

Цитология, 2019, T. 61, № 2, стр. 89-97
Ингибитор CAY10603 гистондеацетилазы HDAC6 вызывает G1/S блок клеточного цикла и способствует старению фибробластов мыши, трансформированных онкогенами E1A и cHa-ras
А. Н. Кукушкин 1, *, С. Б. Светликова 1
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: kan@incras.ru
Поступила в редакцию 27.09.2018
После доработки 14.11.2018
Принята к публикации 15.11.2018
Аннотация
Для фибробластов мыши, трансформированных онкогенами E1A и cHa-ras, вещество CAY10603 (CAY) является эффективным ингибитором гистондеацетилазы HDAC6, когда происходит деацетилирование субстрата α-тубулина. В низких концентрациях CAY вызывает блок клеточного цикла на границе фаз G1/S этих клеток, замедляет их пролиферацию, но не провоцирует апоптотическую гибель. Обработка CAY в сочетании с бутиратом натрия приводит к ускоренному старению трансформантов E1A+cHa-ras. Также при действии CAY происходит накопление аутофагических вакуолей и аутофагосомного белка LC3 в цитоплазме клеток E1A+cHa-ras, что может свидетельствовать об участии HDAC6 в индукции аутофагии в этих трансформированных клетках.
Гистондеацетилаза HDAC6 относится к классу IIb деацетилаз с преимущественно цитоплазматической локализацией в клетке и она деацетилирует негистоновые белки, такие как тубулин, кортактин, Hsp90, HSF1, Ku70 и др. (Verdel, Khochbin, 1999; Hubbert et al., 2002; Matsuyama et al., 2002; Zhang X. et al., 2007; Aldana-Masangkay, Sakamoto, 2011; Kerr et al., 2012; Li et al., 2013). В качестве энзима, деацетилирущего сократительные белки (тубулин и кортактин), HDAC6 регулирует многие клеточные процессы, в которых участвуют микротрубочки и микрофиламенты: подвижность и поддержание формы клеток, клеточный цикл, аутофагию (Zhang Y. et al., 2003; Zhang X. et al., 2007; Li et al., 2013; Yan et al., 2013). Ингибирование активности HDAC6 и повышение уровня ацетилирования тубулина приводит к стабилизации структуры микротрубочек (Matsuyama et al., 2002; Asthana et al., 2013). Наряду с двумя каталитическими деацетилазными доменами молекула HDAC6 обладает С-концевым доменом с цинковыми пальцами, который способен связывать убиквитин и убиквитинированные белки. Таким образом, HDAC6 вовлечена в деградацию мутантных или агрегированных белков как на протеосомах, так и в аутофаголизосомах, способствуя слиянию аутофагосом и лизосом (Seigneurin-Berny et al., 2001; Hook et al., 2002; Lee et al., 2010). Белок HDAC6 интенсивно экспрессируется во многих опухолевых клетках, обусловливая активацию Ras-сигнального пути и поддержание трансформированного фенотипа (Lee et al., 2010; Namdar et al., 2010; Aldana-Masangkay, Sakamoto, 2011; Kanno et al., 2012), поэтому уровень экспрессии HDAC6 может считаться прогностическим маркером онкогенеза (Zhang Z. et al., 2004). В этой связи представляет интерес возможность использования специфичных для HDAC6 фармакологических ингибиторов (SAHA, тубацина, тубастатина А), которые вызывают гиперацетилирование субстратов HDAC6 и нарушения жизнедеятельности опухолевых клеток (остановку клеточного цикла, апоптоз, деструктивную аутофагию) (Haggarty et al.,2003; Lee et al., 2010; Namdar et al., 2010; Kerr et al., 2012; Bai et al., 2015). Было показано, что ингибиторы HDAC6 усиливают действие ДНК-повреждающих агентов (этопозида, доксорубицина) по продуцированию двуцепочечных разрывов ДНК, что приводит к гибели трансформированные клетки, тогда как нормальные клетки выживают, репарируя такие разрывы ДНК (Lee et al., 2010; Namdar et al., 2010).
Одним из новых специфических ингибиторов HDAC6 является вещество CAY10603 (N-[4-[3-[[[7-(Hydroxyamino)-7-oxoheptyl]amino]carbonyl]-5-isoxazolyl]phenyl]-1,1-dimethylethylester), которое, как показано недавно, способно подавлять пролиферацию некоторых опухолевых клеток и вызывать их апоптотическую гибель: аденокарциномы легкого (Wang et al., 2016), медуллобластомы мозжечка и глиобластомы (Dhanyamraju et al., 2015). Однако это вещество может использоваться и в более широких медицинских целях, например, оно может подавлять развитие малярийного плазмодия или паразита овец Lucilia cuprina (Trenholme et al., 2014; Bagnall et al., 2017), ослаблять воспалительную реакцию легочного эндотелия (Yu et al., 2016). Таким образом, пока накоплено немного данных о воздействии CAY на трансформированные клетки, и мы надеемся внести свой вклад в этот вопрос.
