

Цитология, 2019, T. 61, № 11, стр. 895-901
Влияние 2-аминоэтоксидифенил-бората на депозависимый вход Са2+ в перитонеальных макрофагах
Л. С. Миленина 1, *, З. И. Крутецкая 1, **, В. Г. Антонов 2, Н. И. Крутецкая 1
1 Кафедра биофизики Санкт-Петербургского государственного университета
199034 Санкт-Петербург, Россия
2 Кафедра клинической биохимии и лабораторной диагностики Военно-Медицинской академии им. C.М. Кирова
194044 Санкт-Петербург, Россия
* E-mail: cozzy@mail.ru
** E-mail: z.krutetskaya@spbu.ru
Поступила в редакцию 07.06.2019
После доработки 17.07.2019
Принята к публикации 19.07.2019
Аннотация
Депозависимый вход Са2+ является универсальным механизмом регулируемого входа Са2+ в клетки эукариот. Для выяснения фармакологических характеристик депозависимого входа Са2+, исследовали влияние 2-аминоэтоксидифенил-бората (2-APB) на депозависимый вход Са2+ в перитонеальных макрофагах крысы, вызываемый ингибиторами эндоплазматических Са2+-АТФаз тапсигаргином и циклопьязониковой кислотой, а также дисульфидсодержащими иммуномодуляторами глутоксимом и моликсаном. С использованием флуоресцентного Са2+-зонда Fura-2AM впервые показано, что в перитонеальных макрофагах крысы, как и в клетках других типов, 2-APB оказывает дозозависимое модулирующее влияние на депозависимый вход Са2+. В концентрации 25 мкМ 2-APB вызывает потенциацию входа Са2+, в то время как в концентрациях 50 и 100 мкМ эффективно подавляет депозависимый вход Са2+ в макрофаги. Кроме того, полученные данные дополнительно подтверждают, что вход Са2+, индуцируемый глутоксимом или моликсаном, происходит по депозависимому механизму.
Депозависимый или “емкостной” вход Са2+, впервые описанный Джеймсом Патни более тридцати лет назад, является повсеместным механизмом регулируемого входа Са2+ в клетки эукариот, активируемым при опустошении внутриклеточных Са2+-депо (Putney, 1990, 2017). Депозависимый вход Са2+ участвует в регуляции широкого спектра клеточных процессов (экзоцитоз, экспрессия генов, рост и пролиферация клеток и др.) в норме и патологии (Putney, 2011; Prakriya, Lewis, 2015).
Функциональной единицей депозависимого входа Са2+ является мультимолекулярный белковый комплекс (store-operated calcium influx complex, SO-CIC), компоненты которого обладают высокой мобильностью, и взаимодействия между ними жестко регулируются (Vaca, 2010; Moreno, Vaca, 2012). Основными компонентами комплекса, необходимыми и достаточными для активации депозависимого входа Са2+, являются Са2+-каналы Orai1 в плазмалемме и Са2+-сенсор STIM1 в мембране Са2+-депо (Prakriya, Lewis, 2015). При опустошении Са2+-депо, STIM1 олигомеризуется, транслоцируется в участки эндоплазматического ретикулума, расположенные у плазмалеммы, и прямо взаимодействует с белками Orai1, вызывая депозависимый вход Са2+ (Nwokonko et al., 2017; Nguyen et al., 2018; Lunz et al., 2019).
После обнаружения важной роли депозависимых Са2+-каналов в патогенезе тяжелых заболеваний человека, таких как тяжелый комбинированный иммунодефицит (severe combined immunodeficiency, SCID), назальный полипоз, ревматоидный артрит, эктодермальная дисплазия, тромбоз, острый панкреатит, аутоиммунные и аллергические заболевания (Shaw, Feske, 2013; Lacruz, Feske, 2015; Feske, 2019) возрос интерес исследователей к разработке низкомолекулярных блокаторов депозависимых Са2+-каналов.
