rbbp7 (CAF1 histone-binding proteins). Этот комплекс функционирует как метилтрансфераза для H3K27, способствуя его триметилированию и формированию сайта связывания для канонического PRC1 (рис. 1). Структурное и функциональное разнообразие PRC2 достигается с помощью взаимодействия корового комплекса со вспомогательными белками, которые модифицируют его ферментную активность и направляют к различным генам-мишеням (Pasini et al., 2010). Выявлено два различных по составу комплекса PRC2 (Hauri et al., 2016). PRC2.1 формируется при альтернативном связывании корового комплекса с одним из трех белков PCL (Polycomb-like homologs): Phf1, Phf19, или Mtf2. PRC2.2 образуется при участии белков Aebp2 и Jarid2, которые регулируют связывание корового комплекса с хроматином и его ферментную активность.
PRC1 формирует несколько комплексов, которые подразделяются на канонические (cPRC1) и неканонические (ncPRC1). Все комплексы PRC1 имеют общую коровую субъединицу, состоящую из белка Ring1 (Ring1A или Ring1B) c активностью убиквитиновой лигазы Е3, убиквитилирующей лизин 119 гистона Н2А (Н2АК119ub), и одного из белков Pcgf (Pcgf1-6, Polycomb group ring-finger domain proteins). Ring1 обладает слабой E3-лигазной активностью, тогда как гетеродимер Ring1-Pcgf, формирующий минимальный коровый комплекс PRC1, убиквитилирует H2AK119 с намного более высокой активностью (Buchwald et al., 2006; Bentley et al., 2011). Считается, что H2AK119ub ингибирует активность РНК-полимеразы II в стадии элонгации и вызывает транскрипционную репрессию (Simon, Kingston, 2009). Компонент Pcgf детерминирует полный состав и биохимические свойства минимального корового комплекса. cPRC1 содержат в коровой части Pcgf2/4, один из белков, содержащих хромодомен (Cbx2, Cbx4, Cbx6-8), который распознает Н3К27ме3, и один из белков Phc (Phc1−Phc3, Polyhomeotic homologous protein), который содержит домен SAM (sterile alpha motif), опосредующий репрессию, вызываемую PcG (Robinson et al., 2012) (рис. 1). Неканонический PRC1 включает в свой состав Yy1, Rybp (Ring1 and Yy1-binding protein) или паралог Yy1 белок Yaf2, которые опосредуют связывание хроматина с Pcgf1, Pcgf3/5 или Pcgf6 и формируют соответственно ncPRC1.1, ncPRC1.3/PRC1.5 или ncPRC1.6. Дополнительно неканонический PRC1 включает скелетные белки Dcaf7 и Wdr5, содержащие домен WD40.
Каждый комплекс PRC1 может включать вспомогательные белки, которые уточняют его ДНК-связывающую активность и регуляторную функцию. Неканонический PRC1 не связывает H3K27me3, но обладает более высокой способностью убиквитилировать Н2А, чем канонический PRC1. H2AК119ub служит специфической меткой для распознавания PRC2 (Schwartz, Pirotta, 2013). Триметилируя H3K27, PRC2 создает специфический сайт для связывания каноническим PRC1, которое опосредуется хромодоменом Cbx.
Каноническая модель взаимодействия PRC2 и PRC1 не описывает два важных события: 1) механизм взаимодействия РRC2 с ДНК и 2) механизм взаимодействия между PRC2 и неканоническим PRC1, который не включает белков Cbx, распознающих H3K27me3 (Schwartz, Pirotta, 2014). Современные публикации показывают, что гетеродимеры Ring−Pcgf создают основу всех типов комплексов PRC1 путем убиквитилирования H2A. H2AK119ub рекрутирует PRC2, создавая возможность триметилировать H3K27 и рекрутировать канонический PRC1, формируя связь между PRC1 и PRC2 (Kalb et al., 2014) (рис. 2).
Рис. 2.
Неканонический механизм рекрутирования к ДНК и взаимодействия комплексов PRC1 и PRC2. Первоначально неканонический PRC1 (ncPRC1), содержащий связывающий ДНК белок Yy1 и вспомогательный белок Rybp, рекрутируется к хроматину и убиквитилирует лизин 119 гистона Н2А (Н2А119ub), формируя сайт связывания на хроматине для PRC2. PRC2 распознает H2AK119ub c помощью вспомогательного белка Jarid2 и в свою очередь триметилирует Н3К27 (Н3К27me3), создавая сайт связывания для канонического PRC1(cPRC1), который распознается с помощью белка Сbx.
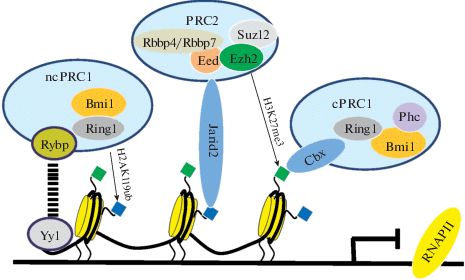
Обнаружено, что отсутствие активности ключевого компонента комплекса PRC1 (AtBMI1) у растений вызывает снижение уровня H3K27me3 в генах созревания семян, что дает основание предположить, что AtBMI1 участвует в инициации репрессии этих генов (Yang et al., 2013). У позвоночных первый шаг к репрессии, опосредованной PcG, также включает физическое взаимодействие PRC1 с хроматином с последующим рекрутированием PRC2 (Blackledge et al., 2014; Cooper et al., 2014; Kalb et al., 2014). Эти результаты поддерживают идею, что первоначально ncPRC1, содержащий связывающий ДНК белок Yy1 и вспомогательный белок Rybp, рекрутируется к хроматину и убиквитинилирует Н2АК119, формируя сайт связывания на хроматине для PRC2. PRC2 распознает H2AK119ub c помощью вспомогательного белка Jarid2 и, в свою очередь, триметилирует Н3К27, создавая сайт связывания для канонического PRC1 (рис. 2). Такая последовательность рекрутирования и взаимодействия комплексов PRC1 и PRC2 переворачивает каноническую модель взаимодействия этих комплексов наоборот (Merini, Calonje, 2015) (рис. 2).
PRC1 и PRC2 играют различную функциональную роль в индукции дифференцировки СК: неканонический PRC1 вызывает транскрипционную репрессию близколежащих генов, PRC2 необходим для инициации дифференцировки, тогда как канонический PRC1 обеспечивает отдаленные взаимодействия благодаря прямой связи с PRC2 через H3K27me3 (Riising et al., 2014).
Функциональная роль белков семейства PcG. PcG формируют часть высоко консервативной машины клеточной памяти вместе с белками семейства Trithorax (TrxG), поддерживая репрессивное или активное состояние генов. Действуя антагонистически, белки этих семейств регулируют у зрелых организмов экспрессию ключевых генов клеточной дифференцировки и самоподдержания, участвуя в механизмах множества клеточных функций, таких как инактивация Х-хромосомы, геномный импринтинг, контроль клеточного цикла, регуляция самоподдержания и дифференцировки СК, и возникновение рака. Такая обширная зона контроля достигается разнообразием комплексов PcG, которые формируются в зависимости от клеток-мишеней и изменяют их активность путем модификации гистонов и ремоделирования хроматина (Schuettengruber et al., 2017).
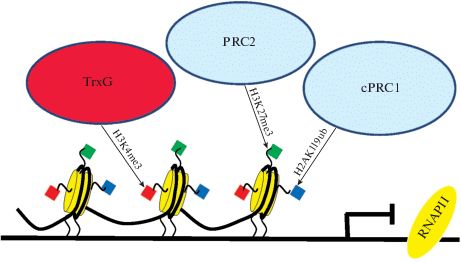
Каким образом белки семейства PcG поддерживают идентичность клеток и реализацию программ самоподдержания и дифференцировки СК? Как в ЭСК, так и в соматических СК промоторы генов, поддерживающих мультипотентность и линейную дифференцировку, содержат “бивалентные” домены, включающие активную (Н3К4ме3) и репрессивную метку (Н3К27ме3) (Shema et al., 2016) (рис. 3). Эти модификации катализируются, соответственно, белками семейств Trithorax (TrxG) и PcG (Piunti, Shilatifard, 2016). В активно делящихся самоподдерживающихся ЭСК гены линейной дифференцировки не транскрибируются, но их репрессия отменяется при индукции линейной дифференцировки, что связано с утратой репрессивной метки Н3К27ме3. Активация экспрессии дифференцировочных генов сочетается с торможением транскрипции генов плюрипотентности (OCT4, SOX2, NANOG) в ЭСК путем повышения уровня репрессивной метки Н3К27ме3 в их промоторах. Такая регуляция позволяет дифференцировку СК в один клеточный тип, но предотвращает де- и трансдифференцировку.
Рис. 3.
Модификации хроматина белками семейств Trithorax (TrxG) и PcG, опосредующие формирование “бивалентных доменов”. Экспрессия генов дифференцировки и плюрипотентности связана с бивалентными доменами в их промоторах, которые содержат “бивалентные” домены, включающие активную (Н3К4ме3) и репрессивную (Н3К27ме3) метку хроматина. Эти модификации катализируются, соответственно, белками семейств Trithorax (TrxG) и PcG. В состоянии активной пролиферации гены линейной дифференцировки не транскрибируются, что связано с присутствием в их промоторах супрессивной метки Н3К27ме3, утрата которой при индукции дифференцировки сопровождается активацией их экспрессии. Наоборот, в состоянии активной дифференцировки тормозится экспрессия генов плюрипотентности, что связано с повышением уровня метилирования их промоторов по Н3К27ме3. H2AK119ub – сайт связывания хроматина cPRC1.
При индукции дифференцировки Cbx7 в комплексе PRC1 заменяется на другие белки того же семейства (Cbx2, Cbx4, Cbx8). Неспособность клетки сделать такую замену вызывает ее гиперпролиферацию и последующий лейкоз (Klauke et al., 2013). Белки Pcgf определяют специфику связывания комплексов PRC1 с тем или иным промотором в ЭСК и, таким образом, регулируют идентичность клеток, выбор клеточной судьбы и репрограммирование (Zdzieblo et al., 2014). Немного известно о роли отдельных белков комплексов PRC1 в регуляции дифференцировки соматических СК. Вспомогательные белки Epop, Mtf2, Phf19 и Jarid2 экспрессируются на высоком уровне в недифференцированных клетках, тогда как белки Phf1 и Aebp2 – в дифференцированных (Kloet et al., 2016). Эти белки могут избирательно направлять комплексы PRC2 путем распознавания модификаций гистонов или через связывание различных последовательностей ДНК (Pasini et al., 2010; Ballare et al., 2012). Инактивация Jarid2 отменяет взаимодействие PRC2 со специфическими мишенями и дифференцировку ЭСК.