В настоящей работе мы изучали влияние ингибитора CAY10603 (CAY) в низких фармакологических концентрациях на клеточные функции модели опухолевых клеток – линии эмбриональных фибробластов мыши, трансформированных онкогенами E1A и cHa-ras, предполагая возможности замедления их пролиферации или гибели. Мы планировали исследовать воздействие CAY на клеточный цикл и на процессы апоптоза и аутофагии в этих трансформированных клетках, а также сравнить действие этого специфичного ингибитора HDAC6 с нашими предыдущими результатами по старению клеток E1A+cHa-ras, полученными для общего ингибитора HDAC – бутирата натрия.
МАТЕРИАЛ И МЕТОДИКА
Клетки. Линия эмбриональных фибробластов мыши, трансформированных онкогенами E1A и cHa-ras, была получена ранее (Поспелова и др., 1990). Клетки на чашках выращивали в среде DMEM, содержащей 10% эмбриональной сыворотки теленка (ЭТС) до плотности 70%. Ингибитор HDAC6 CAY (Santa Cruz Biotech., США) растворяли в 95%-ном этаноле в концентрации 1.12 мМ. Клетки обрабатывали CAY в конечных концентрациях 0.5–2.5 мкМ в течение 24, 48 или 72 ч. К контрольным клеткам добавляли соответствующие количества этанола – растворителя CAY. Также клетки обрабатывали 4 мM бутиратом натрия (Sigma, США) 72 ч.
Для оценки пролиферативной активности клетки E1A+cHa-ras высевали на чашки Петри диаметром 60 мм. Когда их количество достигало 35 × 104, их обрабатывали 2.5 мкМ CAY в течение 24, 48 или 72 ч. Не обработанные клетки служили контролем. Затем клетки снимали и считали в камере Горяева. Кривые роста строили на основании 4 независимых подсчетов.
Проточная цитометрия. Метод использовали для анализа распределения клеток по фазам клеточного цикла. Процедуру подготовки клеток проводили, как описано ранее (Иготти и др., 2016). Число апоптозных клеток оценивали по относительной площади субдиплоидного пика распределения на гистограммах проточной цитометрии, используя прибор Coulter Epicks XL (Bechman, США). Относительные размеры клеток определяли по показателю светорассеивания.
Жизнеспособность клеток определяли с помощью MTT-теста (Sigma) в 96-луночных планшетах, как изложено ранее (Иготти и др., 2016), с 6 повторами для каждой точки. Количество продукта реакции MTT определяли на планшетном спектрофотометре Multiscan-EX (Thermo Scientific., США) при длине волны 570 нм. Достоверность количественных данных оценивали по t-критерию Стьюдента.
Активность каспазы-3 анализировали в 40 мкг экстрактов из клеток E1A+cHa-ras, культивированных 72 ч, в 96-луночных планшетах с 0.2 мМ колориметрического субстрата Ac-DEVD-pNA (Calbiochem, США), как описано ранее (Abramova et al., 2010), с 5 повторами для каждой точки. Количество продукта каспазной реакции определяли на планшетном спектрофотометре Multiscan-EX при длине волны 405 нм. Для ингибирования каспазной активности in vitro использовали 0.5 мМ ингибитор ZVAD (Selleck Chemicals, США).
Старение клеток оценивали по активности β-галактозидазы, ассоциированной со старением (SA-β-gal) (Гордеев и др., 2015). Клетки E1A+cHa-ras выращивали на покровных стеклах 72 ч. Окраску клеток выявляли с помощью микроскопа Pascal LSM5 (Zeiss, Германия) в проходящем свете.
Аутофагию в клетках детектировали in vitro с помощью набора CYTO-ID (Enzo Life Sci., Швейцария), который содержал флуоресцентный краситель, специфично связывающийся с аутофагическими вакуолями. Инкубацию живых клеток с красителем CYTO-ID проводили согласно протоколу производителя, после чего использовали проточную флуориметрию при длине волны 488 нм. Эксперименты повторяли 4 раза.