Из различных фармакологических модуляторов депозависимых Са2+-каналов лучше других исследован 2-аминоэтоксидифенил-борат (2-APB). 2-APB широко использовался в качестве инструмента исследования механизмов функционирования депо-зависимых Са2+-каналов (Putney, 2001; Bootman et al., 2002; Putney, 2010). Показано, что 2-APB модулирует депозависимый вход Са2+ в клетках разных типов (Putney, 2010; Jairaman, Prakriya, 2013; Bird, Putney, 2018).
Ранее (Курилова и др., 2008, 2011) нами было впервые показано, что дисульфидсодержащие иммуномодуляторы глутоксим® (динатриевая соль окисленного глутатиона с d-металлом в наноконцентрации) и моликсан® (комплекс глутоксима и нуклеозида инозина) (ФАРМА-ВАМ, Санкт-Петербург) увеличивают внутриклеточную концентрацию Са2+ ([Ca2+]i), вызывая мобилизацию Са2+ из тапсигаргинчувствительных Са2+-депо и последующий депозависимый вход Са2+ в перитонеальные макрофаги крыс.
Учитывая важную роль депозависимых Са2+-каналов в функционировании клеток иммунной системы и для выяснения фармакологических характеристик депозависимого входа Са2+ в макрофагах, представлялось целесообразным исследовать влияние 2-APB на депозависимый вход Са2+, вызываемый ингибиторами эндоплазматических Са2+-АТ-Фаз тапсигаргином (Thastrup et al., 1989) и циклопьязониковой кислотой (ЦПК) (Goeger et al., 1988) и иммуномодуляторами глутоксимом и моликсаном, в перитонеальных макрофагах крысы.
МАТЕРИАЛ И МЕТОДИКА
Выделение и культивирование перитонеальных макрофагов крыс. Эксперименты проводили на культивируемых резидентных перитонеальных макрофагах крыс линии Wistar. Содержание животных и все манипуляции выполняли в соответствии с нормативными документами и требованиями Приказа Минздрава России № 267 от 19.06.03 “Об утверждении правил лабораторной практики в Российской Федерации”.
Резидентные макрофаги выделяли из перитонеальной полости крыс массой 180–250 г по традиционному методу, сразу после выделения клетки имели сферическую форму (диаметр 10–20 мкм) (Conrad, 1981; Randriamampita, Trautmann, 1987). Суспензию клеток помещали в бакпечатки с кварцевыми стеклами (10 × 10 мм) и культивировали клетки в течение 1–3 сут при 37°С в среде 199 (рН 7.2), содержащей 20% сыворотки крови быка, глутамин (3%), пенициллин (100 ед./мл) и стрептомицин (100 мг/мл). Тестом на α-нафтилэстеразу подтверждали, что по меньшей мере 96% клеток в монослоях являлись макрофагами (Monahan et al., 1981).
Эксперименты проводили при комнатной температуре (22–24°С) через 1–2 сут после начала культивирования клеток. Кварцевые стекла с клетками помещали в экспериментальную камеру, заполненную физиологическим раствором следующего ионного состава (мМ): NaCl – 140, KCl – 5, CaCl2 – 1, MgCl2 – 1, и HEPES-NaOH – 5, рН 7.3–7.4. Бескальциевая среда отличалась тем, что содержала 0 мМ CaCl2 и 1 мМ ЭГТА.
Измерение [Ca2+]i проводили с использованием флуоресцентного зонда Fura-2AM (Sigma-Aldrich, США). Макрофаги инкубировали в течение 45 мин в физиологическом растворе, содержащем 2 мкМ Fura-2AM, при 22–24°С. Стекла с окрашенными клетками отмывали физиологическим раствором и переносили в экспериментальную камеру флуоресцентного микроскопа Leica DM 4000B (Leica Microsystems, Германия). Возбуждение флуоресценции объекта производили при длинах волн 340 и 380 нм через объектив микроскопа. Для выделения соответствующих участков спектра использовали узкополосные оптические фильтры. Эмиссию регистрировали при длине волны 510 нм при помощи специализированной видеокамеры Leica DFC340FX. Для управления экспериментом использовали систему обработки изображения ImageJ (плагин Micro-Manager 1.4).
