

Цитология, 2019, T. 61, № 3, стр. 185-197
Роль иммунных реакций в гомеостазе и патологии печени
Н. Д. Газатова 1, К. А. Юрова 1, Н. М. Тодосенко 1, М. А. Вульф 1, Л. С. Литвинова 1, *
1 Балтийский федеральный университет имени Иммануила Канта
236041 г. Калининград, Россия
* E-mail: larisalitvinova@yandex.ru
Поступила в редакцию 19.11.2018
После доработки 30.11.2018
Принята к публикации 10.12.2018
Аннотация
Метаболические реакции и процессы тканевой регенерации в здоровой печени протекают с привлечением компонентов воспаления. В сложном микроокружении иммунная система печени должна быть толерантной к нетоксичным веществам и в то же время быстро реагировать на антигены инфекционного и неинфекционного генеза. Дисрегуляция иммунных механизмов приводит к патологическому воспалению и нарушению тканевого гомеостаза органа, характеризующегося прогрессирующим развитием фиброза и формированием печеночной недостаточности. В обзоре систематизированы данные об участии (роли) воспалительных реакций в поддержании нормального иммунного гомеостаза печени и в формировании патологии органа.
Согласно современным представлениям, печень, наряду с выполнением метаболических и детоксикационных функций, участвует в регуляции реакций врожденного и адаптивного иммунитета (рис. 1). Так, в печени обнаруживают большое количество популяций резидентных иммунных клеток (Crispe, 2009; Nemeth et al., 2009; Markose et al., 2018) и образование целого ряда биологически активных молекул: белков острой фазы, компонентов комплемента, цитокинов, хемокинов и др. (таблица 1, рис. 1). В состоянии физиологической нормы печень постоянно подвергается нагрузке алиментарными факторами и продуктами бактерий-комменсалов с воспалительным потенциалом (рис. 2).
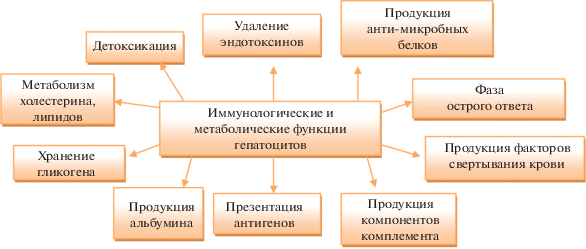
Таблица 1.
Синтез белков острой фазы в печени
| Белки, синтезируемые в фазу острого ответа | Примеры | Функции |
|---|---|---|
| Белки комплемента | Компоненты комплемента – C3, C4 и C9 | Усиление фагоцитоза, регуляция хемотаксиса лейкоцитов, индукция дегрануляции клеток, повышение проницаемости сосудов и лизис бактериальных клеток |
| Железосвязывающие белки | Гаптоглобин, гемопексин, ферритин и гепсидин | Уменьшение свободного железа в сыворотке; антимикробные функции |
| Антимикробные белки | Антимикробный пептид 2, гепсидин | Антимикробная активность |
| Факторы свертывания крови | Фибриноген, протромбин, фактор VIII, фактор IX и фактор Виллебранда | Коагуляция крови |
| Провоспалительные белки | IL-6, липополисахарид (LSP)-связывающий белок, фосфолипаза A 2 | Усиление провоспалительных сигналов и усиление острофазного ответа |
| Лектины, пентаксины, фиколины и коллектины | C-реактивный белок, маннозосвязывающий лектин (МСЛ), коллектин печени 1 (CL-L1), фиколин-2 и сывороточный амилоид P(SAP) | Активация и усиление фагоцитоза |
| Ингибиторы протеаз | α2-Макроглобулин, α1-антихимотрипсин и α1-антитрипсин |
Противовоспалительные функции через ингибирование коагуляции, нейтрофилов и тучных клеток |
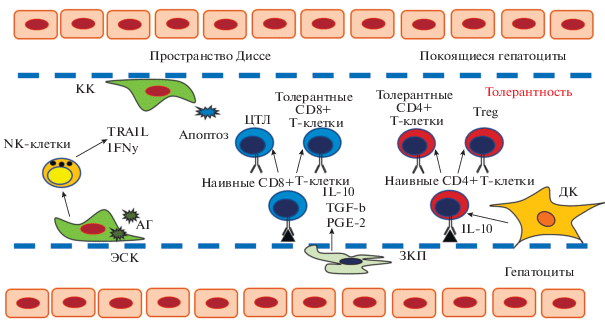
Рис. 2.
Иммунная регуляция локального гомеостаза в печени – норма (адаптировано из Markose et al., 2018). В норме большая часть паренхимы печени представлена покоящимися гепатоцитами. Непаренхиматозные клетки находятся в синусоидах, за исключением ЗКП (в пространстве Диссе). Антигенпрезентирующие клетки (КК, ЭСК, ДК), располагаясь внутри синусоидов, удаляют антигены из кровообращения путем фагоцитоза. Патогены активируют также другие клетки врожденного иммунитета (NK-клетки). ЭСК, взаимодействуя с Т-клетками, способствуют генерации толерантных Т-клеток и инициируют апоптоз активированных Т-лмфоцитов. Медиаторы (IL-10, TGFβ и PGE2), секретируемые АПК также способствуют развитию толерантности печени. Сокращения: КК – клетки Купфера, ЭСК – эндотелиальные синусоидальные клетки, ДК – дендритные клетки, ЗКП – звездчатые клетки.
Непрерывно протекающие метаболические и тканерегенеративные процессы, в сочетании с регулярным воздействием микробных продуктов, приводят к стойкому, строго регулируемому воспалению в здоровом органе, которое необходимо для протекания нормальных физиологических событий, таких как эмбриональная имплантация (Thouas et al., 2015), развитие плода (Wang, Knaut, 2014), инволюционные послеродовые и тканевые регенеративные механизмы (Leoni et al., 2015), гемодинамические изменения, проницаемость капилляров, миграция лейкоцитов в ткани и секреция целого спектра провоспалительных медиаторов и др. (Robinson et al., 2016).