Иммуноблотинг. Клеточные экстракты получали, как описано ранее (Кукушкин и др., 2016). Концентрацию белка определяли методом Брэдфорд (Bradford, 1976). Белки (50 мкг) из клеточных экстрактов разделяли электрофорезом в 10%-ном ПААГ в 25 мМ трис-глициновом буфере, рН 8.5, содержащем 0.1% SDS. Маркерами мол. массы служили цветные белки из набора SeeBluePlus2 (Invitrogen, США). После электрофореза белки переносили на PVDF-мембрану (Millipore, США) с порами 0.45 мкм. Интересующие белки выявляли на мембране с помощью антител кролика к α-тубулину, ацетилированному по лизину-40, (cs-5335, Cell Signaling, США) или антител мыши к α-тубулину (T5168, Sigma). В качестве вторых антител использовали антитела козла против иммуноглобулинов кролика (GAR, A0545, Sigma) или антитела кролика против иммуноглобулинов мыши (RAM, A9044, Sigma), конъюгированные с пероксидазой хрена. Сигналы от антител на мембранах выявляли стандартным методом усиления хемолюминесценции (ECL, Sigma) с помощью гель-документирующей системы PXi6 Access (Syngene, Великобритания). Иммуноблотинг с антителами повторяли не менее 3 раз.
Иммунофлуоресцентную микроскопию проводили, как описано в предыдущей работе (Кукушкин и др., 2016). Использовали антитела кролика к белку LC3 (PM036, MBL, США) или к ацетилированному α-тубулину (см. выше). Вторые антитела козла против иммуноглобулинов кролика были конъюгированы с флуорохромом AlexaFluor 488 (Invitrogen, США). Ядра клеток окрашивали флуоресцентным красителем DAPI (Invitrogen, США). Препараты просматривали на конфокальном микроскопе Leica TCS SP5 (США). Эксперименты по иммунофлуоресценции повторяли не менее 3 раз для каждого белка.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
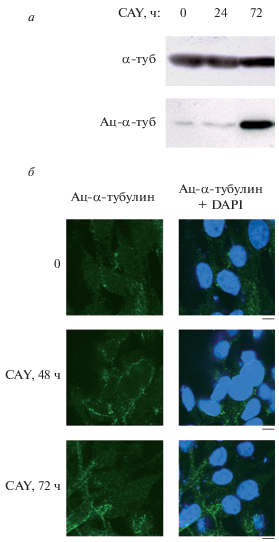
В E1A+cHa-ras-трансформированных фибробластах мыши α-тубулин, который является основным компонентом микротрубочек и одним из субстратов гистондеацетилазы HDAC6, конститутивно экспрессируется на высоком уровне независимо от условий культивирования клеток (рис. 1а), поэтому белок α-тубулин традиционно используется для контроля количества белка во время электрофореза и последующего иммуноблотинга. Ацетилированный по лизину-40 α-тубулин слабо представлен в контрольных клетках E1A+cHa-ras (рис. 1а, 1б), что может быть обусловлено заметной деацетилазной активностью HDAC6 (данные не показаны). Однако при культивировании этих клеток со специфическим ингибитором HDAC6 0.5 мкМ CAY в течение 48 или 72 ч наблюдается значительное накопление ацетилированной формы α-тубулина в цитоплазме, что видно как на иммуноблотинге (рис. 1а), так и при иммунофлуоресценции (рис. 1б). При более коротком культивировании (24 ч) в присутствии CAY ацетил-α-тубулин еще не успевает накопиться (рис. 1а). Таким образом, этот ингибитор HDAC6 подтверждает свою специфичность в E1A+cHa-ras-трансформированных фибробластах на примере субстрата α-тубулина.
Рис. 1.
Содержание α-тубулина в E1A+cHa-ras-трансформированных фибробластах, определяемое с помощью иммуноблотинга (а) и иммунофлуоресценции (б). а – иммуноблот α-тубулина (α-туб) и его ацетилированной по лизину-40 формы (Ац-α-туб ) из экстрактов E1A+cHa-ras клеток, обработанных 0.5 мкМ CAY в течение 24 и 72 ч. 0 – Контрольные клетки, культивированные 24 ч с добавлением соответствующих количеств этанола (растворителя CAY). На дорожки 10%-ного ПААГ наносили по 50 мкг белка клеточных экстрактов, проводили электрофорез и перенос на PVDF-мембраны, которые инкубировали с антителами к α-туб и Ац-α-туб. б – Иммунофлуоресценция ацетилированного α-туб в клетках E1A+cHa-ras контрольных (0) и обработанных 0.5 мкМ CAY в течение 48 и 72 ч. Клетки выращивали в среде DMEM с 10%-ной сывороткой, обрабатывали CAY, фиксировали параформальдегидом, инкубировали с первичными антителами к ацетилированному α-тубулину и (или) c DAPI для окраски ядер, а затем – со вторыми антителами с флуорохромом AlexaFluor 488. Длина масштабного отрезка в правом нижнем углу микрофотографий – 5 мкм.