Результатом измерений являлось отношение интенсивностей флуоресценции Fura-2AM при облучении светом с длиной волны 340 нм к интенсивности флуоресценции при облучении светом с длиной волны 380 нм (Ratio (F340/F380)), где F340 – интенсивность флуоресценции Fura-2AM, связанного с Са2+, а F380 – интенсивность флуоресценции Fura-2AM, не связанного с Са2+, отражающее изменения [Ca2+]i в клетках во время измерений (Bruce, Elliott, 2000; Xie et al., 2002). Для избежания фотовыгорания измерения проводили через каждые 20 с, облучая объект в течение 2 с. В экспериментах применяли объектив 10× с апертурой 8 мм. Значения [Ca2+]i рассчитывали по уравнению Гринкевича (Grynkiewicz et al., 1985). Статистический анализ проводили с применением t-критерия Стьюдента. Данные представлены в виде среднего и стандартного отклонения. Каждую регистрацию получали для группы из 40–50 клеток и на рисунках представлены результаты типичных экспериментов из 6–7 независимых экспериментов. Достоверными считали различия при Р ≤ 0.05.
Использованные реактивы: все реактивы приобретали в фирме Sigma-Aldrich, США. Маточные растворы Fura-2AM (1 мМ), ЦПК (10 мМ) и тапсигаргина (0.5 мМ) готовили в диметилсульфоксиде. Маточный раствор 2-APB (25 мМ) готовили в этиловом спирте. Препараты глутоксим и моликсан предоставлялись фирмой ФАРМА-ВАМ (Санкт-Петербург, Россия). Маточные растворы глутоксима (50 мг/мл) и моликсана (50 мг/мл) готовили в воде.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние 2-APB на депозависимый вход Са2+, индуцируемый ингибиторами эндоплазматических Са2+-АТФаз. В контрольных экспериментах мы обнаружили, что добавление 0.5 мкМ тапсигаргина к макрофагам в бескальциевой среде, вызывает увеличение [Ca2+]i, отражающее мобилизацию Са2+ из внутриклеточных Са2+-депо (рис. 1а). В среднем увеличение [Ca2+]i во время фазы мобилизации составило 30 ± 8 нМ (n = 7). При последующем введении в наружную среду 2 мМ Са2+ наблюдали депозависимый вход Са2+ в цитозоль (рис. 1а). В среднем увеличение [Ca2+]i во время входа Са2+ составило 112 ± 21 нМ (n = 7). Аналогичные результаты мы получили при использовании 10 мкМ ЦПК (рис. 1б). В среднем увеличение [Ca2+]i во время фазы мобилизации Са2+ из депо, вызываемой ЦПК, составило 31 ± 9 нМ (n = 7), а во время входа Са2+ – 99 ± 21 нМ (n = 7) (рис. 1б).
Рис. 1.
Влияние 2-аминоэтоксидифенил-бората (2-APB) на депозависимый вход Са2+, индуцируемый тапсигаргином (0.5 мкМ) (а) и циклопьязониковой кислотой (ЦПК, 10 мкМ) (б) в перитонеальных макрофагах крысы. Объяснения к рис. 1–2: макрофаги стимулировали указанным реагентом в номинально бескальциевой среде, затем вход Са2+ инициировали введением в наружную среду 2 мМ Са2+; на фоне развившегося входа Са2+ добавляли 25, 50 или 100 мкМ 2-аминоэтоксидифенил бората (2-АPB); F340/F380 – отношение интенсивностей флуоресценции Fura-2AM при длинах волн возбуждающего излучения 340 и 380 нм, отн. ед.