В случае необходимости элиминации гепатотропных патогенов, злокачественных клеток или токсических продуктов метаболической активности, в паренхиме печени происходит активация дополнительных механизмов воспаления (нередко чрезмерных), что неизбежно связано с нарушением тканевого гомеостаза органа и может приводить к прогрессии фиброза с последующим формированием цирроза и печеночной недостаточности (Arroyo et al., 2016; Lange, Moreau, 2018). Таким образом, контролируемое воспаление в печени имеет важное значение для поддержания гомеостаза тканей и органов, тогда как дисрегуляция этих механизмов приводит к развитию патологии.
ИММУННАЯ СТРУКТУРА ПЕЧЕНИ
Печень функционирует как важный буфер между содержимым кишечника и системной циркуляцией – 80% кровоснабжения печени обеспечивается портальной веной. Кровь, поступающая из портальной вены, содержит большое количество безопасных пищевых антигенов и антигенов микрофлоры кишечника (Atif et al., 2018). Печень, подвергаясь высокой иммуногенной нагрузке, сохраняет при этом контроль за патогенными инфекциями и злокачественными клетками (Wieland et al., 2015; Chu et al., 2018; Hou et al, 2018).
Венозная кровь из портальной вены, попадая в печень, смешивается с оксигенированной кровью из печеночной артерии и стекает через печеночные синусоиды в центральные вены через пластинки гепатоцитов. Синусоиды выстланы специализированными печеночными синусоидальными эндотелиальными клетками, которые содержат многочисленные фенестры, обеспечивая движение крови через слой этих клеток к базальным гепатоцитам. Такая структура печени позволяет быстро переносить различные вещества из крови в гепатоциты и способствует удалению и деградации иммуногенных молекул (в частности, бактериальных эндотоксинов) (Atif et al., 2018). Рецепторы распознавания паттернов (pattern-recognition receptors, PRR, РРП), экспрессируемые гепатоцитами и резидентными макрофагами печени (клетки Купфера, КК), связываются с патоген-ассоциированными молекулярными паттернами (PAMPs – patogen associated molecular patterns), а также с молекулярными фрагментами, ассоциированными с повреждениями (DAMPs – damage-associated molecular patterns), которые присутствуют в больших количествах в крови, поступающей из воротной вены (Takeuchi, Akira, 2010; Yu et al., 2018). Таким образом, опасные для организма микроорганизмы и эндогенные молекулы фагоцитируются и утилизируются впоследствии гепатоцитами и КК без образования медиаторов воспаления, которые обычно сопровождают РРП-сигнализацию. Этот механизм детоксикации крови, поступающей из кишечника, защищает весь организм от чрезмерной иммунной активации и формирует уникальную иммунологическую среду в печени.
Таким образом, портальный кровоток низкого давления, фенестрированный эндотелий и отсутствие базальной мембраны облегчают взаимодействия между резидентными иммунными клетками и негемопоэтическими клетками печени (Robinson et al., 2016). Резидентные иммунные клетки базируются в синусоидах печени и субэндотелиальных пространствах Диссе, где происходит образование лимфы, попадающей затем в лимфатические сосуды вдоль портального тракта. Популяция этих клеток включает в себя антигенпрезентирующие клетки (АПК), миелоидные клетки, а также лимфоидные клетки врожденного и адаптивного иммунитета (Norris et al., 1998; Kelly et al., 2014; Robinson et al., 2016). Несмотря на то, что некоторые популяции резидентных иммунных клеток печени хорошо изучены, их полный спектр до сих пор не выяснен.
МИЕЛОИДНЫЕ ПОПУЛЯЦИИ КЛЕТОК ПЕЧЕНИ
Макрофаги печени, или КК, составляют до 90% от общей численности резидентных макрофагов в организме, образуя почти 1/3 непаренхимных клеток в печени (Bilzer et al., 2006; MacParland et al., 2018) (рис. 2). КК в большом количестве экспрессируют рецепторы распознавания паттернов PAMPs и DAMPs, комплемента и Fc-рецепторы, что обеспечивает им высокую фагоцитарную активность и способность к продукции цитокинов с провоспалительным спектром действия (Su et al., 2000; van Egmond et al., 2000). КК играют важную роль в иммунной регуляции, репарации и регенерации паренхимы печени (Elsegood et al., 2015), а также обладают способностью реагировать на различные медиаторы, взаимодействовать с толл-подобными рецепторами (Toll-like receptor, TLR) (Wu et al., 2010), RIG-I-подобными (RIG-I-like receptors, RLR) и NOD-подобными рецепторами (Nod-like-receptor, NLR) (Miura et al., 2013).
В здоровой печени представлены также популяции дендритных клеток (ДК): миелоидные и плазмоцитоидные. ДК печени описываются как фенотипически незрелые; однако в некоторых случаях ДК могут стимулировать сильные реакции Т-клеток (Thomson, Knolle, 2010). Относительно недавно было показано, что субпопуляция ДК в печени человека (CD141+) является мощным продуцентом цитокинов и активатором Т-клеток (Kelly, 2014).
В здоровой печени также присутствуют миелоидные супрессорные клетки (myeloid-derived suppressor cells, MDSC, МлСК) (Chen et al., 2011), их количество возрастает при хронических заболеваниях печени (Pallett, 2015). МлСК обладают способностью подавлять активацию Т-лимфоцитов за счет продукции IL-10, трансформирующего фактора роста β (TGFβ) и аргиназы (Gabrilovich, 2009). Гранулоцитарные клетки, в частности нейтрофилы, как правило, отсутствуют в ткани здоровой печени; их обнаруживают только в ответ на инфекцию и воспаление (Markose et al., 2018). Нейтрофилы и их предшественники имеют несколько общих фенотипических маркеров с МлСК.
ЛИМФОИДНЫЕ ИММУННЫЕ КЛЕТКИ ПЕЧЕНИ
Согласно современным представлениям, печень в условиях физиологической нормы, рассматривается в качестве иммунного органа, обогащенного клетками врожденного иммунитета (КК, γδ-Т-лимфоциты, натуральные киллеры (NK, natural killer), натуральные киллеры Т-клетки (NKT, включая инвариантные iNKT-клетки) и др.) и адаптивного иммунитета (популяции CD4+- и CD8+-Т-клеток, В‑лимфоциты) (Kenna et al., 2003, 2004; Dusseaux et al., 2011; Kurioka et al., 2016; Hunter et al., 2018) (рис. 2).