Роль PcG в возникновении рака, вероятно, основана на нарушении его способности регулировать пролиферацию и дифференцировку СК. Известно, что мышиные стволовые кроветворные клетки (СКК) в отсутствие Bmi1 теряют мультипотентность, тогда как усиленная экспрессия Bmi1 в СКК возвращает им мультипотентность (Rizo et al., 2008; Sauvageau, Sauvageau, 2010). Нейральные СК с делецией BMI1 теряют способность к самоподдержанию и дифференцировке, что связано с активацией экспрессии генов локуса INK4 (Molofsky et al., 2003; Hirabayashi et al., 2009). Супрессия BMI1 в клетках молочной железы нарушает их рост (Pietersen et al., 2008). Удаление BMI1 в популяции клеток тонкого кишечника, локализующихся в положении +4, вызывает полное исчезновение крипт тонкого кишечника (Sangiorgi, Capecchi, 2008). С другой стороны, Bmi1 является протоонкогеном и сверхпродуцируется при многих злокачественных заболеваниях, включая лимфому, острый миелоидный лейкоз, рак молочной железы, яичников, поджелудочной железы, эндометрия, шейки матки, толстого кишечника, прямой кишки, печени, легких, мозжечка и глиобластому (Hosen et al., 2007; Honig et al., 2010; Guo et al., 2011).
Белок Ezh2 (Enhancer of Zeste homology 2) играет роль каталитического компонента PRC2. Секвенирование генома ЭСК в сочетании с иммунопреципитацией хроматина (ChiP-on-chip) показывает, что 20% генов, оккупированных PRC2, связаны с регуляцией выбора клеточной судьбы и дифференцировки. Эти гены относятся как к семействам генеральных транскрипционных факторов HOX, SOX, FOX, так и к сигнальным путям, ассоциированным с клеточной дифференцировкой, включая Wnt, BMP, Notch (Boyer et al., 2006).
Индукция клеточной дифференцировки возможна при отмене репрессии, опосредованной PRC2 и потере H3K27me3 в бивалентных доменах дифференцировочных генов ЭСК (Bernstein et al., 2006; Mikkelsen et al., 2007). Уровень Ezh2 и H3K27me3 в дифференцировочных генах снижается при эпидермальной (Ezhkova et al., 2009) и мышечной дифференцировке (Caretti et al., 2004). Ezh2 и деметилаза Utx формируют эпигенетический переключатель, контролирующий уровень H3K27me3 в промоторах регуляторных генов жировой и костной дифференцировки в мезенхимных стволовых клетках (Петров и др., 2016). Индуцированная экспрессия гена Ezh2 в МСК повышает уровень транскриптов Ezh2 и H3K27me3 при сочетанном снижении уровня транскрипции гена RUNX2 и поздних маркеров костной дифференцировки, остеопонтина и остеокальцина (Hemming et al., 2014). С одной стороны, активность переключателя Ezh2/Utx регулируется при взаимодействии PRC1/PRC2 и включает Bmi1 в качестве ключевого члена PRC1. C другой стороны, как Ezh2, так и Utx контролируются белками семейства продукта гена ретинобластомы (pRb) (Bracken, Helin, 2009; Herz et al., 2010). Инактивирующие мутации гена RB приводят к увеличению активности белков Ezh2 и Eed. Их продукция так же, как и продукция Bmil, значительно возрастает в опухолях различной тканевой специфичности (Bracken et al., 2003; Bracken, Helin, 2009; Cao et al., 2011). К тому же, Ezh2 и Bmi1 способны метилировать H3K27 и супрессировать p16Ink4a только в клетках с активным pRb (Kotake et al., 2007).
СТРУКТУРНАЯ ОРГАНИЗАЦИЯ ПЖ ЧЕЛОВЕКА
ПЖ представляет собой небольшой вспомогательный орган в мужской репродуктивной системе, который находится в центре внимания биологов и врачей из-за ведущей роли в заболеваемости и смертности от тканеспецифического рака (Prajapati et al., 2013). ПЖ значительно более чувствительна к онкогенной трансформации, чем другие половые органы, РПЖ диагностируется в течение жизни у каждого седьмого лица мужского пола (Siegel et al., 2016).
ПЖ является железистым органом, развитие и функции которого регулируются действием андрогенов на тканеспецифические стромальные и эпителиальные клетки (Prins, Putz, 2008). В ПЖ человека выделяют периферическую, переходную и центральную зоны (Timms et al., 2008). РПЖ первично возникает в периферической зоне (Abate-Shen, Shen, 2000). Число стромальных клеток в ПЖ, представляющих собой в основном гладкомышечные клетки, дважды превышает число эпителиальных клеток, что связано с насосной функцией органа, периодически выбрасывающего секретируемую жидкость (Bartsch, Rohr, 1980). Выделяют три основных типа эпителиальных клеток ПЖ: базальные, люминальные и нейроэндокринные, которые отличаются между собой по маркерам (рис. 4). Базальные клетки представляют собой плоские клетки, расположенные на базальной мембране и формирующие базальный слой эпителиальных структур ПЖ. Эти клетки продуцируют высокомолекулярные цитокератины (ЦК) 5 и 14 (Brawer et al., 1985), СD44 (Alam et al., 2004), интегрины α6β1 (Bello-DeOcampo et al., 2001) и транскрипционный фактор р63 из семейства р53 (Signerotti et al., 2000).
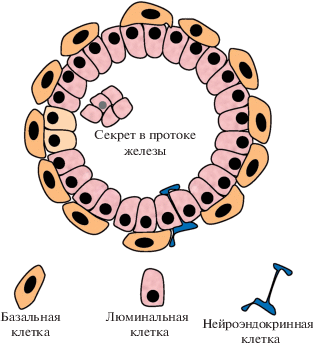
Основным типом эпителиальных клеток ПЖ являются люминальные клетки, которые формируют над базальными клетками круг столбчатых клеток, экспрессирующих андрогенный рецептор (AR), низкомолекулярные ЦК8 и ЦК18, секретирующие в железистые протоки простат-специфический антиген (ПСA), простат-специфическую фосфатазу, и другие тканеспецифические продукты (рис. 4) (Verhagen et al., 1992; Abate-Shen, Shen, 2000). Нейроэндокринные клетки, составляющие менее 1% всех эпителиальных клеток ПЖ, располагаются во всех ее эпителиальных слоях (рис. 4), продуцируют хромогранин, синаптофизин, кальцитонин и нейрон- специфическую энолазу, но не AR (Parimi et al., 2014). В процессе дифференцировки базальные клетки превращаются в люминальные. Между этими типами клеток существуют промежуточные временно амплифицирующиеся клетки, которые продуцируют CD24, ЦК5, ЦК8, интегрины α2β1, но не AR (Wang, Hayward, 2001). Маленькая популяция базальных клеток была идентифицирована в качестве стволовых клеток ПЖ у человека и мышей. Эти клетки экспрессируют TROP2 (tumor associated calcium signal transducer 2), p63, CD133 (Goldstein et al., 2008; Trerotola et al., 2010; Lee et al., 2014).
СТВОЛОВЫЕ И ОПУХОЛЕВЫЕ СТВОЛОВЫЕ КЛЕТКИ ПЖ
Гипотезы формирования опухолевых стволовых клеток. Опухолевая ткань гетерогенна и формируется различными популяциями опухолевых клеток. Существует две модели, объясняющие происхождение гетерогенности опухолевой ткани. Первая модель основана на представлении о клональной эволюции опухоли. Такая модель предполагает, что опухолевые клетки появляются в ходе долгосрочного накопления различных генетических и эпигенетических нарушений в генах, регулирующих контрольные точки клеточного цикла, апоптоз и клеточное старение. В результате возникает автономная и иммортальная клеточная популяция клеток с туморогенным потенциалом (Nowell et al., 1976) (рис. 5). Вторая гипотеза основана на модели ОСК, обладающих, подобно нормальным СК, способностью к самоподдержанию и дифференцировке (рис. 5). Дифференцировка ОСК представляет собой однонаправленный процесс, в результате которого формируются опухолевые клетки, не обладающие способностью к самоподдержанию, с короткой продолжительностью жизни (Bonett, Dick, 1997; Marjanovic et al., 2013). ОСК способны к асимметричному делению, в результате которого формируется ОСК с материнскими свойствами и нестволовая опухолевая клетка, теряющая способность к самоподдержанию. ОСК формируют небольшую популяцию клеток опухолевой ткани, которая, тем не менее, генерирует ее основные свойства: рост, прогрессию, инвазию, метастазирование и устойчивость к противоопухолевой терапии (Bao et al., 2006; Li et al., 2008).
Рис. 5.
Модели гетерогенности опухоли. НС-ОК – не стволовая опухолевая клетка, ОСК – опухолевая стволовая клетка.
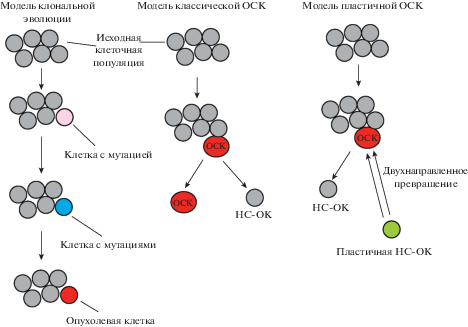
В дополнение к вышеописанным классическим моделям существует пластическая модель ОСК, которая предполагает, что постмитотические нестволовые опухолевые клетки могут дедифференцироваться и приобретать статус ОСК (рис. 5) (Marjanovic et al., 2013). Возможный механизм дедифференцировки включает мутацию, подобную мутации гена PAX5, продукт которого в нормальных условиях направляет дифференцировку лимфоидных предшественников в зрелые В-клетки. Потеря Pax5 в B-клетках связана с развитием агрессивных лимфом, проявляющих фенотип Т-клеток (Cobaleda et al., 2007).