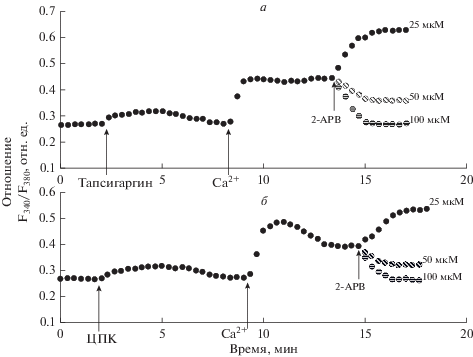
Мы впервые обнаружили, что при добавлении 2-APB на фоне развившегося депозависимого входа Са2+, вызываемого тапсигаргином, действие 2-APB зависит от его концентрации. В концентрации 25 мкМ 2-APB вызывал значительную потенциацию входа Са2+ (на 105.2 ± 25.3%, n = 7) (рис. 1а). В то же время, добавление 2-APB в концентрации 50 или 100 мкМ приводило к подавлению депозависимого входа Са2+ (рис. 1а). Подавление входа Са2+ при действии 50 мкМ 2-APB составило 51.4 ± 14.7% (n = 7), а в концентрации 100 мкМ – 96.0 ± 18.4% (n = 7).
Сходные результаты мы получили при добавлении 2-APB на развившийся депозависимый вход Са2+, индуцированный ЦПК (рис. 1б). При воздействии 25 мкМ 2-APB мы наблюдали потенциацию входа Са2+ на 116.3 ± 23.7% (n = 7). В случае 50 мкМ 2-APB происходило подавление входа Са2+ на 56.5 ± ± 15.3% (n = 7), а при действии 100 мкМ 2-APB – на 97.1 ± 17.5% (n = 7).
Влияние 2-APB на депозависимый вход Са2+, индуцируемый дисульфидсодержащими иммуномодуляторами. В контрольных экспериментах мы обнаружили, что инкубация макрофагов в течение 20 мин с глутоксимом (100 мкг/мл) (рис. 2а) или моликсаном (100 мкг/мл) (рис. 2б) в бескальциевой среде вызывает медленно нарастающее увеличение [Ca2+]i , отражающее мобилизацию Са2+ из внутриклеточных Са2+-депо. В среднем через 20 мин после добавления агентов [Ca2+]i увеличивалась от базального уровня, равного 90 ± 17, до 171 ± 20 нМ (n = 6) для глутоксима и 177 ± 21 нМ (n = 6) для моликсана. При введении в наружную среду 2 мМ Са2+ наблюдали дальнейшее повышение [Ca2+]i, отражающее депозависимый вход Са2+ в цитозоль (рис. 2). В среднем увеличение [Ca2+]i во время входа Са2+ составило 336 ± 34 (n = 6) и 246 ± 31 нМ (n = 6) для глутоксима и моликсана соответственно.
Рис. 2.
Влияние 2-аминоэтоксидифенил бората (2-APB) на депозависимый вход Са2+, вызываемый глутоксимом (100 мкг/мл) (а) или моликсаном (100 мкг/мл) (б) в перитонеальных макрофагах крысы.
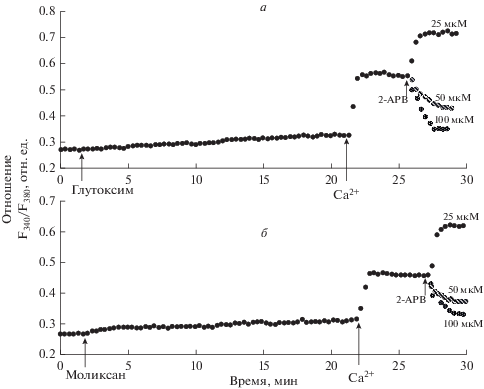
Мы впервые обнаружили, что при добавлении 2-APB на фоне развившегося депозависимого входа Са2+, вызываемого глутоксимом, эффект 2-APB зависит от его концентрации. В концентрации 25 мкМ 2-APB вызывал значительную потенциацию входа Са2+ (на 87.4 ± 19.2%, n = 7) (рис. 2а). В то же время, добавление 2-APB в концентрации 50 или 100 мкМ приводило к значительному подавлению депозависимого входа Са2+ (рис. 2а). Подавление входа Са2+ при действии 50 мкМ 2-APB составило 52.5 ± 12.4% (n = 7), а в концентрации 100 мкМ – 87.2 ± 11.7% (n = 7).