Проточная цитометрия показала, что доля E1A+cHa-ras клеток, находящихся в фазахе G0/G1 клеточного цикла, значительно возрастает после обработки 0.5 мкМ CAY в течение 48–72 ч, соответственно доля S-фазных клеток уменьшается вплоть до 10% (рис. 2а), т.е. имеет место выраженный блок на границе фаз G1/S клеточного цикла. Такой блок наблюдался после 72-часового действия CAY в концентрациях от 0.5 до 2.5 мкМ, что свидетельствует об отсутствии дозового эффекта CAY в данном диапазоне. Ранее подобный блок клеточного цикла при переходе от G1 к фазе S выявлялся для E1A+cHa-ras-трансформированных фибробластов при действии общего неспецифического ингибитора HDAC бутирата натрия в концентрации 4 мМ (Abramova et al., 2006). Кроме того, наблюдалась значительная гипертрофия клеток E1A+cHa-ras в течение 3–5 сут культивирования в присутствии бутирата натрия (Romanov et al., 2010), тогда как в настоящей работе по данным светорассеивания проточной цитометрии CAY за то же время не вызывал подобного эффекта и обработанные клетки мало отличались по размеру от контрольных.
Рис. 2.
Распределение клеток E1A+cHa-ras по фазам клеточного цикла (а) и их пролиферативная активность (б) в присутствии ингибитора CAY. а – клетки культивировали 24–72 ч в присутствии 0.5 мкМ CAY или в его отсутствии (К, контроль, 24 ч); числа в столбиках – доля клеток (%) по результатам одного из экспериментов с помощью проточной цитометрии; б – кривые роста клеток E1A+cHa-ras в контроле (К) и в присутствии 2.5 мкМ CAY. По оси Y – среднее число клеток из 4-х чашек.
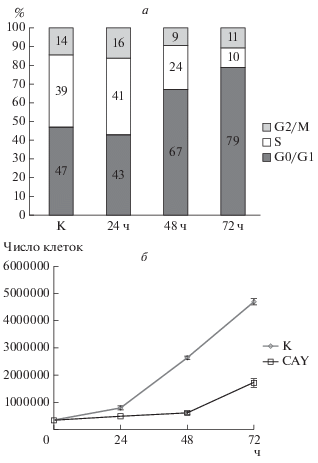
Вызванный CAY блок G1/S клеточного цикла предполагает, вероятно, замедление пролиферации клеток E1A+cHa-ras. Чтобы проверить это, мы построили кривые роста этих клеток, интактных и обработанных CAY (2.5 мкМ) в течение 1–3 сут. Действительно, рост клеток, культивируемых в присутствии CAY, отставал уже через 24 ч и замедлялся в 3–4 раза через 48–72 ч по сравнению с контрольными клетками (рис. 2б). Таким образом, наше предположение о замедлении пролиферации E1A+cHa-ras-трансформированных фибробластов в присутствии CAY экспериментально подтвердилось.
Для того чтобы оценить токсичность CAY в использованных низких концентрациях для клеток E1A+cHa-ras, мы провели MTT-анализ их жизнеспособности, выбрав наибольшую из используемых концентраций CAY – 2.5 мкМ. За 24 ч действия CAY количество живых клеток в среднем снизилась всего до 75% по сравнению с контрольными необработанными клетками, через 48 ч доля живых клеток возрастала до 86% (рис. 3а) за счет некоторого прироста популяции, несмотря на замедление темпов роста в присутствии CAY. Следовательно, выбранные концентрации ингибитора CAY не приводят к значительной гибели клеток E1A+cHa-ras при длительном культивировании. На гистограммах проточной цитометрии этих клеток, культивированных 24–72 ч в присутствии CAY, относительные площади субдиплоидных пиков не превышали 2%, что также говорит о том, что под действием данного ингибитора не происходит апоптотической гибели клеток.
Рис. 3.
Анализ жизнеспособности клеток E1A+cHa-ras в реакции MTT (а) и определение в них активности каспазы-3 (б). а – относительное количество живых клеток в контроле (К, принято за 1) и в клетках, обработанных 2.5 мкМ CAY в течение 24 или 48 ч. б – Относительная активность каспазы-3 в реакции со специфическим субстратом в контрольных клетках (К, принята за 1) и клетках, обработанных 2.5 мкМ CAY в течение 72 ч. Вертикальные отрезки – стандартное отклонение от среднего. Уровень значимости по t-критерию Стьюдента P = 0.001.