Клеточная популяция, обладающая свойствами ОСК, была впервые обнаружена в ткани острой миелоидной лейкемии (Bonett, Dick, 1997) и затем во многих солидных опухолях человека (Visvader, Lindeman, 2008). ОСК экспрессируют специфические маркеры, что позволяет выделять и изолировать такие клетки с помощью проточной цитометрии. Трансплантация ОСК человека иммунодефицитным мышам вызывает рост опухоли, которая не возникает при введении значительно большего количества контрольных клеток, экспрессирующих другие маркеры. ОСК могут также возникать при активирующих мутациях гена BMI1, который в нормальных клетках тормозит активность генов p16Ink4a и p14Arf. Продукты этих генов опосредуют, соответственно, развитие клеточного старения и апоптоза, тогда как их супрессия при сверхпродукции гена BMI1 способствует переходу эпителиальных клеток в ОСК (Valk-Lingbeek et al., 2004; Yu et al., 2011).
Роль эпителиально-мезенхимного перехода (ЭМП) в формировании ОСК. Еще один путь формирования ОСК связан с ЭMП. Этот переход представляет собой высоко консервативную программу развития для перехода эпителия в мезенхиму, которая широко используется в эмбриональной жизни при формировании слоев и органов, при воспалении и регенерации у взрослых животных (Thiery et al., 2009). В ходе ЭМП эпителиальные клетки теряют кадгериновые соединения, базальную полярность и приобретают подвижность. В опухолевой ткани это связано с инвазивностью и метастазированием (Spaderna et al., 2006). Сверхэкспрессия транскрипционных факторов Twist1 и Snail1 в зрелых эпителиальных клетках вызывает ЭМП и экспрессию маркеров CD44hiCD24lo, которые формируют профиль ОСК солидных опухолей (Mani et al., 2008; Morel et al., 2008). Результаты этих и других исследований показывают, что нестволовые опухолевые клетки могут превращаться в ОСК, и такой переход между ОСК и нестволовыми опухолевыми клетки может быть взаимным (рис. 5) (Vermeulen et al., 2010; Scheel, Weinberg, 2011; Chaffer et al., 2011).
Другие пути формирования ОСК. Изучение опухолевых клеток показывает, что в дополнение к популяции CD34+CD38−, клетки, экспрессирующие другие маркеры, также могут вызывать острую лейкемию (Sarry et al., 2011). По-видимому, различные типы зрелых клеток обладают пластичностью, которая дает им возможность трансформироваться в ОСК. Уровень пластичности в различных солидных опухолях различен, наиболее высокой пластичностью характеризуется меланома (Quintana et al., 2008). Популяция клеток меланомы, экспрессирующих гистоновую деметилазу Jarid1B, необходимую для роста опухоли, может спонтанно возникать из клеток, не экспрессирующих Jarid1B (Roesch et al., 2010). Приобретение устойчивости к терапии может также возникать на основе пластичности клеток. Маленькая популяция клеток легочной карциномы, экспрессирующих Jarid1A, показывающая толерантность к опухолевой терапии, может выйти из этого состояния и затем вернуть себе устойчивость к терапии (Sharma et al., 2010). По-видимому, двунаправленные переходы из нестволовых опухолевых клеток в ОСК является общей чертой канцерогенеза (Marjanovic et al., 2013).
Повышение уровня пластичности ОСК может быть связано с торможением активности продукта гена ретинобластомы (pRb). Канцерогенез представляет собой ядерное репрограммирование, направленное на формирование ОСК, и имеет много общего с формированием индуцированных плюрипотентных клеток из соматических дифференцированных клеток (Kohno et al., 2016). Основная общая черта этого феномена – повышенная пластичность клеток, которая может быть вызвана супрессией pRb. Нужно отметить, что функциональная инактивация pRb присутствует во всех типах рака человека (Weinberg, 1995). Функциональный дефицит pRb ведет к формированию незрелого состояния клетки, подобного состоянию СК (Harbour, Dean, 2000; Sage, 2012; Kitajima et al., 2015). Инактивация pRb может вызвать клональную экспансию СК и связана с появлением клеток-предшественников раковых клеток (cells-of-origin; Kohno et al., 2016). Плюрипотентные стволовые клетки имеют короткую фазу G1 и быстро делятся вследствие неактивного pRb, который в активной форме ингибирует активаторы клеточного цикла, например, транкрипционные факторы E2f (Burdon et al., 2002) и, таким образом, супрессирует OCT4 и SOX2, которые вместе с геном NANOG формируют сеть плюрипотентности (Kareta et al., 2015). Инактивация pRb в соматических стволовых клетках изменяет их способность к выбору клеточной судьбы и способствует формированию ОСК (Sage, 2012).
В ткани ретинобластомы, которая образуется в сетчатой оболочке глаза вследствие инактивации pRb, формируются различные клеточные линии (McEvoy et al., 2011), что может быть следствием дедифференцировки и нарушения выбора судьбы ретинальных клеток. Сигнальный путь pRb нарушен в 34 и 74% первичных и метастатических опухолей соответственно (Taylor et al., 2010). Мутации pRb инициируют остеосаркому, гиберному и мелкоклеточную карциному легких. Механистически остеосаркома и гибернома связаны с физическим взаимодействием pRb c Runx2 и Pparγ2 – тканеспецифическими транскрипционными факторами костной и жировой дифференцировки, соответственно. Потеря pRb тормозит костную и жировую дифференцировку и способствует пролиферации остеогенных и адипогенных предшественников (Thomas et al., 2001). Активность pRb включает действие всех членов его семейства (pRb, p130 и p107). Эти белки обладают функциональной избыточностью, и дефицит одного белка компенсируется активностью других членов его семейства.
Ортологи сигнального пути pRb присутствуют в некоторых одноклеточных и во всех многоклеточных организмах и играют ключевую роль в развитии многоклеточных вследствие их центральной роли в регуляции клеточного цикла, выбора судьбы и дифференцировки (Cao et al., 2010; Popov, Petrov, 2014). В настоящее время основной функцией pRb считается регуляция клеточного цикла и опухолевая супрессия (Dyson, 2016). Однако структура эволюционных предшественников молекул pRb более подобна белкам p107/p130, которые играют роль охранников состояния покоя у сложных самоподдерживающихся организмов (Campisi, 2003). Функции генов RB1 и E2F1-3 в развитии в большей степени имеют отношение к дивертисификации клеточного цикла, регуляции апоптоза, обмена веществ и опухолевой супрессии (Cao et al., 2010).
ПРОИСХОЖДЕНИЕ И МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ РПЖ
Клетки-мишени трансформации при РПЖ. По определению клетки-предшественники РПЖ (cells of origin) – это клетки в пределах опухолевой ткани, которые служат мишенями раковой трансформации (Visvader, 2011). Такие клетки отличаются от ОСК, ключевая черта которых – регенерация опухоли (Clevers, 2011). Современная гипотеза предполагает, что профиль экспрессии генов в опухоли в большей степени соответствует таковой в клетках-предшественниках опухоли, чем в клетках других линий опухолевой ткани (Xin, 2013). Прямым подходом для изучения клеточного происхождения опухоли является получение из опухолевой ткани клеточных линий и характеристика их молекулярного профиля. Последующая трансплантация клеток с различным молекулярным профилем иммунодефицитным мышам, у которых возникает опухоль, дает возможность сделать вывод о молекулярном профиле клеток, индуцирующих развитие опухоли. Такой подход был использован вначале для изучения клеток-предшественников ОСК при кроветворных заболеваниях (Krivtsov et al., 2006), а затем и солидных раков, включая РПЖ (Xin et al., 2005). Другим подходом для изучения молекулярного профиля клеток-предшественников является активация онкогенного сигнала в тканеспецифической клеточной линии под контролем линейно-специфического промотора (Baker et al., 2010).
При РПЖ выделяют латентные формы, не требующие активной терапии, и агрессивные формы болезни, которые в отсутствие активной терапии ведут к метастазированию и быстрой смерти (Yegnasubramanian, 2016). Различные варианты клинического течения РПЖ могут быть связаны с клетками с разным молекулярным профилем, из которых возникает РПЖ. Как было описано выше, эпителий ПЖ формируется из базальных, люминальных, промежуточных и нейроэндокринных типов клеток. Из-за отсутствия экспериментальных подходов для изучения нейроэндокринных клеток в настоящее время не удается установить их роль в становлении фенотипа РПЖ. Фенотипически большинство клинических форм ткани при РПЖ состоит из клеток с люминальным фенотипом. С другой стороны, этот рак рецидивирует после антигормональной терапии и превращается в так называемый резистентный к кастрации РПЖ. Поскольку базальные клетки не продуцируют AR и устойчивы к андрогенной терапии, было предположено, что такие формы РПЖ происходят из клеток базального слоя.
В ходе нормальной дифференцировки базальные клетки ПЖ превращаются в люминальные клетки, поэтому в случае трансформации базальных клеток они могут постепенно приобретать люминальный фенотип. Эпителиальная линия, полученная из базальных клеток, после трансформации онкогенами формирует опухолевую ткань из клеток с люминальным фенотипом (Min et al., 2010). В дальнейших экспериментах было показано, что клетки люминального и базального происхождения обладают способностью к самоподдержанию, и что из тех и других может возникать РПЖ (Xin, 2013).
В отличие от рака молочной железы, который может быть подразделен на 5 различных типов на основании профиля экспрессии генов (Perou et al., 2000), при РПЖ не было найдено определенных субтипов (Glinsky et al., 2004). Это свидетельствует о гетерогенности РПЖ на молекулярном уровне. Возможно, выделение различных субтипов РПЖ с отличающимся клиническим течением и прогнозом может быть сделано на основании ассоциированных с ними онкогенных сигнальных путей (Goldstein et al., 2011).