Сходные результаты мы получили в опытах с добавлением 2-APB на развившийся депозависимый вход Са2+, индуцированный моликсаном (рис. 2б). При воздействии 25 мкМ 2-APB наблюдали потенциацию входа Са2+ на 95.3 ± 21.3% (n = 7). При приложении 50 мкМ 2-APB происходило подавление входа Са2+ на 60.5 ± 13.1% (n = 7), а при воздействии 100 мкМ 2-APB – на 90.2 ± 12.8% (n = 7).
Полученные нами результаты согласуются с данными, представленными в литературе. Так, показано, что 50 мкМ 2-APB практически полностью подавляет депозависимый вход Са2+, индуцируемый тапсигаргином в макрофагах из костного мозга мыши (Vaeth et al., 2001), амелобластах мыши (линия LS8) (Nurbaeva et al., 2015) и гладкомышечных клетках легочной артерии крысы (McElroy et al., 2008). Дозозависимое модулирующее влияние 2-APB на депозависимый вход Са2+ показано также на клетках разных типов: B-лимфоцитах цыпленка (линия DT40) (Prakriya, Lewis, 2001; Ma et al., 2002), клетках эмбриональной почки человека (линия HEK293) (De Haven et al., 2008), клетках микроглии крысы (Ohana et al., 2009), T-клетках линии Jurkat человека и лейкемических базофилах крысы (RBL-2H3) (Prakriya, Lewis, 2001). В низких концентрациях (<30 мкМ) 2-APB потенциирует, в то время как в более высоких концентрациях практически полностью подавляет депозависимый вход Са2+. Полагают, что в низких концентрациях 2-APB потенциирует депозависимый вход Са2+ путем прямого расширения поры открытых Orai1 каналов (Xu et al., 2016; Ali et al., 2017). При этом диаметр поры Orai1 каналов увеличивается с 3.8 до 4.6 Å. Обнаружено также, что 2-APB конкурирует со STIM1 за активацию каналов Orai1 (Yamashita et al., 2011). Ингибирование депозависимого входа Са2+ при действии высоких концентраций 2-APB может быть связано с нарушением взаимодействия между белками STIM1 и Orai1 (De Haven et al., 2008; Peinelt et al., 2008; Hendron et al., 2014). В дальнейшем было показано, что 2-APB может подавлять депозависимый вход Са2+, быстро и прямо ингибируя оба молекулярных участника “емкостного” входа Са2+, Orai1 каналы и STIM1 (Wei et al., 2016). Кроме того, 2-APB может опосредованно (более медленно) ингибировать функциональное связывание STIM1 и Orai1 посредством влияния на STIM1, ослабляя взаимодействие между STIM1 и N-терминальным концом канала Orai1 (Wei et al., 2016).
Таким образом, в настоящей работе мы впервые на перитонеальных макрофагах крысы показали, что 2-APB дозозависимо модулирует (активирует или ингибирует) депозависимый вход Са2+, вызываемый ингибиторами эндоплазматических Са2+-АТФаз (тапсигаргин, ЦПК) и иммуномодуляторами глутоксимом и моликсаном. Это комплексное действие 2-APB на депозависимый вход Са2+ в макрофагах может быть связано с его влиянием на оба компонента мультибелкового комплекса депозависимого входа Са2+, на белки STIM1 и Orai1.
Полученные данные свидетельствуют о том, что, как и для клеток других типов, 2-APB является удобным фармакологическим инструментом для изучения депозависимого входа Са2+ в макрофагах. Кроме того, результаты дополнительно подтверждают, что вход Са2+, индуцируемый глутоксимом или моликсаном, происходит по депозависимому механизму.