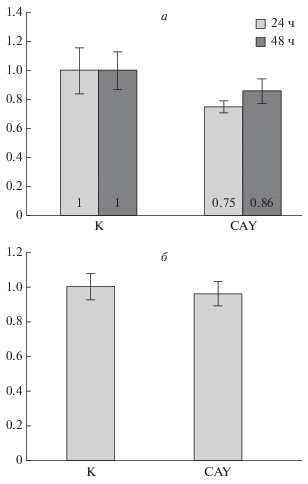
При развитии апоптоза происходит активация каспазного каскада, в частности ключевой каспазы-3. Поэтому с помощью специфичного колориметрического субстрата для каспазы-3 мы проверили ее активность в экстрактах из клеток E1A+cHa-ras, не обработанных (контрольных) и обработанных 2.5 мкМ CAY в течение 3 сут. Активности каспазы-3 оказались практически идентичными (рис. 3б), причем они одинаково снижались до базальной при добавлении протеазного ингибитора ZVAD in vitro (данные не показаны). Таким образом, длительная инкубация клеток с CAY не приводила к какой-либо активации каспазы-3, что также свидетельствует об отсутствии заметного апоптоза при обработке клеток изучаемым ингибитором HDAC6.
Ранее было показано (Зубова и др., 2005), что E1A+cHa-ras-трансформированные фибробласты крысы, обработанные бутиратом натрия (4 мМ) в течение 48 ч и более, обнаруживают признаки ускоренного клеточного старения, выражающегося, в частности, в накоплении β-галактозидазы. Мы решили проверить, может ли специфический ингибитор CAY, подобно бутирату натрия, способствовать старению E1A+cHa-ras-трансформированных фибробластов мыши после 72-часового действия. На рис. 4а видно, что бутират натрия (4 мМ) вызывает заметное накопление β-галактозидазы и старение клеток, тогда как при действии 1 мкМ CAY наблюдали лишь незначительные следы β-галактозидазы. Однако при совместном действии 1 мкМ CAY и 4 мМ бутирата натрия стареющий фенотип был наиболее выражен (рис. 4а). Таким образом, CAY сам по себе не вызывает старения клеток E1A+cHa-ras, но при совместном действии с бутиратом натрия имеет место усиление эффекта и большинство клеток приобретает стареющий фенотип.
Рис. 4.
Старение клеток E1A+cHa-ras (а) и содержание в них белка LC3 (б). а – активность SA-β-галактозидазы, связанной с клеточным старением, в микроскопических препаратах контрольных клеток (К) и клеток, культивированных 72 ч в присутствии 1 мкМ CAY и (или) 4 мМ бутирата натрия (But). б – Иммунофлуоресценция белка LC3 в клетках, обработанных 2.5 мкМ CAY в течение 24 и 72 ч, или в контрольных клетках (К). Ядра окрашены DAPI. Использовали вторые антитела к иммуноглобулинам кролика, конъюгированные с флуорохромом AlexaFluor 488. Длина масштабного отрезка – 5 мкм.
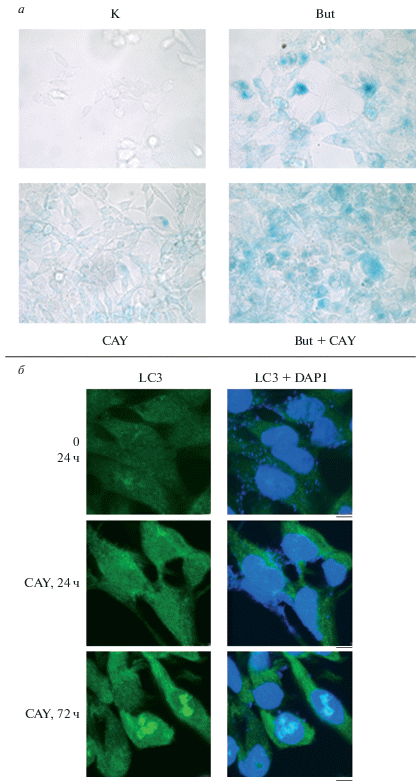
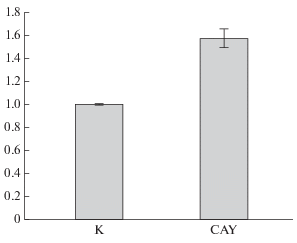
Кроме того, стареющие клетки характеризуются большим содержанием в цитоплазме вакуолей, включая лизосомы и аутофагосомы (Roninson, 2003). Важным маркером аутофагосом является белок LC3, который находится в связанной с мембраной форме LC3-II (Kabeya et al., 2000; Klionsky, 2005; Karim et al., 2007; Mizushima, Yoshimori, 2007). Иммунофлуоресценция с антителами к LC3 показывает незначительное содержание этого белка в цитоплазме контрольных клеток E1A+cHa-ras (рис. 4б). При 1-суточной обработке клеток 2.5 мкМ CAY происходит накопление LC3 в цитоплазме, особенно выраженное в перинуклеарном пространстве. При дальнейшем культивировании клеток в присутствии CAY в течение 72 ч сохраняется высокий цитоплазматический уровень белка LC3, а его избыток может транслоцироваться и депонироваться в ядре (Buckingham et al., 2014; Huang, Liu, 2015) (рис. 4б). Следовательно, CAY может способствовать экспрессии белка LC3 в клетках E1A+cHa-ras, его накоплению в аутофагосомах в связанной с мембранами форме LC3-II (Mizushima, Yoshimori, 2007), а также увеличению количества самих аутофагосом. Чтобы проверить, как CAY может влиять на общий пул аутофагосомных вакуолей в клетках E1A+cHa-ras, мы использовали набор CYTO-ID, который содержит флуоресцентный краситель, специфично и дозозависимо связывающийся с аутофагосомами различной степени развития. После инкубации живых клеток в количестве (4–5) × 105 с этим красителем их анализировали с помощью проточной флуориметрии и по интенсивности сигнала судили о содержании аутофагических вакуолей в клетках. Как видно из рис. 5, обработка клеток 2.5 мкМ CAY в течение 1 сут вызывала прирост количества аутофагосом в среднем более чем в 1.5 раза по сравнению с контрольными необработанными клетками. Таким образом, CAY способствует быстрому развитию аутофагии в E1A+cHa-ras-трансформированных фибробластах.