ОСК предстательной железы. ОСК ПЖ были охарактеризованы как клетки, формирующие субпопуляцию, маркированную CD44+CD24–, и обладающую способностью расти в неадгезивных сфероидах в беcсывороточной среде. Трансплантация клеток с такими маркерами сопровождается ростом опухоли у иммунодефицитных мышей линии NOD/SCID (Hurt et al., 2008). Клетки CD44+CD24– выявляются в различных установленных клеточных линиях, происходящих из ПЖ (Klarmann et al., 2009). Дополнительно к CD44 и CD24, в ОСК ПЖ были найдены маркеры CD49f, CD133, CD166 и интегрины α2β1 (Richardson et al., 2004; Collins et al., 2005; Goldstein et al., 2008; Jiao et al., 2012). ОСК из ПЖ различных пациентов с РПЖ могут отличаться по экспрессия отдельных маркеров вследствие исходной генетической гетерогенности (Collins et al., 2005).
CD133 представляет собой поверхностный гликопротеин с невыясненной биологической функцией, обнаруженный в различных типах соматических стволовых клеток (Yin et al., 1997). Около 1% нормальных клеток базального слоя ПЖ экспрессируют CD133. Клетки, экспрессирующие CD133/α2β1, проявляют высокий пролиферативный потенциал, а после трансплантации бестимусным иммунодефицитным мышам формируют железистые структуры с простатической дифференцировкой (Richardson et al., 2004).
CD44 – гликопротеин, экспрессирующийся в различных клетках и играющий роль рецептора для коллагена, ламинина, фибронектина, Е-селектина, L-селектина и глюкозоаминогликана гиалуроновой кислоты внеклеточного матрикса (Williams et al., 2013). ОСК ПЖ, маркированные CD44, обладают свойствами нормальных СК, в частности повышенной клоногенностью и способностью формировать колонии в жидком агаре (Patrawala et al., 2006).
СD166 является поверхностной молекулой, продукция которой усиливает способность СК ПЖ формировать сфероиды. Экспрессия CD166 повышается при РПЖ у человека, особенно при РПЖ, резистентном к кастрации (Jiao et al., 2012). Изложенные выше данные показывают, что ОСК ПЖ экспрессируют маркеры, подобные таковым в нормальных СК, что дает возможность предположить происхождение ОСК из нормальных СК ПЖ.
РОЛЬ БЕЛКОВ СЕМЕЙСТВА PCG В ИНИЦИАЦИИ И ПРОГРЕССИИ РПЖ
Нарушение уровня метилирования Н3К27. Роль PRC2 в развитии рака ПЖ была обнаружена на модели мышиной лимфомы, которая возникала при взаимодействии PcG и с-Myc и торможении активности гена CDK2N (Jacobs et al., 1999). Затем было показано, что повышение уровня Ezh2 и H3K27me3 при РПЖ свидетельствуют о неблагоприятном исходе заболевания (Varambally et al., 2002). Несколько генов, продукты которых входят в состав комплексов PRC1 и PRC2, сверхэкспрессируются при солидных раках и лейкемиях. При миеломе и карциноме почки была найдена мутация в гене деметилазы Utx (Kdm6A), удаляющей метильную группу из H3K27me3. Была обнаружена активирующая моноаллельная мутация в каталитическом домене гена EZH2 (Y641) при лимфоме (Morin et al., 2010). Эта мутация лишает Ezh2 способности моно- и диметилировать Н3К27, но усиливает ее способность триметилировать Н3К27 и повышает уровень Н3К27ме3 в опухолевой ткани. Эти данные дают основание предположить, что гиперметилирование Н3К27 действует как драйвер при различных солидных опухолях, включая РПЖ (Comet et al., 2016).
Ezh2 может также кооперироваться с другими эпигенетическими модификаторами, такими как метилтрансферазы ДНК, что способствует более длительному подавлению экспрессии генов, опосредованному метилированием (Viré et al., 2006). Таким образом, Ezh2 подавляет экспрессию генов, участвующих в дифференцировке и остановке клеточного цикла, способствуя поддержанию стволовости (Sauvageau, Sauvageau, 2010).
Ezh2 может активировать разные онкогенетические сигнальные пути, например, неканонический сигнальный путь Wnt за счет репрессии Dkk-1 (Dickkopf Wnt signaling pathway inhibitor 1), являющегося ингибитором Lrp (lipoprotein receptor-related protein), который представляет собой корецептор Wnt (Hussain et al., 2009). Ezh2 также участвует в сигнальном пути трансформирующего фактора роста TGF-β1, возможно, индуцирующего ЭМП (Rao et al., 2010).
Идентифицированы многие целевые гены Ezh2. Так как Ezh2 часто функционирует как онкогенетический фактор во многих типах рака, большинство целей Ezh2, идентифицированных в раковых клетках, являются генами опухолевых супрессоров. Ezh2 регулирует экспрессию генов, кодируемых локусом INK4 (Sasaki et al., 2008). Другая важная цель Ezh2 при многих раках – ген Е-кадгерина (CHD1), подавление экспрессии которого существенно для возникновения ЭМП и метастазирования (Wang et al., 2013). Дополнительно, Ezh2 подавляет экспрессию Е-кадгерина, взаимодействуя со Snail (Tong et al., 2012).
Поскольку Ezh2 подавляет дифференцировку нормальных ЭСК путем снижения экспрессии линейно-специфичных факторов (Lee et al., 2006), не удивительно, что он высоко экспрессируется в популяциях ОСК, происходящих из ПЖ (Li et al., 2013), поддерживая их жизнеспособность и ингибируя дифференцировку (Adhikary et al., 2015). Считается, что Ezh2 в составе комплекса PRC2 подавляет клеточные транскрипционные программы, которые опосредуют дифференцировку и апоптоз через GSK-3β и р53 при раке шейки матки (Chen et al., 2016), инсулиновый ростовой фактор при раке языка (Galvis et al., 2015), р21 при раке яичников (Liu et al., 2014) и через IKKα при карциноме носоглотки (Yan et al., 2014). Активность Ezh2 также опосредована транскрипционной активацией онкогенетических сигнальных путей, связанных с жизнеспособностью и пролиферацией ОСК, таких как сигнальный путь Wnt при колоректальном раке (Chen et al., 2016), Notch1 и TGFβ1 при РПЖ (Zhu et al., 2015).
Одна из самых важных биологических ролей Ezh2 в немалигнизированных клетках – регуляция клеточного цикла. Ezh2 необходим для экспрессии генов, участвующих в пролиферации, опосредованной транскрипционными факторами семейства E2F. Подавление активности Ezh2 при глиобластоме приводит к остановке клеточного цикла на границе фаз G0/G1 (Zhang et al., 2015).
Неожиданно оказалось, что инактивирующие мутации генов EZH2, EED, SUZ12 также ассоциированы с некоторыми формами миелодиспластического синдрома, злокачественных опухолей периферических нервов, глиом (Lewis et al., 2013; Lee et al., 2014). Замена лизина на метионин в положении 27 гистона Н3 сопровождается ацетилированием этого сайта и взаимодействием его с белками, содержащими бромодомен. Таким образом, изменяется механизм онкогенного эффекта сайта Н3К27М и принцип поиска противоопухолевых лекарств через торможение его активности (Brooun et al., 2016; Justin et al., 2016).
Cупрессия генов локуса INK4A/ARF. Хотя Bmi1 в контексте PcG регулирует экспрессию множества генов плюрипотентности и дифференцировки, его наиболее изученными мишенями являются гены локуса INK4 (p16Ink4aи p14Arf), торможение активности которых предотвращает апоптоз и клеточное старение (Surface et al., 2010). Белок p16Ink4a выполняет функцию ингибитора циклин-зависимых киназ Cdk4/6 в фазе G1 клеточного цикла. Торможение его активности с помощью Bmil сопровождается активацией комплекса циклин D/Cdk4/6, супрессией белков семейства pRb, и активацией пролиферации клеток. Белок p14Arf функционирует как ингибитор убиквитиновой лигазы Mdm2, инактивирующей опухолевый супрессор p53. Торможение продукции p14Arf, опосредованное Bmi1, сопровождается убиквитинилированием и деградацией p53 в протеасомах, активацией клеточного цикла и увеличением риска злокачественной трансформации.
Bmi1 продуцируется на высоком уровне в СКК, но в ходе дифференцировки он замещается в комплекcах PRC1 на альтернативный белок Mel18 (Morey et al., 2015). В противоположность Bmi1, который репреcсирует экспрессию генов CDK2N и PTEN, активируя сигнальный путь АКТ (Jacobs et al., 1999), Mel18 действует как опухолевый супрессор, ингибируя активность нескольких протоонкогенов, включая MYC (Guo et al., 2007). Возможно, Bmi1 и Mel18 формируют эпигенетический переключатель, регулирующий активность комплекса PRC1 (Сao et al., 2005). Гиперэкспрессия отдельных белков PRC1 также может вызывать супрессию генов локуса INK4A/ARF и способствовать инвазии путем торможения апоптоза и клеточного старения. Например, сверхэкспрессия гена CBX7 была найдена при РПЖ, лейкемиях, раке яичников. Cbx7 проявляет наиболее высокое сродство к Н3К27ме3 по сравнению с другими белками Cbx, и в ходе нормальной дифференцировки СК, по-видимому, заменяется другим белком Cbx с более низким сродством (Bernstein et al., 2006).
ПОДХОДЫ К ДИАГНОСТИКЕ И ТЕРАПИИ РПЖ
В настоящее время общепринятым методом скрининга РПЖ является определение уровня ПСА в сыворотке крови (Strope et al., 2010). Это метод показывает низкие чувствительность, специфичность и прогностическую ценность, а также ведет к гипердиагностике и повышенному уровню оперативного вмешательства при дремлющих формах РПЖ. Большинство из 240 000 пациентов с ежегодно диагностированным РПЖ умирают от других заболеваний, а у значительной части пациентов эта болезнь не становится угрожающей для жизни даже без медицинского вмешательства (Wilt et al., 2012). Для предотвращения одной смерти от РПЖ нужно диагностировать и подвергнуть медицинскому вмешательству от 5 до 48 человек (Barry, 2009; Schroder et al., 2012). Поэтому существует срочная необходимость в разработке методов неинвазивной диагностики агрессивных форм РПЖ. В качестве таких методов можно обсуждать определение в сыворотке крови и моче пациентов метилированных островков CpG в регуляторных областях генов GSTP1, APC, PTGS2, RASSF1A, RARB и других, которые никогда не встречаются при аденоме ПЖ (Aryee et al., 2013).
