

Цитология, 2019, T. 61, № 12, стр. 978-986
Клеточная нейротоксическая модель болезни Паркинсона – динамика дегенерации дофаминергических нейронов под влиянием токсина МФП+
С. А. Сурков 1, Э. Р. Мингазов 1, А. И. Стурова 1, В. Е. Блохин 1, *, М. В. Угрюмов 1
1 Институт биологии развития им. Н.К. Кольцова РАН
119334 Москва, Россия
* E-mail: victor.blokhin@hotmail.com
Поступила в редакцию 09.08.2019
После доработки 19.09.2019
Принята к публикации 20.09.2019
Аннотация
Болезнь Паркинсона (БП) – нейродегенеративное заболевание, характеризующееся гибелью дофаминергических нейронов нигростриатной системы. Молекулярные механизмы, запускающие нейродегенерацию, до сих пор не выяснены и их продолжают изучать как в клинике, так и на моделях in vivo и in vitro. К наиболее перспективным относятся нейротоксические модели in vitro на основе первичной культуры мезенцефалона. В данной работе в течение недели культивировали клетки мезенцефалона эмбрионов мыши, используя для последующей характеристики дифференцирующихся дофаминергических (ДА-ергических) нейронов морфологический, физиологический и биохимический подходы. Через неделю после начала культивирования в среду на 24 ч добавляли МФП+, специфический нейротоксин катехоламинергических нейронов. При этом обнаружено: снижение до 58% числа дофаминергических нейронов, уменьшение общей длины нейритов на 65% на нейрон, снижение более чем в два раза общего содержания дофамина в нейронах и снижение до 85% интенсивности специфического захвата нейронами дофамина. Изучение дегенерации нейронов в динамике показало, что морфологические изменения начинаются с нейритов через 6 ч после введения МФП+. Доказательством специфичности действия МФП+ было значительное снижение числа дегенерировавших ДА-ергических нейронов после одновременного с МФП+ добавления в среду GBR 12909 – ингибитора мембранного переносчика дофамина и МФП+. Всесторонне охарактеризованная нами модель БП может быть успешно использована для изучения клеточных и молекулярных механизмов нейродегенерации и нейропластичности, а также для тестирования потенциальных нейропротекторов.
Прогрессирующая дегенерация нейронов является важнейшей характеристикой нейродегенеративных заболеваний, в частности болезни Паркинсона (БП). Нейродегенерация сопровождается нарушением аксонального транспорта (Chevalier-Larsen et al., 2006; De Vos et al., 2008), нарушением синтеза нейромедиаторов (Hornykiewicz, 2006), протеинопатией (Conway et al., 2000; McLean et al., 2000; Moore et al., 2005), а в конечном итоге деградацией нейритов и тел нейронов (Raff, 2002). Так, гибель дофаминергических (ДА-ергических) нейронов нигростриатной системы является основной причиной нарушения двигательной функции и появления моторных симптомов (Marginelli et al., 2016).
Важной особенностью БП является длительное бессимптомное течение на протяжении многих лет, что обусловлено включением компенсаторных процессов по мере деградации нигростриатной системы (Agid, 1991). Только при достижении определенного уровня деградации нигростриатной системы – гибели 50% ДА-ергических нейронов и снижении содержания ДА в стриатуме на 80%, и после “истощения” компенсаторных резервов мозга появляются первые моторные симптомы (Bezard et al., 1998).
Лечение БП – в основном симптоматическое (медикаментозное), направленное на компенсацию дефицита дофамина (ДА) в нигростриатной системе (Fahn, 2008). Такое лечение позволяет продлить комфортную жизнь пациента, однако не на очень продолжительное время, что обусловлено, в первую очередь, поздней диагностикой этого заболевания.
Из выше изложенного следует актуальность изучения механизмов нейродегенерации и нейропластичности, что особенно важно на доклинической стадии, когда с помощью нейропротекторной терапии можно существенно замедлить гибель нейронов. Однако из-за отсутствия доклинической диагностики БП, пока механизмы дегенерации и нейропластичности можно изучать только на экспериментальных моделях.
Целью данной работы явилось воспроизведение и максимально полная характеристика на клеточном и молекулярном уровнях модели БП на основе культивируемых дофаминергических нейронов среднего мозга эмбрионов мыши в присутствии специфического токсина – 1-метил-4-фенилпиридиния (МФП+).