Рис. 5.
Относительное количество аутофагических вакуолей в клетках E1A+cHa-ras контрольных (К, принято за 1) и обработанных 2.5 мкМ CAY в течение 24 ч. Вертикальные отрезки – стандартное отклонение от среднего. Уровень значимости по t-критерию Стьюдента P = 0.001. Клетки окрашивали красителем CYTO-ID и анализировали с помощью проточной цитофлуориметрии, определяя показатель связывания красителя с аутофагосомами.
В настоящей работе показано, что CAY действительно является специфическим ингибитором HDAC6 в случае деацетилирования ее субстрата α-тубулина в E1A+cHa-ras-трансформированных фибробластах мыши. Его действие вызывает G1/S-блок клеточного цикла, но не приводит к апоптотической гибели клетки mEras. Также CAY вкупе с другим неспецифическим ингибитором HDAC бутиратом натрия может способствовать процессу старения клеток. Кроме того, он влияет на индукцию аутофагических вакуолей в цитоплазме и накоплению в них маркерного белка аутофагосом LC3.
Механизмы влияния CAY на клеточные функции практически не изучены за исключением прямого ингибирующего действия на каталитические центры деацетилазных доменов HDAC6 (Sixto-López et al., 2017), которое приводит к гиперацетилированию ее специфических субстратов, например α-тубулина, и связанного с ним увеличения стабильности микротрубочек в цитоплазме (Asthana et al., 2013). Такая стабилизация способствует укреплению межклеточных контактов, поддержанию целостности клеток легочного эндотелия и подавлению воспалительной реакции посредством ингибирования TNFα-зависимой активации каспазы-3 (Yu et al., 2016). Для клеток аденокарциномы легкого было показано, что CAY понижает уровень белка EGFR и тем самым ингибирует EGFR-сигнальный путь (Wang et al., 2016). Сочетание CAY с противоопухолевым препаратом гефитинибом (ингибитором тирозинкиназы EGFR) вызывает апоптотическую гибель клеток аденокарциномы (Wang et al., 2016). Поскольку во многих опухолевых клетках повышена экспрессия белка HDAC6 и активирован Ras-зависимый сигнальный каскад, что характерно также и для E1A+cHa-ras-трансформированных фибробластов, использование специфических ингибиторов HDAC6, например CAY, приводило к подавлению пролиферации клеток (Dhanyamraju et al., 2015). В нашей работе CAY тоже вызывал G1/S-блок клеточного цикла и подавление роста E1A+cHa-ras-трансформированных фибробластов. Можно предположить, что совместное действие этого ингибитора HDAC6 и какого-либо ДНК-повреждающего агента (этопозида, доксорубицина) будет приводить к гибели этих трансформированных клеток путем деструктивной аутофагии или апоптоза.
Авторы благодарят Н.Д. Аксенова за помощь в проведении проточной цитометрии и В.А. Поспелова за всестороннюю помощь в работе.
Работа выполнена при поддержке РНФ (проект 14-50-00068).
Список литературы
Гордеев С.А., Быкова Т.В., Зубова С.Г., Аксенов Н.Д., Поспелова Т.В. 2015. Антиапоптотический ген bcl-2 препятствует реактивации программы старения, индуцированной ингибитором гистоновых деацетилаз бутиратом натрия в фибробластах крысы, трансформированных онкогенами E1A и c-Ha-Ras. Цитология. 57 (2): 135–143. (Gordeev S.A., Bykova T.V., Zubova S.G., Aksenov N.D., Pospelova T.V. 2015. Antiapoptotic gene bcl-2 prevents cellular senescence program reactivation induced by deacetylase inhibitor sodium butyrate in E1A and c-Ha-Ras transformed rat fibroblasts.Tsitologiya. 57 (2): 135–143.)