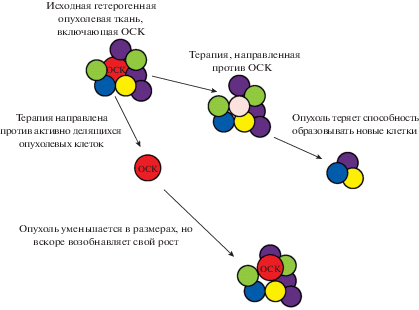
РПЖ происходит из эпителиальных клеток, экспрессирующих AR, поэтому в начальной стадии заболевания антиандрогенная терапия является эффективной для его терапии. В последующем на фоне такой терапии рак неизбежно прогрессирует в более инвазивную и метастатическую форму болезни, которая называется резистентным к кастрации РПЖ (CRPC – castration resistant prostate cancer) (Siegel et al., 2016). В такой форме этот рак устойчив к терапии, которая направлена против быстро делящихся опухолевых клеток, но не ОСК (Ni et al., 2014) (рис. 6). Устойчивость к антиандрогенной терапии основана на нарушении сигнального пути AR (Shiota et al., 2011). В ходе заболевания формируются сплайс-варианты AR без лиганд-связывающего домена, которые экспрессируются конститутивно и не регулируются андрогенами (Li et al., 2012).
Рис. 6.
Схема использования различных подходов при противоопухолевой терапии, включая терапию, направленную на удаление ОСК.
Мишенями для противораковой терапии при раке ПЖ служат ключевые белки комплексов PRC1/PRC2 – Ezh2 и Bmi1. Ezh2 играет критическую роль в инициации и прогрессии РПЖ, поэтому специфическая терапия включает разработку ингибиторов его активности (Wu et al., 2014, 2016). Одним из широко используемых ингибиторов Ezh2 является 3-деазоаденозин (DZN). Недостатком этого препарата является недостаточно высокая специфичность и высокая токсичность (Kikuchi et al., 2012). Препарат EPZ005687 показывает 500-кратную селективность по отношению к Ezh2 по сравнению с другими метилтрансферазами (Hsieh et al., 2016). Препарат GSK343 показывает 1000-кратную селективность к Ezh2 по сравнению с таковой к другим метилтрансферазам и 60-кратную по сравнению с таковой к Ezh1 (Verma et al., 2012). Продолжительное применение ингибиторов Ezh2 сопровождается появлением в гене EZH2 вторичных мутаций (Y111L и Y661D), что приводит к устойчивости к терапии (Gibaja et al., 2016).
Ген BMI1 защищает опухолевые клетки от апоптоза, а эктопическая экспрессия BMI1 спасает кератиноциты от индуцированного стрессом апоптоза (Cui et al., 2007). Наоборот, инактивация Bmi1 усиливает апоптоз в лимфоцитах селезенки и тимуса (Jacobs et al., 1999). ОСК яичников, экспрессирующие высокий уровень Bmi1, показывают повышенную устойчивость к цисплатину и паклитакселю (Zhang et al., 2008). Торможение активности Bmi1 значительно усиливает противоопухолевую эффективность доцетакселя при лечении РПЖ. Bmi1 способствует выживанию опухолевых клеток ПЖ после хемотерапии путем модулирования активности антиоксидантной системы (Cao et al., 2011). Было обнаружено, что в основе резистентности опухолевых клеток к химиотерапии лежит модуляция активности восстановленного глютатиона (GSH) через Bmi1. Торможение активности Bmi1 снижает уровень внутриклеточного GSH и сенсибилизирует опухолевые клетки к цисплатину. Таким образом, опосредованный цисплатином апоптоз способствует образованию реактивных форм кислорода (Wang et al., 2011).
Bmi1 был обнаружен в качестве одного из ключевых регуляторных факторов, определяющих смертельную форму фенотипа при широком спектре устойчивых к терапии опухолей, включая рак ПЖ, молочной железы, легких, яичников и мочевого пузыря (Glinsky et al., 2005). Такой фенотип основан на активируемом Bmi1 консервативном сигнальном пути, включающем активность 11 генов, экспрессия которых определяет стволовость и устойчивость к терапии опухолей различного тканевого происхождения. Bmi1 оккупирует локус опухолевого супрессора PTEN и тормозит его экспрессию (Song et al., 2009). В результате активируется сигнальный путь PI3K/AKT, стабилизируется Snail, который супрессирует продукцию E-кадгерина. Bmi1 связывается с промотором CDH1, который кодирует E-кадгерин (Song et al., 2009). Bmi1 активирует пролиферацию путем повышения экспрессии Cdk2, Cdk4 и циклина D1 (Lee et al., 2008). Наночастицы, несущие siRNA против BMI1 были успешно использованы для торможения роста хеморезистентных раковых клеток (Wang et al., 2011), что вероятно связано с удалением ОСК (Liu et al., 2006).
ЗАКЛЮЧЕНИЕ
Белки семейства Polycomb, структурно организованные в комплексы PRC1 и PRC2, играют ключевую роль в эпигенетической регуляции при формировании и дифференцировке стволовых клеток различной тканевой специфичности. В нормальных стволовых клетках происходит сбалансированной взаимодействие комплексов PRC1 и PRC2. Гиперпродукция отдельных компонентов этих комплексов, например, Ezh2 и Bmi1, является причиной формирования ОСК, злокачественной трансформации и возникновения опухолей в различных тканях, включая РПЖ, который в настоящее время занимает лидирующие позиции в заболеваемости и смертности среди всех онкологических заболеваний. Повышенный уровень белков Ezh2 и Bmi1 опосредует рецидивы опухоли, метастазирование, и устойчивость к противоопухолевой терапии. Эффективность противоопухолевой терапии связана с поиском и применением препаратов, тормозящих гиперпродукцию Ezh2 и Bmi1. Дальнейшая характеристика роли всего семейства PcG при РПЖ и опухолях другой тканевой специфичности будет способствовать их эффективному скринингу, диагностике и терапии.
Список литературы
Петров Н.С., Верещагина Н.А., Сушилова Е.Н., Кропотов А.В., Михеева Н.Ф., Попов Б.В. 2016. Продукт гена BMI1 – ключевой компонент семейства Polycomb – позитивно регулирует жировую дифференцировку мезенхимных стволовых клеток. Цитология. 58 : 83–90. (Petrov N.S., Vereschagina N.A., Sushilova E.N., Kropotov A.V., Miheeva N.F., Popov B.V. 2016. Product of the BMI1 – a key component of Polycomb – positively regulates adipocyte differentiation of mouse mesenchymal stem cells. Tsitologiia. 58 : 83–90).
Abate-Shen C., Shen M.M. 2000. Molecular genetics of prostate cancer. Genes Dev. 14 : 2410–2434.
Adhikary G., Grun D., Balasubramanian S., Kerr C., Huang J.M., Eckert R.L. 2015. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis. 36 : 800–810.
Alam T.N., O’Hare M.J., Laczkó I., Freeman A., Al-Beidh F., Masters J.R., Hudson D.L. 2004. Differential expression of CD44 during human prostate epithelial cell differentiation. J. Histochem. Cytochem. 52 : 1083–1090.
Aryee M.J., Liu W., Engelmann J.C., Nuhn P., Gurel M., Haffner M.C., Esopi D., Irizarry R.A., Getzenberg R.H., Nelson W.G., Luo J., Xu J., Isaacs W.B., Bova G.S., Yegnasubramanian S. 2013. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl. Med. 5 : 169ra10.
Baker C.M., Verstuyf A., Jensen K.B., Watt F.M. 2010. Differential sensitivity of epidermal cell subpopulations to beta-catenin-induced ectopic hair follicle formation. Dev. Biol. 343 : 40–50.
Ballaré C., Lange M., Lapinaite A., Martin G.M., Morey L., Pascual G., Liefke R., Simon B., Shi Y., Gozani O., Carlomagno T., Benitah S.A., Di Croce L. 2012. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 19 : 1257–1265.
Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444 : 756–760.
Barry M.J. 2009. Screening for prostate cancer–the controversy that refuses to die. N. Engl. J. Med. 360 : 1351-1354.
Bartsch G., Rohr H.P. 1980. Comparative light and electron microscopic study of the human, dog and rat prostate. An approach to an experimental model for human benign prostatic hyperplasia (light and electron microscopic analysis) – a review. Urol. Int. 35 : 91–104.
Bello-DeOcampo D., Kleinman H.K., Deocampo N.D., Webber M.M. 2001. Laminin-1 and alpha6beta1 integrin regulate acinar morphogenesis of normal and malignant human prostate epithelial cells. Prostate. 46 : 142–153.
Bentley M.L., Corn J.E., Dong K.C., Phung Q., Cheung T.K., Cochran A.G. 2011. Recognition of UbcH5c and the nucleosome by the Bmi1/Ring1b ubiquitin ligase complex. EM-BO J. 30 : 3285–3297.
Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S.L., Lander E.S. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 125 : 315–326.
Bernstein E., Duncan E.M., Masui O., Gil J., Heard E., Allis C.D. 2006. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 26 : 2560–2569.
Blackledge N.P., Farcas A.M., Kondo T., King H.W., McGouran J.F., Hanssen L.L., Ito S., Cooper S., Kondo K., Koseki Y., Ishikura T., Long H.K., Sheahan T.W., Brockdorff N., Kessler B.M., Koseki H., Klose R.J. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 157 : 1445–1459.
Bonnet D., Dick J.E. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3 : 730–737.
Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Bell G.W., Otte A.P., Vidal M., Gifford D.K., Young R.A., Jaenisch R. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 441 : 349–353.
Bracken A.P., Helin K. 2009. Polycomb group proteins: navigators of lineage pathways led astray in cancer, Nat. Rev. Cancer. 9 : 773–784.
Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. 2003. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22 : 5323–5335.
Brawer M.K., Peehl D.M., Stamey T.A., Bostwick D.G. 1985. Keratin immunoreactivity in the benign and neoplastic human prostate. Cancer Res. 45 : 3663–3667.