МАТЕРИАЛ И МЕТОДИКА
Для получения культуры клеток были использованы плоды мышей линии C57BL/6 на 13-й день эмбрионального периода (Э13). Время зачатия определяли по образованию вагинальной пробки у самок после спаривания с самцами и считали этот день за нулевой день (Э0). На 13-й день беременности у самок под ингаляционным наркозом (изофлуран) извлекали матку и промывали ее охлажденным буфером Хенкса без Са2+ и Mg2+ (Gibco, США) с добавлением антибиотиков гентамицина и амфотерицина (Gibco, США). После декапитации эмбрионов извлекали мозг и в охлажденном стерильном буфере Хенкса без Са2+ и Mg2+ выделяли вентральную область среднего мозга.
Культура клеток. Диссоциацию ткани и культивирование клеток проводили согласно ранее опубликованному протоколу (Pruszak et al., 2009). Выделенную ткань помещали в микропробирку со средой для суспендирования DMEM/F12, содержащей также бессывороточную добавку B-27, 0.5 мМ L-глутамин (все реактивы – Gibco, США). Кусочки ткани механически диссоциировали пипетированием. Полученную суспензию клеток фильтровали через нейлоновый фильтр (диаметр пор – 70 мкм) и центрифугировали 5 мин при 4°С и 200× g. Образовавшийся осадок ресуспендировали в среде культивирования следующего состава: Neurobasal medium, бессывороточная добавка B-27, 0.5 мМ L-глутамина, 100 нг/мл фактора роста нервов и 10 нг/мл мышиного фактора роста фибробластов с добавлением гентамицина (все реактивы – Gibco, США).
Клеточную суспензию окрашивали трипановым синим и проводили подсчет живых клеток в камере Горяева. Выживаемость составляла 80–90%. Затем клетки высаживали в предварительно обработанные поли-L-лизином 24-луночные планшеты (Corning, США) по 400 тыс. клеток на лунку и культивировали в атмосфере с 5% CO2 при 37°C. Среду культивирования заменяли каждые 2 дня. На четвертый день культивирования в среду вводили 10 мкМ цитарабина (Sigma, США) на 24 ч для устранения глии из первичной культуры клеток. Все эксперименты проводили на 7-й день культивирования в среде Neurobasal medium, содержащей 0.5 мМ L-глутамина и бессывороточную добавку N-2 (Gibco, США).
Для моделирования БП in vitro на седьмой день культивирования нейронов среднего мозга в среду культивирования вводили нейротоксин – ион 1-ме-тил-4-фенилпиридиния (MФП+) (Sigma, США) в дозе 10 мкМ на 3, 6 и 24 ч, а в контрольные лунки добавляли аналогичный объем 0.9% NaCl. После окончания инкубации с МФП+ культуру клеток использовали для иммуноцитохимического выявления тирозингидроксилаза-иммунореактивных (ТГ-ИР) нейронов и дофаминового транспортера (ДАТ), а также для определения содержания катехоламинов в клеточном экстракте и оценки обратного захвата дофамина клетками. Для проверки специфичности действия нейротоксина MФП+ в лунки добавляли ингибитор обратного захвата дофамина GBR 12909 (1 мкМ) за 15 мин до введения в культуральную среду МФП+.
Иммуноцитохимия. Для проведения иммуноцитохимической реакции на тирозингидроксилазу (ТГ) культуру нервных клеток отмывали от культуральной среды изотоническим фосфатно-солевым буфером (ФСБ) 10 мин при 4°С, после чего фиксировали в 4%-ном параформальдегиде на 0.2 М ФСБ (pH 7.2–7.4) в течение 1 ч при 4°С. Затем культуру клеток последовательно инкубировали с: (а) 3%-ным бычьим сывороточным альбумином (Sigma, США) и 0.3% тритоном-X100 (Sigma, США) на 0.02 М ФСБ 30 мин при 20°С; (б) кроличьими антителами к ТГ (1 : 5000) (предоставлены проф. Ж. Тибо, Франция) (Arluison et al., 1984) на 0.02 М ФСБ, содержащем 1%-ный бычий сывороточный альбумин и 0.1%-ный Triton X-100 в течение 20 ч при 20°С; (в) биотинилированными козьими антителами к гамма-глобулинам кролика (1 : 400) (Vector Laboratories, США) на 0.02 М ФСБ в течение 2 ч при 20°С; (д) авидин-биотиновым комплексом, связанным с пероксидазой хрена (Vector Laboratories, США), на 0.02 М ФСБ в течение 1 ч при 20°С. После каждой инкубации лунки промывали в 0.02 М ФСБ в течение 30 мин при 20°С. В контроле проводили те же самые инкубации, но без антител к ТГ. Пероксидазу авидин-биотинового комплекса выявляли путем инкубации с 0.05% 3,3'-диаминобензидинтетрогидрохлоридом (Sigma, США) и 0.02% H2O2 на ФСБ при 20°С под визуальным контролем. Культуру клеток заключали в гидрофильную среду Mowiol 4-88 (Sigma, США).