Расположенные в печени популяции лимфоцитов врожденного звена иммунитета являются мощными продуцентами различных медиаторов, оказывая влияние на врожденные и адаптивные иммунные реакции (рис. 2). Репертуар этих клеток этих клеток у человека сильно отличается от такового у животных. Так, у мышей NKT- и NK-клетки составляют до 40 и 10% от общего количества лимфоцитов печени соответственно, тогда как в организме человека, напротив, преобладают NK-клетки (Gao et al., 2008; Robinson et al., 2016).
У людей CD56bright NK-клетки составляют более 50% от общего количества печеночных NK-популяций по сравнению с 10–15% в периферической крови (Moroso et al., 2010). У мышей инвариантные NKT-клетки попадают в печень преимущественно через связывание лимфоцитарного функционального антигена (Lymphocyte function-associated antigen 1, LFA-1) с молекулой межклеточной адгезии 1-го типа (Inter-Cellular Adhesion Molecule 1, ICAM-1) и образуют тканерезидентную популяцию иммунных клеток (Lynch et al., 2015).
В здоровой печени также встречаются клетки, отвечающие за формирование адаптивного иммунитета. Наиболее распространены популяции цитотоксических CD8 Т-клеток, активированных Т-клеток и Т-клеток памяти (Norris et al., 1998). Накопление этих субпопуляций Т-лимфоцитов ассоциировано с их апоптозом и секвестрацией в печени, в связи с чем печень рассматривают в качестве органа-“утилизатора” Т-лимфоцитов (Robinson et al., 2016). В-клетки составляют до 8% от общей популяции лимфоцитов в печени человека (Norris et al., 1998). Субпопуляции В-клеток, в частности CD5+, увеличиваются при гепатотропной вирусной инфекции (Racanelli et al., 2001; Curry et al., 2003).
ГЕМОПОЭТИЧЕСКИЕ КЛЕТКИ-ПРЕДШЕСТВЕННИЦЫ ПЕЧЕНИ
Согласно классическим представлениям, у здоровых людей процесс развития гемопоэтических клеток ограничен костным мозгом. Относительно недавно была выдвинута гипотеза о том, что некоторые популяции иммунных клеток дифференцируются локально в печени (Golden-Mason et al., 2000; Robinson et al., 2016). Гемопоэтические клетки-предшественницы печени донора восстанавливают популяции иммунных клеток после летального облучения у мышей (Jiang et al., 2013). Печень взрослого человека также содержит популяции клеток-предшественниц, которые экспрессируют поверхностные маркеры, характерные для незрелых кроветворных клеток и образуют in vitro мультилинейные гемопоэтические колонии (Golden-Mason et al., 2000). Эти миелоидные и лимфоидные популяции-"предшественницы" могут способствовать развитию фенотипически различных популяций иммунных клеток в печени посредством механизмов локальной дифференцировки (Robinson et al., 2016). После трансплантации печени имеет место химеризм, при котором гемопоэтические стволовые клетки реципиента поступают в донорскую печень, где могут дифференцироваться также в эндотелиальные клетки и гепатоциты (Carbone, Neuberger, 2014).
РОЛЬ НЕГЕМОПОЭТИЧЕСКИХ КЛЕТОК ПЕЧЕНИ В ИММУНОРЕГУЛЯЦИИ
Наряду с иммунными клетками печени, ключевую роль в формировании местного и системного врожденного иммунитета играют негемопоэтические клетки, такие как гепатоциты, синусоидальные эндотелиальные и звездчатые клетки (клетки Ито, ЗКП), для которых характерна экспрессия широкого диапазона РРП (Seki, Brenner, 2008; Wu et al., 2010; Jenne, Kubes, 2013) (рис. 2). Экспрессия этими клетками TLR и молекул к гликопротеину обеспечивает связывание и клиренс портальной крови от PAMPs и регулирует продукцию воспалительных медиаторов (Robinson et al., 2016).
Синусоидальные эндотелиальные клетки и гепатоциты, экспрессируя молекулы главного комплекса гистосовместимости (HLA) II класса, обладают способностью представлять антигены Т-клеткам локально в печени (Crispe, 2009; Thomson, Knolle, 2010). В результате такого взаимодействия Т-клетки образуют популяции функциональных T-эффекторных лимфоцитов в отсутствие CD4 T-клеточной активации и увеличивают иммуногенность патогенов (Böttcher et al., 2013). Гепатоциты мыши способны представлять липидные антигенные компоненты инвариантным NKT-клеткам за счет экспрессии на своей мембране CD1d-дифференцировочных молекул (Yang et al., 2007). У человека экспрессия молекул CD1d на здоровых гепатоцитах не обнаружена и появляется только в ответ на инвазию вирусами гепатита (Yanagisawa et al., 2013).
РОЛЬ МИКРООКРУЖЕНИЯ ПЕЧЕНИ В ИММУНОРЕГУЛЯЦИИ
Анатомическая структура печени, ее резидентный иммунный репертуар и ее постоянная стимуляция патогенами способствуют созданию в этом органе уникальной цитокиновой среды, содержащей основные провоспалительные (интерлейкин 2 (IL-2), IL-7, IL-12, IL-15, интерферон-γ (IFNγ) и др.) и противовоспалительные (IL-10, IL-13 и TGFβ) цитокины/факторы роста (Golden-Mason et al., 2004; Kelly et al., 2006; Tu et al., 2007). Сформированное микроокружение определяет баланс между иммунной толерантностью и реакциями воспаления в здоровой печени. Как уже упоминалось ранее, существенный вклад в формирование уникального микроокружения печени вносят особенности ее кровоснабжения (Wilson et al., 2014). Кроме того, негемопоэтические и гемопоэтические клетки печени постоянно получают сигналы от алиментарных и комменсальных молекул, что вызывает состояние толерантности (Robinson et al., 2016).