Список литературы
Курилова Л.С., Крутецкая З.И., Лебедев О.Е., Крутец-кая Н.И., Антонов В.Г. 2008. Влияние окисленного глутатиона и его фармакологического аналога препарата глутоксим на внутриклеточную концентрацию Са2+ в макрофагах. Цитология. 50(5) : 452–461. (Kurilova L.S., Krutetskaya Z.I., Lebedev O.E., Krutetskaya N.I., Antonov V.G. 2008. The effect of oxidized glutathione and its pharmacological analogue glutoxim on intracellular Са2+ concentration in macrophages. Cell Tissue Biol. 2 : 322–332.)
Курилова Л.С., Крутецкая З.И., Лебедев О.Е., Крутецкая Н.И., Антонов В.Г. 2011. Влияние препарата моликсан на процессы Са2+-сигнализации в макрофагах. Цитология. 53(9) : 708. (Kurilova L.S., Krutetskaya Z.I., Lebedev O.E., Krutetskaya N.I., Antonov V.G. 2011. The effect of drug molixan on Са2+-signalling processes in macrophages. Tsitologiya. 53 : 708.)
Ali Sh., Xu T., Xu X. 2017. CRAC channel gating and its modulation by STIM 1 and 2-aminoethoxydiphenyl borate. J. Physiol. 595 : 3085–3095.
Bird G.S., Putney J.W. 2018. Pharmacology of store-operated calcium entry channels. In: Calcium entry channels in non-excitable cells. CRC Press, 311–324.
Bootman M.D., Collins T.J., Mackenzie L., Roderick H.L., Berridge M.J., Peppiatt C.M. 2002. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16 : 1145–1150.
Bruce J.I.E., Elliott A.C. 2000. Pharmacological evaluation of the role of cytochrome P450 in intracellular calcium signaling in rat pancreatic acinar cells. Brit. J. Physiol. 131 : 761–771.
Conrad R.E. 1981. Induction and collection of peritoneal exudate macrophages. In: Manual of macrophages methodo-logy. New York, Marcell Dekker. 5–11.
De Haven W.I., Smyth J.T., Boyles R.R., Bird G.S., Putney J.W. 2008. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 283 : 19 265–19 273.
Feske St. 2019. CRAC channels and disease – from human CRAC channelopathies and animal models to novel drugs. Cell Calcium. 80 : 112–116.
Goeger D.E., Riley R.T., Dorner J.W., Cole R.J. 1988. Cyclopiazonic acid inhibition of the Ca2+ transport ATPase in rat skeletal muscle sarcoplasmic reticulum vesicles. Biochem. Pharmacol. 37 : 978–981.
Grynkiewicz G., Poenie M., Tsien R.Y. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence pro-perties. J. Biol. Chem. 260 : 3440–3450.
Hendron E., Wang X., Zhou Y., Cai X., Goto J.-I., Mikoshiba K., Baba Y., Kurosaki T., Wang Y., Gill D.L. 2014. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium. 56 : 482–492.
Jairaman A., Prakriya M. 2013. Molecular pharmacology of store-operated CRAC channels. Channels. 7 : 402–414.
Lacruz R.S., Feske St. 2015. Diseases caused by mutations in Orai1 and STIM1. Ann. N.Y. Acad. Sci. 1356 : 45–79.
Lunz V., Romanin C., Frischauf I. 2019. STIM1 activation of Orai1. Cell Calcium. 77 : 29–38.
Ma H.-T., Venkatachalam K., Parys J.B., Gill D.L. 2002. Modi-fication of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. 277 : 6915–6922.
McElroy S.P., Gurney A.M., Drummond R.M. 2008. Pharmacological profile of store-operated Ca2+ entry in intrapulmonary artery smooth muscle cells. Eur. J. Pharmacol. 584 : 10–20.
Monahan R.A., Dvorak H.F., Dvorak A.M. 1981. Ultrastructural localization of nonspecific esterase activity in guinea pig and human monocytes, macrophages and lymphocytes. Blood. 58 : 1089–1099.
Moreno C., Vaca L. 2012. Microdomain organization of SOCE signaling. In: Store-operated Ca2+ entry (SOCE) pathways. Wien, Springer-Verlag. 93–113.