Двойное мечение ТГ и ДАТ проводили по следующей схеме: фиксированную культуру клеток последовательно инкубировали с: (а) блокирующим раствором (3% бычий сывороточный альбумин, 0.3% Triton-X100 на 0.02 М ФСБ) 30 мин при 20°С; (б) смесью овечьих поликлональных антител к ТГ (1 : 750, Millipore, США) с крысиными моноклональными антителами к дофаминовому транспортеру (1 : 500, Abcam, США) на 0.02 М ФСБ, содержащем 1%-ный бычий сывороточный альбумин и 0.1%-ный Triton X-100 в течение 20 ч при 20°C; (в) ослиными антителами к гамма-глобулинам овцы, конъюгированными с красителем Alexa Fluor 488 (1 : 500, Invitrogen, США) на 0.02 М ФСБ в течение 2 ч при 20°C; (г) куриными антителами к гамма-глобулинам крысы, конъюгированными с красителем Alexa Fluor 647 (1 : 200, Invitrogen, США) на 0.02 М ФСБ в течение 2 ч при 20°С. Инкубировали клетки с раствором DAPI в течении 5 мин и отмывали в ФСБ 3 раза по 5 мин. Обработанную антителами клеточную культуру заключали в гидрофильную среду Mowiol 4-88 (Sigma, США).
Анализ изображений. После иммуноцитохимической реакции на ТГ лунки с культурой клеток исследовали с помощью микроскопа Zeiss Observer Z1 (Zeiss, Германия), оснащенного сканирующим координатным столиком и фотокамерой при увеличении объектива ×20/NA 0.4 (EC Plan-Neofluar, Zeiss, Германия) и с использованием программного обеспечения AxioVision 4.8. С помощью модуля MosaiX Axio Vision (Zeiss, Германия) воссоздавали целостное изображение лунки и подсчитывали общее число ТГ-ИР нейронов на лунку с помощью программы ImageJ (Wayne Rasband-NIH, США).
В каждой лунке случайным образом выбирали 100 ТГ-ИР нейронов и определяли длину нейритов по ранее описанной методике (Rønn, et al., 2000). С помощью программы Photoshop CS 5.1 на изображениях ТГ-ИР нейронов создавали сеть параллельных линий, расстояние между которыми соответствовало 20 мкм. Затем подсчитывали количество пересечений нейритов (отростков) с этими линиями, вычисляя длину нейритов (L) по формуле:
где I – количество пересечений нейритов с параллельными линиями, d – расстояние (20 мкм) между параллельными линиями.Иммунофлуоресценцию на ТГ и ДАТ детектировали с помощью инвертированного микроскопа Leica DMI 6000.
Изотопный метод. Измерение обратного захвата дофамина нейронами проводили в искусственной спинно-мозговой жидкости (иСМЖ) следующего состава: NaCl – 126 мМ, KCl – 2.5 мМ, NaH2PO4 – 1.2 мМ, CaCl2 – 2.4 мМ, MgCl2 – 1.2 мМ, NaHCO3 – 25 мМ, HEPES – 20 мМ, глюкоза – 11 мМ с добавлением аскорбиновой кислоты – 0.15 мМ и паргилина-HCl – 0.1 мМ (Sigma, США) при рН 7.4. Культуру клеток как в контроле, так и после инкубации с MФП+, отмывали от среды и инкубировали в иСМЖ 15 мин. при 37°С в присутствии 32 нМ [3H]-дофамина, 33 Ки/ммоль (синтезирован в Институте молекулярной генетики РАН). Реакцию останавливали охлаждением на льду до 0°С и последующей двукратной промывкой охлажденной иСМЖ. Поглощенный клетками [3H]-дофамин экстрагировали в 0.5 мл 96% этанола. Экстракты вносили во флаконы с 10 мл сцинтилляционной жидкости (Liquid scintillator 402, Koch-light LTD, Англия); измерения проводили на счетчике (Tri-Carb 2810 TR, Perkin Elmer). Специфический захват меченого тритием дофамина определяли как разницу между значениями радиоактивности образцов, полученных без использования ингибитора мембранного транспортера дофамина GBR 12909 (общий захват) и с его применением (неспецифический захват).