На состояние цитокиновой среды в печени влияют также высокие уровни жиров и углеводов, поступающих в кровоток из пищи. Углеводы поглощаются гепатоцитами и депонируются в виде гликогена, в то время как жиры, переносимые из кишечника в виде хиломикронов, преобразуются в ряд липопротеинов. Последние распределяют холестерин и триглицериды по всему организму. Важно отметить, что метаболические реакции в печени тесно связаны с процессами воспаления (Tannahill et al., 2013). В частности, триглицериды и холестерин способствуют передаче TLR-сигналов и активации воспаления (Tall, Yvan-Charvet, 2015). Так, в результате метаболической регуляции воспаления при неалкогольной жировой болезни печени регистрируются повышенные уровни провоспалительных цитокинов, которые способствуют развитию фиброза печени (Tilg, Moschen, 2010; Вульф и др., 2018). Провоспалительный профиль на мышиных моделях может быть воспроизведен с помощью диеты с высоким содержанием жиров; частично этот эффект обусловлен сенсибилизацией гепатоцитов к агонистам TLR – насыщенным жирным кислотам (Csak et al., 2011). Метаболические изменения в инфицированных гепатоцитах при вирусных гепатитах В и С способствуют репликации вируса (Bar-Yishay et al., 2011; Filipe, McLauchlan, 2015). Макрофаги и ДК подвергаются метаболическому перепрограммированию при активации антигенами, переходя с окислительного фосфорилирования на аэробный гликолиз (так называемый эффект Варбурга), что способствует синтезу этими клетками провоспалительных медиаторов (O’Neill, Pearce, 2015). При нормоксии переход к аэробному гликолизу в макрофагах приводит к увеличению содержания сукцината, что в свою очередь, активирует продукцию фактора транскрипции HIF1α (фактор, индуцируемый гипоксией 1α) и IL-1β (Tannahill et al., 2013). В связи с особенностями печеночного кровотока, гипоксический ответ в здоровой печени не развивается (Wilson et al., 2014), что свидетельствует об уникальном ответе клеток печени на провоспалительные метаболические сигналы. Более глубокое понимание роли динамического микроокружения печени в поддержании нормального гомеостаза необходимо для интерпретации и оценки спектра изменений при формировании патологии печени.
ВОСПАЛИТЕЛЬНЫЕ ПРОЦЕССЫ В ЗДОРОВОЙ ПЕЧЕНИ. ИММУННАЯ ТОЛЕРАНТНОСТЬ ПЕЧЕНИ
Воспаление и провоспалительные медиаторы, индуцируемые резидентными иммунными клетками и негемопоэтическими клетками печени, играют важную роль в поддержании местного и системного гомеостаза у здоровых людей (рис. 2).
Здоровая печень часто описывается как иммунологически толерантный орган (Protzer et al., 2012, Crispe, 2014; Markose et al., 2018). Впервые это представление возникло в трансплантологии, когда было обнаружено, что аллогенная трансплантация печени у свиней значительно лучше переносится, чем трансплантация других органов (Calne et al., 1969). В частности, эта концепция подтверждается тем фактом, что реципиенты после трансплантации печени не нуждаются в высоком уровне иммуносупрессии (Lerut et al., 2006, Simpson et al., 2006).
Поддержанию иммунной толерантности печени способствуют резидентные миелоидные клетки. Так, КК под воздействием бактериальных эндотоксинов, продуцируют противовоспалительные цитокины, включая IL-10 и простагландины (Markose et al., 2018). Повышение уровня IL-10 ингибирует экспрессию ко-стимулирующих молекул на АПК, предотвращая активацию CD4 T-лимфоцитов, ограничивая тем самым адаптивный иммунный ответ (Robinson et al., 2016; Markose et al., 2018). Презентация антигенных молекул КК способствует увеличению популяции T-регуляторных клеток (Treg), обеспечивающих антиген-специфическую толерантность также за счет продукции IL-10 (Heymann et al., 2015). Миелоидные ДК печени менее эффективно активируют Т-лимфоциты и продуцируют значительно больше IL-10 по сравнению с аналогичными клетками селезенки (Goddard et al., 2004; De Creus et al., 2005; Bamboat et al., 2009). Печеночные плазмоцитоидные ДК фенотипически незрелые и не являются эффективными активаторами Т-клеток ex vivo, однако способны к эффективному представлению антигена после созревания, индуцированного факторами роста или агонистами TLR (Kingham et al., 2007). Также показано, что плазмоцитоидные ДК печени способны к синтезу IL-10 и активации Treg лимфоцитов (Tokita et al., 2008). В дополнение к этим специфичным АПК синусоидальные эпителиальные клетки и гепатоциты также обладают способностью самостоятельно представлять антигены T-лимфоцитам (Thomson, Knolle, 2010). В определенных условиях антигенпрезентирующую функцию могут выполнять и ЗКП, выступая в роли вспомогательных “клеток-наблюдателей” (Ichikawa et al., 2011). Презентация антигенов в печени, в отсутствие ко-стимулирующих молекул и без содействия CD4 Т-лимфоцитов, способствует формированию толерантности Т-клеток, препятствуя их эффективной дифференцировке (Bowen et al., 2004; Wuensch et al., 2010). Толерогенная среда печени также поддерживается регуляторными миелоидными популяциями, в частности, МлСК (Sander et al., 2010; Zimmermann et al., 2015). Последние опосредуют свою активность посредством продукции иммуносупрессивных цитокинов, таких как IL-10 и TGFβ, а также синтеза аргиназы и индоламин-2,3-диоксигеназы (IDO), ограничивая тем самым доступ иммунных клеток к незаменимым аминокислотам, необходимым им для процессов активации и пролиферации (Gabrilovich, Nagaraj, 2009).
СИСТЕМНЫЙ ГОМЕОСТАЗ И ФАЗА ОСТРОГО ОТВЕТА
Ключевой системной функцией печени является синтез сывороточных белков, включая белки комплемента, альбумин, фибриноген, факторы свертывания крови, транспортные белки, ингибиторы протеазы и липопротеины (табл. 1), которые участвуют в системном транспорте питательных веществ, в регуляции осмотического давления крови и действуют как неактивные предшественники нескольких врожденных иммунных медиаторов. Печень также играет центральную роль в обнаружении и формировании реакции на воспалительные сигналы с любых участков тела. На ранних стадиях внепеченочного воспаления цитокины, продуцируемые иммунными клетками, поступают в кровоток и активируют гепатоциты, которые впоследствии инициируют системный острофазный ответ (Markose et al., 2018; Sá-Pereira et al., 2018).