Зубова Ю.Г., Быкова Т.В., Зубова С.Г., Абрамова М.В., Аксенов Н.Д., Поспелов В.А., Поспелова Т.В. 2005. Индукция программы ускоренного старения ингибитором гистоновых деацетилаз бутиратом натрия в нормальных и трансформированных фибробластах крысы. Цитология. 47 (12): 1055–1062. (Zubova Yu.G., Bykova T.V., Zubova S.G., Abramova M.V., Aksenov N.D., Pospelov V.A., Pospelova T.V. 2005. Induction of premature senescence program by an inhibitor of sodium butyrate in normal and transformed rat fibroblasts. Tsitologiya. 47 (12): 1055–1062.)
Иготти М.В., Гнедина О.О., Светликова С.Б., Филиппова Е.А., Поспелов В.А. 2016. Бутират натрия усиливает антипролиферативное действие низких концентраций актиномицина Д. Цитология. 58 (10): 755–762. (Igotti M.V., Gnedina O.O., Svetlikova S.B., Filippova E.A., Pospelov V.A. 2016. Sodium butyrate enhances the antiproliferative effect of low concentrations of actinomycin D. Tsitologiya. 58 (10): 755–762.)
Кукушкин А.Н., Светликова С.Б., Поспелов В.А. 2016. Влияние ингибитора HDAC бутирата натрия на экспрессию репарационных генов Rad51 и XRCC5 в клеточных линиях фибробластов mEras-Waf1+/+ и mEras-Waf1–/–. Цитология. 58 (12): 908–915. (Kukushkin A.N., Svetlikova S.B., Pospelov V.A. 2016. Influence of HDAC inhibitor sodium butyrate on the expression of DNA repair genes Rad51 and XRCC5 in mEras-Waf1+/+ and mEras-Waf1–/– transformed cells. Tsitologiya. 58 (12): 908–915).
Поспелова Т.В., Кислякова Т.В., Медведев А.В., Светликова С.Б., Поспелов В.А. 1990. Характеристика трансформированного фенотипа и экспрессии САТ-плазмид в клетках эмбриональных фибробластов крысы, трансформированных онкогенами E1A + cHa-ras. Цитология. 32 (1): 148–155. (Pospelova T.V., Kislyakova T.V., Medvedev A.V., Svetlikova S.B., Pospelov V.A. 1990. The characteristics of the transformed phenotype and the expression of indicator plasmids in the cells of rat embryonic fibroblasts immortalized by oncogene E1Aad5 and transformed by oncogenes E1Aad5+c-Ha-ras. Tsitologiya. 32 (1): 148–155.)
Abramova M.V., Pospelova T.V., Nikulenkov F.P., Hollander C.M., Fornace A.J.Jr., Pospelov V.A. 2006. G1/S arrest induced by histone deacetylase inhibitor sodium butyrate in E1A + Ras-transformed cells is mediated through down-regulation of E2F activity and stabilization of beta-catenin. J. Biol. Chem. 281: 21 040–21 051.
Abramova M.V., Zatulovskiy E.A., Svetlikova S.B., Pospelov V.A. 2010. HDAC inhibitor-induced activation of NF-kB prevents apoptotic response of E1A+Ras-transformed cells to proapoptotic stimuli. Int. J. Biochem. Cell. Biol. 42: 1847–1855.
Aldana-Masangkay G.I., Sakamoto K.M. 2011. The role of HDAC6 in cancer. J. Biomed. Biotech. 2011: 1–10.
Asthana J., Kapoor S., Mohan R., Panda D. 2013. Inhibition of HDAC6 deacetylase activityincreases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J. Biol. Chem. 288 (31): 22 516–22 526.
Bai J., Lei Y., An G., He L. 2015. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS One. 10 (3): 1–11.
Bagnall N.H., Hines B.M., Lucke A.J., Gupta P.K., Reid R.C., Fairlie D.P., Kotze A.C. 2017. Insecticidal activities of histone deacetylase inhibitors against a dipteran parasite of sheep, Lucilia cuprina. Int. J. Parasitology. 7 : 51–60.
Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt. Biochem. 72: 248‑254.
Buckingham E.M., Carpenter J.E., Jackson W., Grose C. 2014. Nuclear LC3-positive puncta in stressed cells do not represent autophagosomes. Biotechniques. 57: 241–244.