Высокоэффективная жидкостная хроматография. Пробы для измерения концентрации ДА, полученные из образцов культуры клеток, подготавливали методом твердофазной экстракции на оксиде алюминия. Концентрацию ДА и L-ДОФА в пробах определяли методом ВЭЖХ. Разделение осуществляли на обращенно-фазной колонке ReproSil-Pur, ODS-3, 4 × 100 мм с диаметром пор 3 мкм (Dr.Majsch GMBH, Германия) при температуре 30°С и скорости подвижной фазы 1 мл/мин, поддерживаемой жидкостным хроматографом LC-20ADsp (Shimadzu, Япония). В состав мобильной фазы входили: 0.1 М цитратно-фосфатный буфер, 0.3 мМ октансульфат натрия, 0.1 мМ ЭДТА и 9% ацетонитрил (все реактивы – Sigma, США), pH 2.5. Электрохимический детектор Decade II (Antec Leyden, Нидерланды) был укомплектован стеклоуглеродным рабочим электродом (+0.85 V) и хлорсеребряным электродом сравнения. Пики дофамина и L-ДОФА идентифицировали по времени их выхода в стандартном растворе. Содержание катехоламинов и метаболитов рассчитывали методом внутреннего стандарта как отношение площадей пиков в стандартной смеси и в образце, с помощью программного обеспечения LabSolutions (Shimadzu, Япония).
Статистика. Статистическую обработку данных выполняли при помощи программы GraphPad Prism Version 6.0 для Windows (GraphPad Software, США). Статистическую значимость результатов определяли с использованием однофакторного дисперсионного анализа (1-way ANOVA) с последующим использованием теста Тьюки для множественного сравнения средних. Данные представлены как среднее арифметическое ± средняя ошибка.
РЕЗУЛЬТАТЫ
Морфофункциональная характеристика первичной культуры нейронов мезенцефалона из эмбрионов мыши. С помощью иммуногистохимического окрашивания культуры на ТГ показано, что нейроны имеют характерные клеточные тела и разветвленную сеть нейритов (рис. 1а). С помощью двойного иммуномечения полученной клеточной культуры на ТГ и ДАТ была показана полная колокализация ТГ и ДАТ в нашей клеточной культуре (рис. 2). Таким образом, мы убедительно показали наличие в культуре дофаминергических нейронов.
Рис. 1.
Культура тирозингидроксилаза-иммунореактивных (ТГ-ИР) нейронов в контроле (а), после инкубации с МФП+ в концентрации 10 мкм (б) и после совместной инкубации с МФП+ (10 мкМ) и ингибитором GBR-12909 (1 мкМ) (в). Инвертированный микроскоп – Leica DM IL. Масштабные отрезки – 100 мкм и 20 мкм (на большем увеличении).
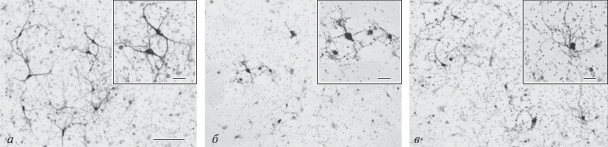
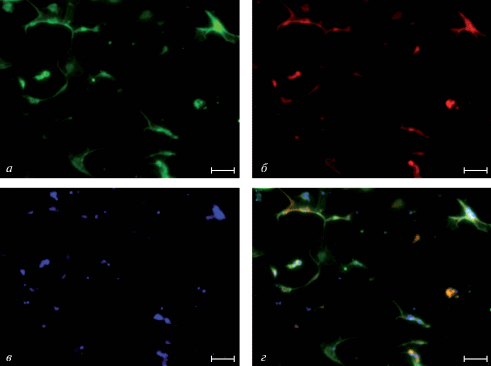
Рис. 2.
Репрезентативное иммунофлуоресцентное мечение, демонстрирующее колокализацию тирозингидроксилазы (ТГ) и дофаминового транспортера (ДАТ) в полученной нейрональной клеточной культуре. а – Иммунофлуоресцентное выявление ТГ, б – иммунофлуоресцентное выявление ДАТ, в – окраска ядер клеток флуоресцентным красителем DAPI, г – совмещение изображений. Масштабные отрезки – 50мкм.