Гепатоциты увеличивают продукцию белков острой фазы, а также синтезируют IL-6, который на системном уровне усиливает воспалительные реакции. Острофазные белки (БОФ), продуцируемые гепатоцитами, имеют прямую эффекторную функцию (табл. 1) и отвечают за формирование системного воспаления, которое способствует обезвреживанию патогенов (Zhou et al., 2016; Lindquist et al., 2018). Кроме того, БОФ запускают целый ряд механизмов в макроорганизме. В частности, воздействуя на костный мозг, они обеспечивают развитие лейкоцитоза; через центральные механизмы – повышение температуры тела, а также способствуют массивной инфильтрации иммунокомпетентных клеток в очаг воспаления (Lindquist et al., 2018).
В то же время острофазная реакция включает ряд механизмов для ограничения чрезмерного воспаления, в том числе снижение активности нейтрофилов ингибиторами протеазы, такими как α2-макроглобулин, подавление C-реактивным белком продукции TNF-α макрофагами (Inatsu et al., 2009), рекрутирование МлСК посредством сывороточного амилоида A (Sander et al., 2010). Безусловно, эти процессы необходимы для ограничения повреждения тканей, вызванного воспалительным процессом и запуска репаративного процесса.
ИММУНИТЕТ ПЕЧЕНИ И ПРОЦЕССЫ ФИБРОГЕНЕЗА/ФИБРОЛИЗА
Как уже упоминалось ранее, при сохранении общей толерогенной среды, иммунная система печени должна быстро реагировать на опухолевые клетки и патогенные микроорганизмы. Некоторые патогены, включая несколько видов вирусов, бактерий и паразитов, являются гепатотропными (Protzer et al., 2012), для борьбы с которыми необходим эффективный местный иммунитет. Во время острого поражения печени, активированные КК становятся мощными продуцентами цитокинов/хемокинов, таких как IL-1, IL-6, TNF-α, MIP-1α и RANTES и др. (Mosher et al., 2001; Gregory, Wing, 2002; Tacke et al., 2009). Этот провоспалительный ответ индуцируется после передачи сигналов через TLR (в частности, TLR3), независимые от MyD88 (Myeloid differentiation primary response gene (88)) (Tu et al., 2008). В здоровой печени взрослого индивидуума есть также мощные провоспалительные CD141+ миелоидные ДК, способные индуцировать продукцию IFN-γ и IL-17 Т-клетками (Kelly et al., 2014). Последние, при наличии воспалительной среды в печени, могут дифференцироваться в популяции функциональных T-эффекторных клеток, способных к элиминации патогенов даже в отсутствие классической поддержки CD4 Т-лимфоцитов (Böttcher et al., 2013).
Реакции острого воспаления в печени приводят к рекрутингу и активации популяций лейкоцитов, а также способствуют индукции фиброзных реакций в очаге повреждения (Baeck, Tacke, 2014; Pellicoro et al., 2014) (рис. 3). Фиброгенез, выявляемый при острой травме, оказывает протекторное действие в отношении выживших гепатоцитов, путем снижения проапоптотической передачи сигналов и повышения устойчивости к ряду токсинов (Bourbonnais et al., 2012). Этот процесс регулируется воспалительными цитокинами и факторами роста, продуцируемыми лейкоцитами, которые мигрируют в поврежденную ткань. TNFα, IL-6, фактор роста тромбоцитов (Platelet-derived growth factor, PDGF) и TGF-β приводят к активации и пролиферации ЗКП, которые являются активными продуцентами компонентов внеклеточного матрикса (ВКМ), включая α-гладкомышечный актин и коллаген I типа (Chae et al., 2018).
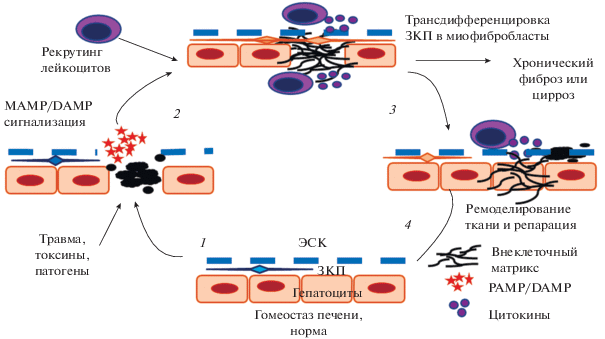
Рис. 3.
Локальный гомеостаз в печени в ответ на повреждение (адаптировано из Robinson et al., 2016). (1) При острой травме, гибели клеток или инфекции апоптотические гепатоциты высвобождают множество DAMPs и (или) PAMPs, которые распознаются соседними гепатоцитами, ЗКП и популяциями резидентных иммунных клеток; активация клеток. (2) Активированные клетки секретируют воспалительные медиаторы, опосредующие рекрутинг лейкоцитов и трансдифференцировку ЗКП в миофибробласты. Последние инициируют фиброз посредством синтеза компонентов внеклеточного матрикса. (3) Воспаление способствует экспрессии факторов фибролиза лейкоцитами и апоптозу миофибробластов. (4) Регенерация поврежденной ткани печени, восстановление гомеостаза. Отсутствие фазы ремоделирования и репарации печени способствует тому, что постоянное воспаление приводит к прогрессирующему развитию фиброза печени и возможному циррозу. Сокращения: те же, что и на рис. 3.
Фиброз печени – патологический процесс, однако он необходим для восстановления гомеостаза тканей после их повреждения (рис. 3). Фиброз становится клинически значимым, когда он приводит к изменению структуры и функции ткани из-за нарушения регуляции или чрезмерного воспаления. Для фиброгенеза, индуцированного большинством гепатотоксических факторов, характерна цикличность процессов повреждения/репарации гепатоцитов (Thouas et al., 2015). В фазе репарации/прекращения острого фиброза печени, КК продуцируют ряд противовоспалительных медиаторов, которые увеличивают экспрессию матричных металлопротеиназ и подавляют синтез их ингибиторов (Thouas et al., 2015), а цитотоксическое действие NK-клеток, направленное на ЗКП, способствует регуляции внутрипеченочного фиброза. Трансдифференцировка активированных ЗКП в миофибробласты изменяет баланс активирующих и ингибирующих лигандов клеточных рецепторов, что приводит к их делеции посредством NK-клеток через TRAIL (TNF-related apoptosis-inducing ligand, TRAIL), NKG2D (natural killer group 2D) и FasL-зависимые механизмы (Radaeva et al., 2006; Glässner et al., 2012). Многогранные взаимодействия субпопуляций иммунных клеток, регулирующие процессы репарации печени, приводят к рассмотрению ее роли в качестве важного компонента врожденного иммунного ответа на повреждение тканей.