Nguyen N.T., Han W., Cao W.-M., Wang Y., Wen S., Huang Y., Li M., Du L., Zhou Y. 2018. Store-operated calcium entry mediated by Orai and STIM. Comprehensive Physiol. 8 : 981–1002.
Nurbaeva M.K., Eckstein M., Snead M.L., Feske S., Lacruz R.S. 2015. Store-operated Ca2+ entry modulates the expression of enamel genes. J. Dent. Res. 94 : 1471–1477.
Nwokonko R.M., Cai X., Loktionova N.A., Wang Y., Zhou Y., Gill D.L. 2017. The STIM−Orai pathway: Conformational coupling between STIM and Orai in the activation of store-operated Ca2+ entry. Adv. Exp. Med. Biol. 993 : 83–98.
Ohana L., Newell E.W., Stanley E.F., Schlichter L.C. 2009. The Ca2+ release-activated Ca2+ current (ICRAC) mediates store-operated Ca2+ entry in rat microglia. Channels. 3 : 129–139.
Peinelt C., Lis A., Beck A., Fleig A., Penner R. 2008. 2-amino-ethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 586 : 3061–3073.
Prakriya M., Lewis R.S. 2001. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethoxydiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 536 : 3–19.
Prakriya M., Lewis R.S. 2015. Store-operated calcium channels. Physiol. Rev. 95 : 1383–1436.
Putney J.W. 1990. Capacitative calcium entry revisited. Cell Calcium. 11 : 611–624.
Putney J.W. 2001. Pharmacology of capacitative calcium entry. Mol. Interv. 1 : 84–94.
Putney J.W. 2010. Pharmacology of store-operated calcium channels. Mol. Interv. 10 : 209–218.
Putney J.W. 2011. The physiological function of store-operated calcium entry. Neurochem. Res. 36 : 1157–1165.
Putney J.W. 2017. Store-operated calcium entry: An historical overview. Adv. Exp. Med. Biol. 981 : 205–214.
Randriamampita C., Trautmann A. 1987. Ionic channels in murine macrophages. Cell. Biol. 105 : 761–769.
Shaw P. J., Feske St. 2013. Physiological and pathophysiological functions of SOCE in the immune system. Front. Biosci. 4 : 2253–2268.
Thastrup O., Dawson A.P., Scharff O., Foder B., Cullen P. J., Drobak B. K., Bjerrum P. J., Christensen S. B., Hanley M. R. 1989. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions. 27 : 17–23.
Vaca L. 2010. SOCIC: The store-operated calcium influx complex. Cell Calcium. 47 : 199–209.
Vaeth M., Zee I., Concepcion A.R., Maus M., Shaw P., Portal-Celhay C., Zahra A., Kozhaya L., Weidinger C., Philips J., Unutmaz D., Feske S. 2001. Ca2+-signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells. J. Immunol. 195 : 1202–1217.
Wei M., Zhou Y., Sun A., Ma G., He L., Zhou L., Zhang S., Liu J., Zhang S.L., Gill D.L., Wang Y. 2016. Molecular mechanisms underlying inhibition of STIM1−Orai1-mediated Ca2+ entry induced by 2-aminoethoxydiphenyl borate. Pflugers Arch. 468 : 2061–2074.
Xie Q., Zhang Y., Zhai C., Bonanno J.A. 2002. Calcium influx factor from cytochrome P-450 metabolism and secretion-like coupling mechanisms for capacitative calcium entry in corneal endothelial cells. J. Biol. Chem. 277 : 16 559–16 566.
Xu X., Ali Sh., Li Y., Yu H., Zhang M., Lu J., Xu T. 2016. 2-ami-noethoxydiphenyl borate potentiates CRAC current by directly dilating the pore of open Orai1. Sci. Rep. 6 : 29304.
Yamashita M., Somasundaram A., Prakriya M. 2011. Competitive modulation of Ca2+ release-activated Ca2+ channel gating by STIM1 and 2-aminoethyldiphenyl borate. J. Biol. Chem. 286 : 9429–9442.
Дополнительные материалы отсутствуют.