Dhanyamraju P.K., Holz P.S., Finkernagel F., Fendrich V., Lauth M. 2015. Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway. Mol. Cancer Ther. 14: 727–739.
Haggarty S.J., Koeller R.M., Wong J.C., Grozinger C.M., Schreiber S.L. 2003. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA. 100: 4389–4394.
Hook S.S., Orian A., Cowley S.M., Eisenman R.N. 2002. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA. 99: 13425–13430.
Huang R., Liu W. 2015. Identifying an essential role of nuclear LC3 forautophagy. Autophagy. 11: 852-853.
Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.-F., Yao T.-P. 2002. HDAC6 is a microtubule-associateddeacetylase. Nature. 417: 455–458.
Kabeya Y., Mirushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19: 5720–5728.
Kanno K., Kanno S., Nitta H., Uesugi N., Sugai T., Masuda T., Wakabayashi G., Maesawa C. 2012. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncology Rep. 28: 867–873.
Karim M.R., Kanazawa T., Daigaku Y., Fujimura S., Miotto G., KadowakiM. 2007. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 3: 553–560.
Kerr E., Holohan C., McLaughlin K.M., Majkut J., Dolan S., Redmond K., Riley J., McLaughlin K., Stasik I., Crudden M., Van Schaeybroeck S., Fenning C., O’Connor R., Kiely P., Sgobba M., Haigh D., Johnston P.G., Longley D.B. 2012. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Diff. 19: 1317–1327.
Klionsky D.J. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118: 7–18.
Lee J.-Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y.-S., Pandey U.B., Kaushik S., Tresse E., Lu J., Taylor J.P., Cuervo A.M., Yao T.-P. 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29: 969–980.
Li Y., Shin D., Kwon S.H. 2013. Histone deacetylase 6 plays a role as a distinct regulatorof diverse cellular processes. FEBS J. 280: 775–793.
Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D.A., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., Yoshida M. 2002. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21: 6820–6831.
Mizushima N., Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy. 3 (6): 542–545.
Namdar M., Perez G., Ngo L., Marks P.A. 2010. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA. 107: 20 003–20 008.
Roninson I.B. 2003. Tumor cell senescence in cancer treatment. Cancer Res. 63: 2705–2715.
Romanov V.S., Abramova M.V., Svetlikova S.B., Bykova T.V., Zubova S.G., Aksenov N.D., Fornace A.J.Jr., Pospelova T.V., Pospelov V.A. 2010. p21Waf1 is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 9: 3945–3955.
Seigneurin-Berny D., Verdel A., Curtet S., Lemercier C., Garin J., Rousseaux S., Khochbin S. 2001. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 21: 8035–8044.
Sixto-López Y., Bello M., Rodríguez-Fonseca R.A., Rosales-Hernández M.C., Martínez-Archundia M., Gómez-Vidal J.A., Correa-Basurto J. 2017. Searching the conformational complexity and binding properties of HDAC6 through docking and molecular dynamic simulations. J. Biomol. Struct. Dyn. 35: 2794–2814.
Trenholme K., Marek L., Duffy S., Pradel G., Fisher G., HansenF.K., Skinner-Adams T.S., Butterworth A., Ngwa C.J., Moecking J., Goodman C.D., McFadden G.I., Sumanadasa S.D.M., Fairlie D.P., Avery V.M., Kurz T., Andrews K.T. 2014. Lysine acetylation in sexual stage malaria parasites is a target forantimalarial small molecules. Antimicrob. Agents Chemother. 58: 3666–3678.
Verdel A., Khochbin S. 1999. Identification of a new family of higher eukaryotic histone deacetylases. J. Biol. Chem. 274: 2440–2445.
Wang Z., Tang F., Hu P., Wang Y., Gong J., Sun S., Xie C. 2016. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncology Rep. 36: 589–597.
Yan J., Seibenhener M.L., Calderilla-Barbosa L., Diaz-Meco M.-T., Moscat J., Jiang J., Wooten M.W., Wooten M.C. 2013. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One. 8: 1–10.
Yu J., Ma M., Ma Z., Fu J. 2016. HDAC6 inhibition prevents TNF-α-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget. 7: 54 714–54 722.
Zhang X., Yuan Z., Zhang Y., Yong S., Salas-Burgos A., Koomen J., Olashaw N., Parsons J.T., Yang X.-J., Dent S.R., Yao T.-P., Lane W.S., Seto E. 2007. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 27: 197–213.
Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. 2003. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo.EMBO J. 22 (5): 1168–1179.
Zhang Z., Yamashita H., Toyama T., Sugiura H., Omoto Y., Ando Y., Mita K., Hamaguchi M., Hayashi S., Iwase H. 2004. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 10: 6962–6968.
Дополнительные материалы отсутствуют.
Инструменты
Цитология