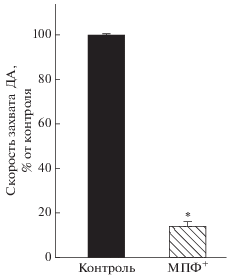
Количество ТГ-ИР нейронов на лунку составило 487 ± 56 нейронов (рис. 3а). Средняя длина нейритов в клеточной культуре нейронов составила 628 ± ± 76 мкм (рис. 3б). С помощью ВЭЖХ было определено, что содержание дофамина в культуре нейронов на лунку равно 0.69 ± 0.06 пмоль (рис. 4), а содержание L-ДОФА – 0.95 ± 0.28 пмоль. Наше исследование также показало, что культивируемые мезенцефальные нейроны специфически захватывают меченый дофамин, причем специфический захват составил 82% от общего захвата дофамина (рис. 5).
Рис. 3.
Количество тирозингидроксилаза-иммунореактивных (ТГ-ИР) нейронов (а) и длина нейритов (б) в контроле и через 24 ч после введения МФП+ (10 мкМ) или МФП+ (10 мкМ) совместно с ингибитором дофаминового переносчика GBR-12909 (1 мкМ). *Достоверные отличия от контроля (P < 0.05); **достоверные различия между экспериментальными группами (Р < 0.05).
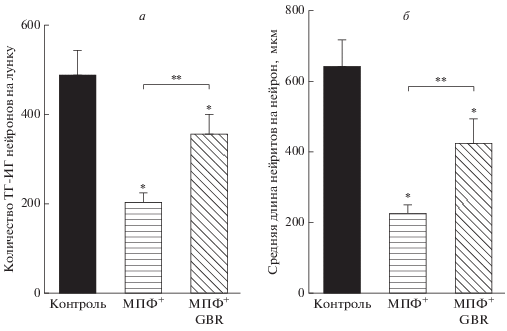
Рис. 4.
Содержание дофамина (ДА) в культуре дофаминергических нейронов в контроле и через 24 ч после введения МФП+ (10 мкМ) или МФП+ (10 мкМ) совместно с ингибитором мембранного переносчика дофамина GBR-12909 (1 мкМ). **Достоверные различия между экспериментальными группами (Р < 0.05).
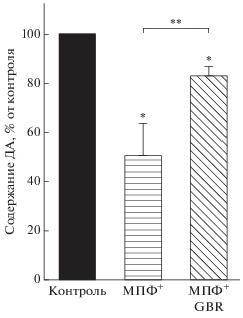
Рис. 5.
Специфический захват дофамина (ДА) в культуре дофаминергических нейронов в контроле и через 24 ч после начала инкубации с 10 мкМ МФП+.
Морфологические и функциональные показатели дофаминергических нейронов в культуре после воздействия МФП+. Моделирование БП in vitro осуществлялось путем добавления в культуральную среду 10 мкМ специфического нейротоксина МФП+. Через 1 сут инкубации с МФП+ наблюдалось снижение числа ТГ-ИР нейронов на 58% (рис. 3а). Добавление 1 мкМ GBR-12909 (селективный ингибитор обратного захвата дофамина) приводило к увеличению количества ТГ-ИР нейронов на 56% по сравнению с количеством нейронов после инкубации с МФП+ (рис. 3а), но не достигало уровня ТГ-ИР нейронов в контроле.
Добавление нейротоксина в культуру клеток вызывало не только уменьшение числа выживших нейронов, но и уменьшение длины нейритов на 65% относительно контроля (рис. 3б). Добавление к среде культивирования GBR-12909 приводило к снижению средней длины нейритов только на 34% по сравнению с контролем (рис. 3б).
Помимо указанных морфологических изменений наблюдалось снижение содержания внутриклеточного ДА в нейронах на 52% после инкубации культуры клеток с 10 мкМ МФП+ в течение 24 ч (рис. 4). При инкубации одновременно с МФП+ и GBR-12909 содержание дофамина было снижено в гораздо меньшей степени (на 18%), чем после инкубации только с МФП+. Следует также отметить, что воздействие на клеточную культуру МФП+ вызывало значительное (до 85%) снижение специфического захвата ДА нейронами (рис. 5).