Brooun A., Gajiwala K.S., Deng Y.L., Liu W., Bolaños B., Bingham P., He Y.A., Diehl W., Grable N., Kung P.P., Sutton S., Maegley K.A., Yu X., Stewart A.E. 2016. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 7 : 11 384.
Buchwald G., van der Stoop P., Weichenrieder O., Perrakis A., van Lohuizen M., Sixma T.K. 2006. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 25 : 2465–2474.
Burdon T., Smith A., Savatier P. 2002. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell. Biol. 12 : 432–438.
Campisi J. 2003. Cancer and ageing: rival demons? Nat. Rev. Cancer. 3 : 339–349.
Cao L., Bombard J., Cintron K., Sheedy J., Weetall M.L., Davis T.W. 2011. BMI1 as a novel target for drug discovery in cancer. J. Cell Biochem. 112 : 2729–2741.
Cao L., Peng B., Yao L., Zhang X., Sun K., Yang X., Yu L. 2010. The ancient function of RB-E2F pathway: Insights from its evolutionary history. Biol. Direct. 5 : 55.
Cao R., Tsukada Y., Zhang Y. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 20 : 845–854.
Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 298 : 1039–1043.
Caretti G., Di Padova M., Micales B., Lyons G.E., Sartorelli V. 2004. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 18 : 2627–2638.
Chaffer C.L., Brueckmann I., Scheel C., Kaestli A.J., Wiggins P.A., Rodrigues L.O., Brooks M., Reinhardt F., Su Y., Polyak K., Arendt L.M., Kuperwasser C., Bierie B., Weinberg R.A. 2011. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA. 108 : 7950–7955.
Chen Q., Zheng P.S., Yang W.T. 2016. EZH2-mediated repression of GSK-3β and TP53 promotes Wnt/β-catenin signaling-dependent cell expansion in cervical carcinoma. Oncotarget. 7 : 36115–36129.
Clevers H. 2011. The cancer stem cell: Premises, promises and challenges. Nat. Med. 17 : 313–319.
Cobaleda C., Schebesta A., Delogu A., Busslinger M. 2007. Pax5: The guardian of B cell identity and function. Nat. Immunol. 8 : 463–470.
Collins A.T., Berry P.A., Hyde C., Stower M.J., Maitland N.J. 2005. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65 : 10946–10951.
Comet I., Riising E.M., Leblanc B., Helin K. 2016. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer. 16 : 803–810.
Cooper S., Dienstbier M., Hassan R., Schermelleh L., Sharif J., Blackledge N.P., De Marco V., Elderkin S., Koseki H., Klose R., Heger A., Brockdorff N. 2014. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 7 : 1456–1470.
Cui H., Hu B., Li T., Ma J., Alam G., Gunning W.T., Ding H.F. 2007. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am. J. Pathol. 170 : 1370–1378.
Dyson N.J. 2016. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 30 : 1492–1502.
Ezhkova E., Pasolli H.A., Parker J.S., Stokes N., Su I.H., Hannon G., Tarakhovsky A., Fuchs E. 2009. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 136 : 1122–1135.
Galvis L.A., Holik A.Z., Short K.M., Pasquet J., Lun A.T., Blewitt M.E., Smyth I.M., Ritchie M.E., Asselin-Labat M.L. 2015. Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development. 142 : 1458–1469.
Gibaja V., Shen F., Harari J., Korn J., Ruddy D., Saenz-Vash V., Zhai H., Rejtar T., Paris C.G., Yu Z., Lira M., King D., Qi W., Keen N., Hassan A.Q., Chan H.M. 2016. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 35 : 558–566.
Glinsky G.V., Berezovska O., Glinskii A.B. 2005. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Invest. 115 : 1503–1521.
Glinsky G.V., Glinskii A.B., Stephenson A.J., Hoffman R.M., Gerald W.L. 2004. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Invest. 113 : 913–923.
Goldstein A.S., Lawson D.A., Cheng D., Sun W., Garraway I.P., Witte O.N. 2008. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. USA. 105 : 20882–20887.
Goldstein A.S., Zong Y., Witte O.N. 2011. A two-step toward personalized therapies for prostate cancer. Sci. Transl. Med. 3 : 72ps7.
Guo B.H., Feng Y., Zhang R., Xu L.H., Li M.Z., Kung H.F., Song L.B., Zeng M.S. 2011. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol. Cancer. 10 : 10.
Guo W.J., Datta S., Band V., Dimri G.P. 2007. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol. Biol. Cell. 18 : 536–546.
Gutiérrez L., Oktaba K., Scheuermann J.C., Gambetta M.C., Ly-Hartig N., Müller J. 2012. The role of the histone H2A ubiquitinase Sce in Polycomb repression. Development. 139 : 117–127.
Hadorn E. 1968. Transdetermination in cells. Sci. Am. 219 : 110–114.
Harbour J.W., Dean D.C. 2000. Chromatin remodeling and Rb activity. Curr. Opin. Cell. Biol. 12 : 685–689.
Hauri S., Comoglio F., Seimiya M., Gerstung M., Glatter T., Hansen K., Aebersold R., Paro R., Gstaiger M., Beisel C. 2016. A high-density map for navigating the human Polycomb complexome. Cell Rep. 17 : 583–595.
Hemming S., Cakouros D., Isenmann S., Cooper L., Menicanin D., Zannettino A., Gronthos S. 2014. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells. 32 : 802–815.
Herz H.M., Madden L.D., Chen Z., Bolduc C., Buff E., Gupta R., Davuluri R., Shilatifard A., Hariharan I., Bergmann A. 2010. The H3K27me3 demethylase dUTX a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol. Cell. Biol. 30 : 2485–2497.
Hirabayashi Y., Suzki N., Tsuboi M., Endo T.A., Toyoda T., Shinga J., Koseki H., Vidal M., Gotoh Y. 2009. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 63 : 600–613.
Honig A., Weidler C., Häusler S., Krockenberger M., Buchholz S., Köster F., Segerer S.E., Dietl J., Engel J.B. 2010. Overexpression of polycomb protein BMI-1 in human specimens of breast, ovarian, endometrial and cervical cancer. Anticancer Res. 30 : 1559–1564.
Hosen N., Yamane T., Muijtjens M., Pham K., Clarke M.F., Weissman I.L. 2007. Bmi-1-green fluorescent protein-knock-in mice reveal the dynamic regulation of bmi-1 expression in normal and leukemic hematopoietic cells. Stem Cells. 25 : 1635–1644.
Hsieh Y.Y., Lo H.L., Yang P.M. 2012. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am. J. Cancer Res. 6 : 1661–1680.
Hurt E.M., Kawasaki B.T., Klarmann G.J., Thomas S.B., Farrar W.L. 2008. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer. 98 : 756–765.
Hussain M., Rao M., Humphries A.E., Hong J.A., Liu F., Yang M., Caragacianu D., Schrump D.S. 2009. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 69 : 3570–3578.
Jacobs J.J., Kieboom K., Marino S., DePinho R.A., van Lohuizen M. 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 397 : 164–168.
Jiao J., Hindoyan A., Wang S., Tran L.M., Goldstein A.S., Lawson D., Chen D., Li Y., Guo C., Zhang B., Fazli L., Gleave M., Witte O.N., Garraway I.P., Wu H. 2012. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS One. 7 : e42564.
Justin N., Zhang Y., Tarricone C., Martin S.R., Chen S., Underwood E., De Marco V., Haire L.F., Walker P.A., Reinberg D., Wilson J.R., Gamblin S.J. 2016. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7 : 11316.
Kalb R., Latwiel S., Baymaz H.I., Jansen P.W., Müller C.W., Vermeulen M., Müller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 21 : 569–571.
Kareta M.S., Gorges L.L., Hafeez S., Benayoun B.A., Marro S., Zmoos A.F., Cecchini M.J., Spacek D., Batista L.F., O’Brien M., Ng Y.H., Ang C.E., Vaka D., Artandi S.E., Dick F.A., Brunet A., Sage J., Wernig M. 2015. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. 16 : 39–50.
Kennison J.A. 1995. The Polycomb and Trithorax group proteins of Drosophila: trans-regulators of homeotic gene function. Annu. Rev. Genet. 29 : 289–303.
Kikuchi J., Takashina T., Kinoshita I., Kikuchi E., Shimizu Y., Sakakibara-Konishi J., Oizumi S., Marquez V.E., Nishimura M., Dosaka-Akita H. 2012. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer. 78 : 138–143.
Kitajima S., Kohno S., Kondoh A., Sasaki N., Nishimoto Y., Li F., Abdallah Mohammed M.S., Muranaka H., Nagatani N., Suzuki M., Kido Y., Takahashi C. 2015. Undifferentiated State Induced by Rb-p53 Double Inactivation in Mouse Thyroid Neuroendocrine Cells and Embryonic Fibroblasts. Stem Cells. 33 : 1657–1669.
Klarmann G.J., Hurt E.M., Mathews L.A., Zhang X., Duhagon M.A., Mistree T., Thomas S.B., Farrar W.L. 2009. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis. 26 : 433–446.
Klauke K., Radulović V., Broekhuis M., Weersing E., Zwart E., Olthof S., Ritsema M., Bruggeman S., Wu X., Helin K., Bystrykh L., de Haan G. 2013. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat. Cell Biol. 15 : 353–362.
Kloet S.L., Makowski M.M., Baymaz H.I., van Voorthuijsen L., Karemaker I.D., Santanach A., Jansen P.W.T.C., Di Croce L., Vermeulen M. 2016. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 23 : 682–690.
Kohno S., Kitajima S., Sasaki N., Takahashi C. 2016. Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World J. Stem Cells. 8 : 170–184.
Kotake Y., Cao R., Viatour P., Sage J., Zhang Y., Xiong Y. 2007. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 21 : 49–54.
Krivtsov A.V., Twomey D., Feng Z., Stubbs M.C., Wang Y., Faber J., Levine J.E., Wang J., Hahn W.C., Gilliland D.G., Golub T.R., Armstrong S.A. 2006. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 442 : 818–822.
Lee D.K., Liu Y., Liao L., Wang F., Xu J. 2014. The prostate basal cell (BC) heterogeneity and the p63-positive BC differentiation spectrum in mice. Int. J. Biol. Sci. 10 : 1007–1017.
Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Koseki H., Fuchikami T., Abe K., Murray H.L., Zucker J.P., Yuan B., Bell G.W., Herbolsheimer E., Hannett N.M., Sun K., Odom D.T., Otte A.P., Volkert T.L., Bartel D.P., Melton D.A., Gifford D.K., Jaenisch R., Young R.A. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 125 : 301–313.
Lee W., Teckie S., Wiesner T., Ran L., Prieto Granada C.N., Lin M., Zhu S., Cao Z., Liang Y., Sboner A., Tap W.D., Fletcher J.A., Huberman K.H., Qin L.X., Viale A., Singer S., Zheng D., Berger M.F., Chen Y., Antonescu C.R., Chi P. 2014. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 46 : 1227–1232.
Lewis P.W., Müller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D. 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 340 : 857–861.
Li K., Liu C., Zhou B., Bi L., Huang H., Lin T., Xu K. 2013. Role of EZH2 in the growth of prostate cancer stem cells isolated from LNCaP cells. Int. J. Mol. Sci. 14 : 11981–11993.
Li X., Lewis M.T., Huang J., Gutierrez C., Osborne C.K., Wu M.F., Hilsenbeck S.G., Pavlick A., Zhang X., Chamness G.C., Wong H., Rosen J., Chang J.C. 2008. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer. Inst. 100 : 672–679.
Li Y., Hwang T.H., Oseth L.A., Hauge A., Vessella R.L., Schmechel S.C., Hirsch B., Beckman K.B., Silverstein K.A., Dehm S.M. 2012. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 31 : 4759–4767.
Liu L., Andrews L.G., Tollefsbol T.O. 2006. Loss of the human polycomb group protein BMI1 promotes cancer-specific cell death. Oncogene. 25 : 4370–4375.
Liu T., Hou L., Huang Y. 2014. EZH2-specific microRNA-98 inhibits human ovarian cancer stem cell proliferation via regulating the pRb-E2F pathway. Tumour Biol. 35 : 7239–7247.
Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M., Campbell L.L., Polyak K., Brisken C., Yang J., Weinberg R.A. 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 133 : 704–715.
Marjanovic N.D., Weinberg R.A., Chaffer C.L. 2013. Cell plasticity and heterogeneity in cancer. Clin. Chem. 59 : 168–179.
McEvoy J., Flores-Otero J., Zhang J., Nemeth K., Brennan R., Bradley C., Krafcik F., Rodriguez-Galindo C., Wilson M., Xiong S., Lozano G., Sage J., Fu L., Louhibi L., Trimarchi J., Pani A., Smeyne R., Johnson D., Dyer M.A. 2011. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 20 : 260–275.
Merini W., Calonje M. 2015. PRC1 is taking the lead in PcG repression. Plant J. 83 : 110–120.
Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., Lee W., Mendenhall E., O’Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E.S., Bernstein B.E. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 448 : 553–560.
Min J., Zaslavsky A., Fedele G., McLaughlin S.K., Reczek E.E., De Raedt T., Guney I., Strochlic D.E., Macconaill L.E., Beroukhim R., Bronson R.T., Ryeom S., Hahn W.C., Loda M., Cichowski K. 2010. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 16 : 286–294.
Molofsky A.V., Pardal R., Iwashita T., Park I.K., Clarke M.F., Morrison S.J. 2003. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 425 : 962–967.
Morel A.P., Lièvre M., Thomas C., Hinkal G., Ansieau S., Puisieux A. 2008. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 3 : e2888.
Morey L., Santanach A., Blanco E., Aloia L., Nora E.P., Bruneau B.G., Di Croce L. 2015. Polycomb Regulates Mesoderm Cell Fate-Specification in Embryonic Stem Cells through Activation and Repression Mechanisms. Cell Stem Cell. 17 : 300–315.
Morin R.D., Johnson N.A., Severson T.M., Mungall A.J., An J., Goya R., Paul J.E., Boyle M., Woolcock B.W., Kuchenbauer F., Yap D., Humphries R.K., Griffith O.L., Shah S., Zhu H., Kimbara M., Shashkin P., Charlot J.F., Tcherpakov M., Corbett R., Tam A., Varhol R., Smailus D., Moksa M., Zhao Y., Delaney A., Qian H., Birol I., Schein J., Moore R., Holt R., Horsman D.E., Connors J.M., Jones S., Aparicio S., Hirst M., Gascoyne R.D., Marra M.A. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42 : 181–185.
Müller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O’Connor M.B., Kingston R.E., Simon J.A. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 111 : 197–120.
Nowell P.C. 1976. The clonal evolution of tumor cell populations. Science. 194 : 23–28.
Ni J., Cozzi P., Hao J., Duan W., Graham P., Kearsley J., Li Y. 2014. Cancer stem cells in prostate cancer chemoresistance. Curr Cancer Drug Targets. 14 : 225–240.
Parimi V., Goyal R., Poropatich K., Yang X.J. 2014. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2 : 273–285.
Pasini D., Cloos P.A., Walfridsson J., Olsson L., Bukowski J.P., Johansen J.V., Bak M., Tommerup N., Rappsilber J., Helin K. 2010. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 464 : 306–310.
Patrawala L., Calhoun T., Schneider-Broussard R., Li H., Bhatia B., Tang S., Reilly J.G., Chandra D., Zhou J., Claypool K., Coghlan L., Tang D.G. 2006. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 25 : 1696–1708.
Perou C.M., Sørlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A., Fluge O., Pergamenschikov A., Williams C., Zhu S.X., Lønning P.E., Børresen-Dale A.L., Brown P.O., Botstein D. 2000. Molecular portraits of human breast tumours. Nature. 406 : 747–752.
Pietersen A.M., Evers B., Prasad A.A., Tanger E., Cornelissen-Steijger P., Jonkers J., van Lohuizen M. 2008. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr. Biol. 18 : 1094–1099.
Piunti A., Shilatifard A. 2016. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 352 : aad9780.
Popov B., Petrov N. 2014. pRb-E2F signaling in life of mesenchymal stem cells: Cell cycle, cell fate, and cell differentiation. Genes & Diseases. 1 : 174–187.
Prajapati A., Gupta S., Mistry B., Gupta S. 2013. Prostate stem cells in the development of benign prostate hyperplasia and prostate cancer: Emerging role and concepts. Biomed. Res. Int. 2013 : 107954.
Prins G.S., Putz O. 2008. Molecular signaling pathways that regulate prostate gland development. Differentiation. 76 : 641–659.
Quintana E., Shackleton M., Sabel M.S., Fullen D.R., Johnson T.M., Morrison S.J. 2008. Efficient tumour formation by single human melanoma cells. Nature. 456 : 593–598.
Rao Z.Y., Cai M.Y., Yang G.F., He L.R., Mai S.J., Hua W.F., Liao Y.J., Deng H.X., Chen Y.C., Guan X.Y., Zeng Y.X., Kung H.F., Xie D. 2010. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis. 31 : 1576–1583.
Richardson G.D., Robson C.N., Lang S.H., Neal D.E., Maitland N.J., Collins A.T. 2004. CD133, a novel marker for human prostatic epithelial stem cells. J. Cell. Sci. 117 : 3539–3545.
Riising E.M., Comet I., Leblanc B., Wu X., Johansen J.V., Helin K. 2014. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell. 55 : 347–360.
Rizo A., Dontje B., Vellenga E., de Haan G., Schuringa J.J. 2008. Long-term maintenance of human hematopoietic stem/progenitor cells by expression of BMI1. Blood. 111 : 2621–2630.
Robinson A.K., Leal B.Z., Chadwell L.V., Wang R., Ilangovan U., Kaur Y., Junco S.E., Schirf V., Osmulski P.A., Gaczynska M., Hinck A.P., Demeler B., McEwen D.G., Kim C.A. 2012. The growth-suppressive function of the polycomb group protein polyhomeotic is mediated by polymerization of its sterile alpha motif (SAM) domain. J. Biol. Chem. 287 : 8702–8713.
Roesch A., Fukunaga-Kalabis M., Schmidt E.C., Zabierowski S.E., Brafford P.A., Vultur A., Basu D., Gimotty P., Vogt T., Herlyn M. 2010. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 141 : 583–594.
Sage J. 2012. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 26 : 1409–1420.
Sangiorgi E., Capecchi M.R. 2008. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 40 : 915–920.
Sasaki M., Yamaguchi J., Itatsu K., Ikeda H., Nakanuma Y. 2008. Over-expression of polycomb group protein EZH2 relates to decreased expression of p16 INK4a in cholangiocarcinogenesis in hepatolithiasis. J. Pathol. 215 : 175–183.
Sarry J.E., Murphy K., Perry R., Sanchez P.V., Secreto A., Keefer C., Swider C.R., Strzelecki A.C., Cavelier C., Récher C., Mansat-De Mas V., Delabesse E., Danet-Desnoyers G., Carroll M. 2011. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Invest. 121 : 384–395.
Sauvageau M., Sauvageau G. 2010. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 7 : 299–313.
Scheel C., Weinberg R.A. 2011. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer. 129 : 2310–2314.
Schröder F.H., Hugosson J., Roobol M.J., Tammela T.L., Ciatto S., Nelen V., Kwiatkowski M., Lujan M., Lilja H., Zappa M., Denis L.J., Recker F., Páez A., Määttänen L., Bangma C.H., Aus G., Carlsson S., Villers A., Rebillard X., van der Kwast T., Kujala P.M., Blijenberg B.G., Stenman U.H., Huber A., Taari K., Hakama M., Moss S.M., de Koning H.J., Auvinen A. 2012. Prostate-cancer mortality at 11 years of follow-up. N. Engl. J. Med. 366 : 981–990.
Schwartz Y.B., Pirrotta V. 2013. A new world of Polycombs: Unexpected partnerships and emerging functions. Nat. Rev. Genet. 14 : 853–864.
Schwartz Y.B., Pirrotta V. 2014. Ruled by ubiquitylation: a new order for polycomb recruitment. Cell Rep. 8 : 321–325.
Schuettengruber B., Bourbon H.M., Di Croce L., Cavalli G. 2017. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell. 171 : 34–57.
Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A., Wong K.K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J. 2010. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 141 : 69–80.