ВОСПАЛЕНИЕ И РЕГЕНЕРАЦИЯ ПЕЧЕНИ
Способность печени полностью или частично восстанавливаться после травмы необходима для поддержания основных функций органа при контроле метаболизма и ксенобиотической детоксикации. Эта регенеративная способность печени обусловлена пролиферацией зрелых гепатоцитов в ответ на различные экологические сигналы (Malato et al., 2011; Hou et al., 2018). Регенерационный потенциал печени обусловлен медиаторами воспаления (такими как IL-1α, TNFα и IL-6), факторами роста (фактор роста гепатоцитов и фактор эпидермального роста) и резидентными популяциями иммунных клеток (Selzner et al., 2003; Michalopoulos, 2007; Chae et al., 2018). Так, у мышей с низким уровнем IL-6 нарушена регенерация печени и снижена пролиферация гепатоцитов; эти дефекты устраняются сразу после введения IL-6 (Cressman et al., 1996). Снижение регенерации печени наблюдается также у мышей, получавших антитела против TNFα. Прорегенерационный эффект TNFα требует экспрессии индуцибельной синтазы оксида азота, которая блокирует потенциальный проапоптотический сигналинг TNFα (Rai et al., 1998). Активация КК обусловлена рекрутингом нейтрофилов в печень в ответ на воспалительные сигналы, через ICAM-1-зависимый путь. Белки комплемента – C3 и C5 – также играют важную роль в прайминге КК и способствуют пролиферации гепатоцитов после частичной гепатэктомии (Strey et al., 2003).
Резидентные иммунные клетки могут также оказывать ингибирующее действие на процессы регенерации печени (Gao et al., 2009). Так, снижение активности NK-клеток способствует усилению регенерации печени за счет уменьшения продукции IFNγ, который ингибирует клеточный цикл гепатоцитов и их пролиферацию (Sun, Gao, 2004; Shen et al., 2008). Истощение NKT-клеток у трансгенных мышей, инфицированных вирусом гепатита B, также усиливает регенерацию печени из-за снижения продукции IFNγ и TNFα (Dong et al., 2007). Эти популяции резидентных иммунных клеток действуют по принципу отрицательной обратной связи, регулируя регенерацию печени и восстанавливая ее гомеостаз.
ТЕРАПЕВТИЧЕСКИЕ СТРАТЕГИИ, НАПРАВЛЕННЫЕ НА ВОССТАНОВЛЕНИЕ ИММУННОГО ГОМЕОСТАЗА ПРИ ЗАБОЛЕВАНИЯХ ПЕЧЕНИ
Как уже упоминалось ранее, реакции воспаления в паренхиме печени являются важным аспектом функционирования здорового органа, тогда как процессы фиброзирования необходимы для восстановления ее нормального гомеостаза в результате повреждения (Robinson et al., 2016).
При длительной (хронической) активации механизмов врожденного иммунитета под действием различных факторов: хронической инфекции, повреждения тканей, избыточного потребления алкоголя или жирной пищи, роста опухоли и др., происходит формирование патологического (чрезмерного) воспаления, прогрессирование фиброза печени до цирроза, что опосредует, в целом, нарушение функций и архитектоники органа (Brenner et al., 2013; Li et al., 2016). Постоянные провоспалительные сигналы от популяций негемопоэтических и резидентных иммунных клеток поддерживают в активированном состоянии ЗКП, трансформированные в миофибробласты (Seki, Schwabe et al., 2015), что препятствует их старению и снижает способность NK-клеток индуцировать их апоптоз (Gao et al., 2009).
В то же время хроническое воспаление в печени, ассоциированное с действием большинства гепатотоксических факторов, приводит к нарушению механизмов иммунной толерантности органа и имеет важное клиническое значение, обусловливая развитие патологических процессов в паренхиме печени (Karlmark et al., 2009; Heymann et al., 2015). Так, распространение бактериальных продуктов при декомпенсированном циррозе может привести к провоспалительному цитокиновому каскаду (шторму) и последующей системной органной недостаточности (Bernardi et al., 2015). Терапевтические стратегии, направленные на поддержание нормальных толерогенных механизмов в печени, способствующие прекращению воспаления, регрессии фиброза и регенерации гепатоцитов, могут усилить действие иммунодепрессантов (Carbone, Neuberger, 2014). Потенциал таких подходов четко продемонстрирован в клиническом испытании, где была использована комбинация гранулоцитарного колониестимулирующего фактора (GM-CSF) и эритропоэтина для усиления регенерации печени, что улучшало выживаемость у пациентов с прогрессирующим циррозом на 12 мес. и значительно уменьшало показатели тяжести заболевания (Kedarisetty et al., 2015).
В контексте вышесказанного следует отметить двойственную роль механизмов, обеспечивающих, с одной стороны, иммунологическую толерантность, с другой – реакции воспаления в печени. Нарушение тонкой грани между этими процессами приводит к формированию патологии. В частности, толерогенные механизмы в печени могут способствовать сохранению патогенов, таких как вирус гепатита С, и потенцировать рост первичных и метастатических опухолей, которые со временем создают состояние дисрегуляционного воспаления. Презентация патологических антигенов в печени может активно подавлять иммунные реакции, вызывая тем самым состояние иммунной толерантности к патогену или опухоли (Bowen et al., 2004; Wuensch et al., 2010; Protzer et al., 2012; Makarova-Rusher et al., 2015). В то же время, активация компонентов врожденного иммунитета на молекулы PAMPs или DAMPs, высвобождаемые при инфекционном процессе или опухолевом росте, с одной стороны, способствует формированию эффективного воспаления, с другой – опосредует стимуляцию механизмов, направленных на минимизацию повреждения тканей. Последние обеспечиваются за счет увеличения популяций иммунорегуляторных клеток (МлСК), способствующих развитию опухолей (Gabrilovich, Nagaraj, 2009; Kitamura et al., 2015), эпигенетических и метаболических изменений, приводящих к толерантности иммунных клеток (Saeed et al., 2014), индукции негативных регуляторов провоспалительных сигнальных путей (Ivashkiv, Donlin, 2014) и истощения Т-клеток (Park, Rehermann, 2014; Makarova-Rusher et al., 2015).