При инкубации культуры клеток с МФП+ в течение 3 ч не было отмечено изменений ни числа ТГ-ИР нейронов, ни длины и строения их нейритов (рис. 6).
Рис. 6.
Изменение длины нейритов ТГ-ИР нейронов через 3 и 6 ч после добавления 10 мкМ МФП+. *Достоверные различия между группами (P < 0.05).
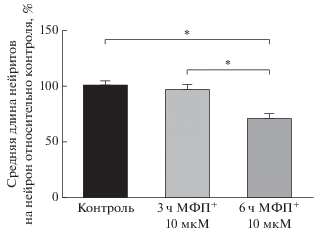
При инкубации культуры клеток с МФП+ в течение 6 ч число ТГ-ИР нейронов не изменялось по сравнению с контролем. Однако в этом случае уменьшалась общая дина нейритов на нейрон на 30% (рис. 6). Также были замечены морфологические изменения нейронов. Появились характерные участки фрагментации нейритов, которые выглядят как “варикозные” расширения (рис. 7).
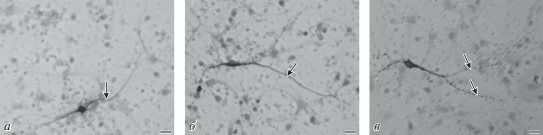
Рис. 7.
Морфология ТГ-ИР нейронов в контроле (а), через 3 ч (б) и через 6 ч (в) после введения МФП+ (10 мкМ). Через 3 ч после добавления в культуру МФП+ морфологических изменений в нейронах обнаружено не было. Через 6 ч у нейронов инкубированных с МФП+ появились изменения в морфологии, в частности появление характерных “варикозных” расширений и фрагментации на концах нейритов. Масштабный отрезок – 20 мкм.
ОБСУЖДЕНИЕ
Многие разработанные ранее модели БП in vitro на основе перевиваемых клеточных линий, в частности нейробластом человека и мыши (Amazzal et al., 2007; Constantinescu et al., 2007), обладают существенными недостатками. Действительно, дифференцирующиеся нейроны на этих моделях не воспроизводят в полной мере ДА-ергический фенотип, поскольку они также обладают некоторыми признаками холинергического фенотипа (Kovalevich, Langford, 2013). Этого недостатка лишены модели на основе диссоциированной первичной культуры эмбриональных ДА-ергических нейронов среднего мозга грызунов. Однако число работ, проведенных с использованием первичных нейрональных культур, существенно уступает числу работ, выполненных на моделях с использованием нейробластом, что объясняется техническими трудностями и высокой затратностью культивирования первичных ДА-ергически нейронов.
Использование предложенной и исчерпывающе охарактеризованной в этой работе модели первичной культуры ДА-ергических нейронов среднего мозга может позволить существенно продвинуться в изучении молекулярных механизмов нейродегенерации и нейропластичности при БП, начатом нами ранее на моделях БП in vivo (Ugrumov et al., 2011; Kozina et al., 2014; Mingazov et al., 2018). В основе данной модели in vitro, как и в основе модели in vivo, лежит нейротоксическое действие токсина МФП+ на ДА-ергические нейроны, приводящее к оксидативному стрессу, а в конечном итоге к гибели нейронов путем апоптоза, что было показано в ранее опубликованных работах (Fallon et al., 1997; Kaul et al., 2003; Nakai et al., 2003; Zawada et al., 2011).
Культивирование нейронов среднего мозга для последующего моделирования БП требует определенных условий. Так, эмбриональные предшественники нейронов должны пройти процесс дифференцировки, а культура клеток должна быть избавлена от глии. В нашей работе на 7-й день культивирования нейроны уже имели хорошо развитую сеть нейритов, содержащих ключевой фермент синтеза дофамина – ТГ. В предыдущей работе было также показано, что, наряду с ТГ, в нейронах первичной культуры среднего мозга грызунов также экспрессируется и второй фермент синтеза ДА – декарбоксилаза ароматических L-аминокислот (Yan et al., 2001). И, наконец, в нашей работе были получены прямые доказательства того, что культивируемые нейроны синтезируют ДА и способны к его специфическому обратному захвату с помощью мембранного транспортера дофамина. Таким образом, нами была получена клеточная культура нейронов, экспрессирующих основные признаки ДА-ергического фенотипа.