Shema E., Jones D., Shoresh N., Donohue L., Ram O., Bernstein B.E. 2016. Single-molecule decoding of combinatorially modified nucleosomes. Science. 352 : 717–721.
Shiota M., Yokomizo A., Fujimoto N., Naito S. 2011. Androgen receptor cofactors in prostate cancer: Potential therapeutic targets of castration-resistant prostate cancer. Curr. Cancer Drug Targets. 11 : 870–881.
Siegel R.L., Miller K.D., Jemal A. 2016. Cancer statistics, 2016. CA Cancer J. Clin. 66 : 7–30.
Signoretti S., Waltregny D., Dilks J., Isaac B., Lin D., Garraway L., Yang A., Montironi R., McKeon F., Loda M. 2000. p63 is a prostate basal cell marker and is required for prostate development. Am. J. Pathol. 157 : 1769–1775.
Simon J.A., Kingston R.E. 2009. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10 : 697–708.
Song L.B., Li J., Liao W.T., Feng Y., Yu C.P., Hu L.J., Kong Q.L., Xu L.H., Zhang X., Liu W.L., Li M.Z., Zhang L., Kang T.B., Fu L.W., Huang W.L., Xia Y.F., Tsao S.W., Li M., Band V., Band H., Shi Q.H., Zeng Y.X., Zeng M.S. 2009. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Invest. 119 : 3626–3636.
Spaderna S., Schmalhofer O., Hlubek F., Berx G., Eger A., Merkel S., Jung A., Kirchner T., Brabletz T. 2006. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 131 : 830–840.
Strope S.A., Andriole G.L. 2010. Prostate cancer screening: current status and future perspectives. Nat. Rev. Urol. 7 : 487–493.
Surface L.E., Thornton S.R., Boyer L.A. 2010. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell. 7 : 288–298.
Timms B.G. 2008. Prostate development: A historical perspective. Differentiation. 76 : 565–577.
Taylor B.S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B.S., Arora V.K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J.E., Wilson M., Socci N.D., Lash A.E., Heguy A., Eastham J.A., Scher H.I., Reuter V.E., Scardino P.T., Sander C., Sawyers C.L., Gerald W.L. 2010. Integrative genomic profiling of human prostate cancer. Cancer Cell. 18 : 11–22.
Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. 2009. Epithelial-mesenchymal transitions in development and disease. Cell. 139 : 871–890.
Thomas D.M., Carty S.A., Piscopo D.M., Lee J.S., Wang W.F., Forrester W.C., Hinds P.W. 2001. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol. Cell. 8 : 303–316.
Tong Z.T., Cai M.Y., Wang X.G., Kong L.L., Mai S.J., Liu Y.H., Zhang H.B., Liao Y.J., Zheng F., Zhu W., Liu T.H., Bian X.W., Guan X.Y., Lin M.C., Zeng M.S., Zeng Y.X., Kung H.F., Xie D. 2012. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene. 31 : 583–594.
Trerotola M., Rathore S., Goel H.L., Li J., Alberti S., Piantelli M., Adams D., Jiang Z., Languino L.R. 2010. CD133, Trop-2 and alpha2beta1 integrin surface receptors as markers of putative human prostate cancer stem cells. Am. J. Transl. Res. 2 : 135–144.
Valk-Lingbeek M.E., Bruggeman S.W., van Lohuizen M. 2004. Stem cells and cancer; the Polycomb connection. Cell. 118 : 409–418.
Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G., Otte A.P., Rubin M.A., Chinnaiyan A.M. 2002. The Polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 419 : 624–629.
Verhagen A.P., Ramaekers F.C., Aalders T.W., Schaafsma H.E., Debruyne F.M., Schalken J.A. 1992. Colocalization of basal and luminal cell-type cytokeratins in human prostate cancer. Cancer Res. 52 : 6182–6187.
Verma S.K., Tian X., LaFrance L.V., Duquenne C., Suarez D.P., Newlander K.A., Romeril S.P., Burgess J.L., Grant S.W., Brackley J.A., Graves A.P., Scherzer D.A., Shu A., Thompson C., Ott H.M., Aller G.S., Machutta C.A., Diaz E., Jiang Y., Johnson N.W., Knight S.D., Kruger R.G., McCabe M.T., Dhanak D., Tummino P.J., Creasy C.L., Miller W.H. 2012. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 3 : 1091–1096.
Vermeulen L., De Sousa E Melo F., van der Heijden M., Cameron K., de Jong J.H., Borovski T., Tuynman J.B., Todaro M., Merz C., Rodermond H., Sprick M.R., Kemper K., Richel D.J., Stassi G., Medema J.P. 2010. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell. Biol. 12 : 468–476.
Viré E., Brenner C., Deplus R., Blanchon L., Fraga M., Didelot C., Morey L., Van Eynde A., Bernard D., Vanderwinden J.M., Bollen M., Esteller M., Di Croce L., de Launoit Y., Fuks F. 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 439 : 871–874.
Visvader J.E. 2011. Cells of origin in cancer. Nature. 469 : 314–322.
Visvader J.E., Lindeman G.J. 2008. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer. 8 : 755–768.
Wang C., Liu X., Chen Z., Huang H., Jin Y., Kolokythas A., Wang A., Dai Y., Wong D.T., Zhou X. 2013. Polycomb group protein EZH2-mediated E-cadherin repression promotes metastasis of oral tongue squamous cell carcinoma. Mol. Carcinog. 52 : 229–236.
Wang E., Bhattacharyya S., Szabolcs A., Rodriguez-Aguayo C., Jennings N.B., Lopez-Berestein G., Mukherjee P., Sood A.K., Bhattacharya R. 2011. Enhancing chemotherapy response with Bmi-1 silencing in ovarian cancer. PLoS One. 6 : e17918.
Wang R., Taylor A.B., Leal B.Z., Chadwell L.V., Ilangovan U., Robinson A. K., Schirf V., Hart P.J., Lafer E.M., Demeler B., Hinck A.P., McEwen D.G., Kim C.A. 2010. Polycomb group targeting through different binding partners of RING1B C-terminal domain. Structure. 18 : 966–975.
Wang Y., Hayward S., Cao M., Thayer K., Cunha G. 2001. Cell differentiation lineage in the prostate. Differentiation. 68 : 270–279.
Weinberg R.A. 1995. The retinoblastoma protein and cell cycle control. Cell. 81 : 323–330.
Williams K., Motiani K., Giridhar P.V., Kasper S. 2013. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. (Maywood). 238 : 324–338.
Wilt T.J., Brawer M.K., Jones K.M., Barry M.J., Aronson W.J., Fox S., Gingrich J.R., Wei J.T., Gilhooly P., Grob B.M., Nsouli I., Iyer P., Cartagena R., Snider G., Roehrborn C., Sharifi R., Blank W., Pandya P., Andriole G.L., Culkin D., Wheeler T. 2012. Prostate Cancer Intervention versus Observation for Localized Prostate Cancer. Radical prostatectomy versus observation for localized prostate cancer. N. Engl. J. Med. 367 : 203–213.
Wu C., Jin X., Yang J., Yang Y., He Y., Ding L., Pan Y., Chen S., Jiang J., Huang H. 2016. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget. 7 : 3440–3452.
Wu Y., Yu J., Liu Y., Yuan L., Yan H., Jing J., Xu G. 2014. Delivery of EZH2-shRNA with mPEG-PEI nanoparticles for the treatment of prostate cancer in vitro. Int. J. Mol. Med. 33 : 1563–1569.
Xin L. 2013. Cells of origin for cancer: An updated view from prostate cancer. Oncogene. 32 : 3655–3663.
Xin L., Lawson D.A., Witte O.N. 2005. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA. 102 : 6942–6947.
Yan M., Zhang Y., He B., Xiang J., Wang Z.F., Zheng F.M., Xu J., Chen M.Y., Zhu Y.L., Wen H.J., Wan X.B., Yue C.F., Yang N., Zhang W., Zhang J.L., Wang J., Wang Y., Li L.H., Zeng Y.X., Lam E.W., Hung M.C., Liu Q. 2014. IKKα restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat. Commun. 5 : 3661.
Yang C., Bratzel F., Hohmann N., Koch M., Turck F., Calonje M. 2013. VAL- and AtBMI1-mediated H2Aub initiate the switch from embryonic to postgerminative growth in Arabidopsis. Curr. Biol. 23 : 1324–1329.
Yegnasubramanian S. 2016. Prostate cancer epigenetics and its clinical implications. Asian J. Androl. 18 : 549–558.
Yin A.H., Miraglia S., Zanjani E.D., Almeida-Porada G., Ogawa M., Leary A.G., Olweus J., Kearney J., Buck D.W. 1997. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 90 : 5002–5012.
Yu C.C., Lo W.L., Chen Y.W., Huang P.I., Hsu H.S., Tseng L.M., Hung S.C., Kao S.Y., Chang C.J., Chiou S.H. 2011. Bmi-1 Regulates Snail Expression and Promotes Metastasis Ability in Head and Neck Squamous Cancer-Derived ALDH1 Positive Cells. J. Oncol. 2011. pii: 609259.
Zdzieblo D., Li X., Lin Q., Zenke M., Illich D.J., Becker M., Müller A.M. 2014. Pcgf6, a polycomb group protein, regulates mesodermal lineage differentiation in murine ESCs and functions in iPS reprogramming. Stem Cells. 32 : 3112–3125.
Zhang J., Chen L., Han L., Shi Z., Zhang J., Pu P., Kang C. 2015. EZH2 is a negative prognostic factor and exhibits pro-oncogenic activity in glioblastoma. Cancer Lett. 356 : 929–936.
Zhang S., Balch C., Chan M.W., Lai H.C., Matei D., Schilder J.M., Yan P.S., Huang T.H., Nephew K.P. 2008. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 68 : 4311–4320.
Zhu J., Yang D.R., Sun Y., Qiu X., Chang H.C., Li G., Shan Y., Chang C. 2015. TR4 Nuclear Receptor Alters the Prostate Cancer CD133+ Stem/Progenitor Cell Invasion via Modulating the EZH2-Related Metastasis Gene Expression. Mol. Cancer Ther. 14 : 1445–1453.
Дополнительные материалы отсутствуют.
Инструменты
Цитология