Таким образом, регуляция механизмов, опосредующих процессы воспаления и иммунной толерантности в печени, с использованием различных подходов медикаментозной терапии, основанных на применении таргетных молекул, может привести к уменьшению выраженности патологического процесса в паренхиме печени или вызвать элиминацию патогенов.
ЗАКЛЮЧЕНИЕ
Поддержание нормального гомеостаза печени, а также защита организма от патогенов, опухолей и повреждения тканей, осуществляются посредством запуска в печени механизмов воспаления, которые инициируют, опосредуют и устраняют системные и местные иммунные реакции, способствуя, в то же время, формированию патологии печени. Разнообразный репертуар популяций иммунных клеток в печени, наряду с провоспалительным потенциалом негемопоэтических клеток, играет центральную роль, как в гомеостатическом, так и в патологическом воспалении печени. Комплексные воспалительные и иммунорегуляторные механизмы в печени необходимы для поддержания органного и системного гомеостаза, а также для мобилизации комплементарных воспалительных механизмов для защиты от инфекции, метастазов и повреждения тканей. Чрезмерная или дисрегуляционная воспалительная активность печени сопровождается формированием патологии органа.
На наш взгляд, терапевтические стратегии лечения заболеваний печени, в том числе хронических диффузных (включая стеатоз, фиброз и цирроз), должны быть направлены на восстановление ее иммунологического гомеостаза.
Работа выполнена при финансовой поддержке Программы повышения конкурентоспособности и субсидии “Организация проведения научных исследований 20.4986.2017/ВУ” БФУ им. И. Канта.
Список литературы
Вульф М.А., Кириенкова Е.В., Скуратовская Д.А., Левада E.В., Волкова Л.В., Затолокин П.А., Газатова Н.Д., Литвинова Л.С. 2018. Факторы, способствующие развитию неалкогольной жировой болезни печени и инсулинорезистентности при ожирении. Биомедицинская химия. 64 (5) : 444–450. (Vulf M.A., Kirienkova E.V., Skuratovskaia D.A., Levada E.V., Volkova L.V., Zatolokin P.A., Gazatova N.D., Litvinova L.S. 2018. Factors governing development of nonalcoholic fatty liver disease and insulin resistance in obesity. Biomed. Khim. 64 (5) : 444–450).
Arroyo V., Moreau R., Kamath P.S., Jalan R., Ginès P., Nevens F., Fernández J., To U., García-Tsao G., Schnabl B. 2016. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Primers. 2: 16041.
Atif M., Warner S., Oo Y.H. 2018. Linking the gut and liver: crosstalk between regulatory T-cells and mucosa-associated invariant T-cells. Hepatol. Int. 12 : 305–314.
Baeck C., Tacke F. 2014. Balance of inflammatory pathways and interplay of immune cells in the liver during homeostasis and injury. EXCLI J. 13 : 67–81.
Bamboat Z.M., Stableford J.A., Plitas G., Burt B.M., Nguyen H.M., Welles A.P. 2009. Human liver dendritic cells promote T‑cell hyporesponsiveness. J. Immunol. 182 : 1901–1911.
Bar-Yishay I., Shaul Y., Shlomai A. 2011. Hepatocyte metabolic signalling pathways and regulation of hepatitis B virus expression. Liver Int. 31 : 282–290.
Bernardi M., Moreau R., Angeli P., Schnabl B., Arroyo V. 2015. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 63 : 1272–1284.
Bilzer M., Roggel F., Gerbes A.L. 2006. Role of Kupffer cells in host defense and liver disease. Liver Int. 26 : 1175–1186.
Böttcher J.P., Schanz O., Wohlleber D., Abdullah Z., Debey-Pascher S., Staratschek-Jox A. 2013. Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep. 3 : 779–795.
Bourbonnais E., Raymond V.-A., Ethier C., Nguyen B.N., El-Leil M.S., Meloche S. 2012. Liver fibrosis protects mice from acute hepatocellular injury. Gastroenterology. 142 : 130–139.
Bowen D.G., Zen M., Holz L., Davis T., McCaughan G.W., Bertolino P. 2004. The site of primary T-cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J. Clin. Invest. 114 : 701–712.
Brenner C., Galluzzi L., Kepp O., Kroemer G. 2013. Decoding cell death signals in live inflammation. J. Hepatol. 59 : 583–594.
Calne R.Y., Sells R.A., Pena J.R., Davis D.R., Millard P.R., Herbertson B.M. 1969. Induction of immunological tolerance by porcine liver allografts. Nature. 223 : 472–476.
Carbone M., Neuberger J.M. 2014. Autoimmune liver disease, autoimmunity and liver transplantation. J. Hepatol. 60 : 210–223.
Chae M.S., Moon K.U., Chung H.S., Park C.S., Lee J., Choi J.H., Hong S.H. 2018. Serum interleukin-6 and tumor necrosis factor-α are associated with early graft regeneration after living donor liver transplantation. PLoS One. 13 : e0195262.
Chen S., Akbar S.M.F, Abe M., Hiasa Y., Onji M. 2011. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin. Exp. Immunol. 166 : 134–142.
Chu H., Williams B., Schnabl B. 2018. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2 : 43–51.
Cressman D.E., Greenbaum L.E., DeAngelis R.A., Ciliberto G., Furth E.E., Poli V. 1996. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 274 : 1379–1383.
Crispe I.N. 2009. The liver as a lymphoid organ. Annu. Rev. Immunol. 27 : 147–163.
Crispe I.N. 2014. Immune tolerance in liver disease. Hepatology. 60 : 2109–2117.
Csak T., Ganz M., Pespisa J., Kodys K., Dolganiuc A., Szabo G. 2011. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 54 : 133–144.
Curry M.P., Golden-Mason L., Doherty D.G., Deignan T., Norris S., Duffy M. 2003. Expansion of innate CD5pos B cells expressing high levels of CD81 in hepatitis C virus infected liver. J. Hepatol. 38 : 642–650.