Получение первичной клеточной культуры нейронов среднего мозга, экспрессирующих ДА-ергический фенотип, позволило перейти к моделированию in vitro БП c помощью МФП+ – специфического токсина ДА-ергических нейронов. Действие этого нейротоксина оценивали в первую очередь по следующим показателям: числу выживших ТГ-ИР (ДА-ергических) нейронов на лунку, общей длине нейритов на нейрон, способности нейронов синтезировать и специфически захватывать из окружающей среды ДА. Введение МФП+ в культуральную среду по морфологическим показателям приводило к снижению числа ДА-ергических нейронов на 58% и уменьшению длины их нейритов.
Особый интерес представляет наблюдаемое нами уменьшение длины нейритов ДА-ергических нейронов под действием МФП+, поскольку считается, что дегенерация нейронов начинается именно с терминалей аксонов, распространяясь ретроградно вплоть до тела нейрона (Raff et al., 2002; Li et al., 2009). Поэтому нас заинтересовала оценка времени начала деградации нейритов на нашей модели. Так, трехчасовое воздействие МФП+ на культуру не привело к морфологическим изменениям ДА-ергических нейронов на клеточном уровне, хотя это не исключает развития нейродегенеративного изменения на молекулярном уровне. Значительные морфологические изменения ДА-ергических нейронов отмечены через 6 ч после введения МФП+. Наиболее ярким изменением было уменьшение длины нейритов ДА-ергических нейронов на 30% длины по сравнению с контролем. Более того, показано, что начальные этапы дегенерации ТГ-ИР нейронов сопровождаются фрагментацией нейритов.
Важно отметить, что токсическое действие МФП+, проявляющееся в гибели нейронов и уменьшении длины нейритов, частично предотвращается добавлением в среду GBR 12909 – ингибитора обратного захвата ДА. Это наблюдение подтверждает представления о том, что МФП+ захватывается в ДА-ергические нейроны из окружающей среды с помощью мембранного переносчика ДА.
Наряду с морфологическими показателями действия МФП+ на культивируемые ДА-ергические нейроны, были использованы и биохимические показатели, характеризующие изменения метаболизма ДА. Так, введение МФП+ в культуральную среду приводило к снижению содержания ДА в культуре клеток на 58% и L-ДОФА на 78%, что, вероятно, в основном отражает дегенерацию ДА-ергических нейронов. Однако наблюдаемое нами большее снижение содержания L-ДОФА по сравнению с ДА можно рассматривать как косвенный показатель влияния МФП+ на содержание и активность ТГ в выживших ДА-ергических нейронах. Из приведенных данных следует, что изменение внутриклеточного содержания ДА в культивируемых нейронах может рассматриваться как важный показатель токсического действия МФП+ на ДА-ергические нейроны.
Важным инструментом демонстрации дегенерации ДА-ергических нейронов является также оценка обратного захвата ДА. В нашей работе отмечено существенное снижение скорости обратного захвата ДА клетками после инкубации с МФП+. В большей степени это обусловлено гибелью нейритов и тел нейронов. Эти данные подтверждают адекватность используемой клеточной модели патогенезу БП.
Таким образом, нами была исчерпывающе охарактеризована, на клеточном и отчасти на молекулярном уровнях, первичная культура дофаминергических нейронов в норме и при моделировании БП. Это позволит использовать ее для изучения клеточных и молекулярных механизмов нейродегенерации и нейропластичности, а также для оценки эффективности лекарственных средств с нейропротекторными свойствами.
Список литературы
Amazzal L., Lapotre A., Quignon F., Bagrel D. 2007. Mangiferin protects against 1-methyl-4-phenylpyridinium toxicity mediated by oxidative stress in N2A cells. Neurosci. Lett. 418 : 159–164.
Agid Y. 1991. Parkinson’s disease: Pathophysiology. Lancet. 337 : 1321–1324.
Arluison M., Dietl M., Thibault J. 1984. Ultrastructural morphology of dopaminergic nerve terminals and synapses in the striatum of the rat using tyrosine hydroxylase immunocytochemistry: A topographical study. Brain Res. Bull. 13 : 269–285.
Bezard E., Gross C.E. 1998. Compensatory mechanisms in experimental and human parkinsonism: Towards a dynamic approach. Prog. Neurobiol. 55 : 93–116.
Chevalier-Larsen E., Holzbaur E.L. 2006. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta. 1762 : 1094–1108.
Constantinescu R., Constantinescu A. T., Reichmann H., Janetzky B. 2007. Neuronal differentiation and long-term culture of the human neuroblastoma line SH-SY5Y. In: Neuropsychiatric disorders an integrative approach. Wien, Springer-Verlag, 17–28.
Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. Jr. 2000. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA. 97 : 571–576.
De Vos K.J., Grierson A.J., Ackerley S., Miller C.C. 2008. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31 : 151–173.
Fallon J., Matthews R.T., Hyman B.T., Beal M.F. 1997. MPP+ produces progressive neuronal degeneration which is mediated by oxidative stress. Exp. Neurol. 144 : 193–198.
Fahn S. 2008. The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov. Disord. 23 : 497–508.
Hornykiewicz O. 2006. The discovery of dopamine deficiency in the parkinsonian brain. In: Parkinson’s Disease and Related Disorders. Wien, Springer-Verlag, 9–15.
Kaul S., Kanthasamy A., Kitazawa M., Anantharam V., Kanthasamy A.G. 2003. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: Relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 18 : 1387–1401.
Kovalevich J., Langford D. 2013. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. In: Neuronal cell culture: Methods and protocols. N.Y., Springer, 9–21.
Kozina E.A., Khakimova G.R., Khaindrava V.G., Kucheryanu V.G., Vorobyeva N.E., Krasnov A.N., Georgieva S.G., Kerkerian-Le Goff L., Ugrumov M.V. 2014. Tyrosine hydroxylase expression and activity in nigrostriatal dopaminergic neurons of MPTP-treated mice at the presymptomatic and symptomatic stages of parkinsonism. J. Neurol. Sci. 340 : 198–207.
Li L.H., Qin H.Z., Wang J.L., Wang J., Wang X.L., Gao G.D. 2009. Axonal degeneration of nigra-striatum dopaminergic neurons induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J. Int. Med. Res. 37 : 455–463.
Magrinelli F., Picelli A., Tocco P., Federico A., Roncari L., Smania N., Zanette G., Tamburin S. 2016. Pathophysiology of motor dysfunction in Parkinson’s disease as the rationale for drug treatment and rehabilitation. Parkinsons Dis. 2016 : 3–21.
McLean P.J., Kawamata H., Ribich S., Hyman B.T. 2000. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 275 : 8812–8816.
Mingazov E.R., Khakimova G.R., Kozina E.A., Medvedev A.E., Buneeva O.A., Bazyan A.S., Ugrumov M.V. 2018. MPTP mouse model of preclinical and clinical Parkinson’s disease as an instrument for translational medicine. Mol. Neurobiol. 55 : 2991–3006.
Moore D.J., West A.B., Dawson V.L., Dawson T.M. 2005. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 28 : 57–87.
Nakai M., Mori A., Watanabe A., Mitsumoto Y. 2003. 1-methyl-4-phenylpyridinium (MPP+) decreases mitochondrial oxidation-reduction (REDOX) activity and membrane potential (Deltapsi(m)) in rat striatum. Exp. Neurol. 179 : 103–110.
Pruszak J., Just L., Isacson O., Nikkhah G. 2009. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. In: Current protocols in stem cell biology, 1st Edition. John Wiley & Sons Inc., 2D.5.1–2D.5.21.
Raff M.C., Whitmore A.V., Finn J.T. 2002. Axonal self-destruction and neurodegeneration. Science. 296 : 868–871.
Rønn L.C., Ralets I., Hartz B.P., Bech M., Berezin A., Berezin V., Møller A., Bock E. 2000. A simple procedure for quantification of neurite outgrowth based on stereological principles. J. Neurosci. Methods. 100 : 25–32.
Ugrumov M.V., Khaindrava V.G., Kozina, E.A., Kucheryanu V.G., Bocharov E.V., Kryzhanovsky G.N., Kudrin V.S., Narkevich V.B., Klodt P.M., Rayevsky K.S., Pronina T.S. 2011. Modeling of presymptomatic and symptomatic stages of parkinsonism in mice. Neuroscience. 181 : 175–188.
Yan J., Studer L., McKay R.D. 2001. Ascorbic acid increases the yield of dopaminergic neurons derived from basic fibroblast growth factor expanded mesencephalic precursors. J. Neurochem. 76 : 307–311.
Zawada W.M., Banninger G.P., Thornton J., Marriott B., Cantu D., Rachubinski A.L., Das M., Griffin W.S., Jones S.M. 2011. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J. Neuroinflammation. 8 : 129.
Дополнительные материалы отсутствуют.