De Creus A., Abe M., Lau A.H., Hackstein H., Raimondi G., Thomson A.W. 2005. Low TLR4 expression by liver dendritic cells correlates with reduced capacity to activate allogeneic T-cells in response to endotoxin. J. Immunol. 174 : 2037–2045.
Dong Z., Zhang J., Sun R., Wei H., Tian Z. 2007. Impairment of liver regeneration correlates with activated hepatic NKT cells in HBV transgenic mice. Hepatology. 45 : 1400–1412.
Dusseaux M., Martin E., Serriari N., Péguillet I., Premel V., Louis D. 2011. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood.117 : 1250–1259.
Elsegood C.L., Chan C.W., Degli-Esposti M.A., Wikstrom M.E., Domenichini A., Lazarus K. 2015. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology. 62 : 1272–1284.
Filipe A., McLauchlan J. 2015. Hepatitis C virus and lipid droplets: finding a niche. Trends Mol. Med. 21 : 34–42.
Gabrilovich D.I., Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9 : 162–174.
Gao B., Jeong W.-I., Tian Z. 2008. Liver: an organ with predominant innate immunity. Hepatology. 47 : 729–736.
Gao B., Radaeva S., Park O. 2009. Liver natural killer and natural killer T-cells: immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 86 : 513–528.
Glässner A., Eisenhardt M., Krämer B., Körner C., Coenen M., Sauerbruch T. 2012. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Invest. 92 : 967–977.
Goddard S., Youster J., Morgan E., Adams D.H. 2004. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Amer. J. Pathol. 164 : 511–519.
Golden-Mason L., Curry M.P., Nolan N., Traynor O., McEntee G., Kelly J. 2000. Differential expression of lymphoid and myeloid markers on differentiating hematopoietic stem cells in normal and tumor-bearing adult human liver. Hepatology. 31 : 1251–1256.
Golden-Mason L., Kelly A.M., Doherty D.G., Traynor O., McEntee G., Kelly J. 2004. Hepatic interleuklin 15 (IL-15) expression: implications for local NK/NKT cell homeostasis and development. Clin. Exp. Immunol. 138 : 94–101.
Gregory S.H., Wing E.J. 2002. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J. Leukoc. Biol. 72 : 239–248.
Heymann F., Peusquens J., Ludwig-Portugall I., Kohlhepp M., Ergen C., Niemietz P. 2015. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 62 : 279–291.
Hou C.T., Chen Y.L., Lin C.C., Chou C.T., Lin K.H., Lin P.Y., Hsu Y.L., Chen C.B., Lin H.C., Ko C.J., Wang S.H., Weng L.C., Hsieh C.E. 2018. Portal venous velocity affects liver regeneration after right lobe living donor hepatectomy. PLoS One. 13 : e0204163.
Hunter S., Willcox C.R., Davey M.S., Kasatskaya S.A., Jeffery H.C., Chudakov D.M., Oo Y.H., Willcox B.E. 2018. Human liver infiltrating γδ-T-cells are composed of clonally expanded circulating and tissue-resident populations. J. Hepatol. 69 : 654–665.
Ichikawa S., Mucida D., Tyznik A.J., Kronenberg M., Cheroutre H. 2011. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 186 : 5549–5555.
Inatsu A., Kinoshita M., Nakashima H., Shimizu J., Saitoh D., Tamai S. 2009. Novel mechanism of C-reactive protein for enhancing mouse liver innate immunity. Hepatology. 49: 2044–2054.
Ivashkiv L.B., Donlin L.T. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol. 14 : 36–49.
Jenne C.N., Kubes P. 2013. Immune surveillance by the liver. Nat. Immunol. 14 : 996–1006.
Jiang X., Chen Y., Wei H., Sun R., Tian Z. 2013. Characterizing the lymphopoietic kinetics and features of hematopoietic progenitors contained in the adult murine liver in vivo. PLoS One. 8 : e76762.
Karlmark K.R., Weiskirchen R., Zimmermann H.W., Gassler N., Ginhoux F., Weber C. 2009. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 50 : 261–274.
Kedarisetty C.K., Anand L., Bhardwaj A., Bhadoria A.S., Kumar G., Vyas A.K. 2015. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology. 148 : 1362–1370.e7.
Kelly A.M., Golden-Mason L., Traynor O., Geoghegan J., McEntee G., Hegarty J.E. 2006. Changes in hepatic immunoregulatory cytokines in patients with metastatic colorectal carcinoma: implications for hepatic anti-tumour immunity. Cytokine. 35 : 171–179.
Kelly A., Fahey R., Fletcher J.M., Keogh C., Carroll A.G., Siddachari R. 2014. CD141+ myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 60 : 135–142.
Kenna T., Golden-Mason L., Norris S., Hegarty J.E., O’Farrelly C., Doherty D.G. 2004. Distinct subpopulations of gamma delta T-cells are present in normal and tumor-bearing human liver. Clin. Immunol. 113 : 56–63.
Kenna T., Golden-Mason L., Porcelli S.A., Koezuka Y., Hegarty J.E., O’Farrelly C. 2003. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J. Immunol. 171 : 1775–1779.
Kingham T.P., Chaudhry U.I., Plitas G., Katz S.C., Raab J., De Matteo R.P. 2007. Murine liver plasmacytoid dendritic cells become potent immunostimulatory cells after Flt-3 ligand expansion. Hepatology. 45 : 445–454.
Kitamura T., Qian B.-Z., Pollard J.W. 2015. Immune cell promotion of metastasis. Nat. Rev. Immunol. 15 : 73–86.
Kurioka A., Walker L.J., Klenerman P., Willberg C.B. 2016. MAIT cells: new guardians of the liver. Clin. Transl. Immunology. 5 : e98.
Lange C.M., Moreau R. 2018. Immunodysfunction in acute-on-chronic liver failure. Visc. Med. 34 : 276–282.
Leoni G., Neumann P.-A., Sumagin R., Denning T.L., Nusrat A. 2015. Wound repair: role of immune-epithelial interactions. Mucosal Immunol. 8 : 959–968.
Lerut J., Sanchez-Fueyo A. 2006. An appraisal of tolerance in liver transplantation. Amer. J. Transplant. 6 : 1774–1780.
Li S., Hong M., Tan H.Y., Wang N., Feng Y. 2016. Insights into the role and interdependence of oxidative stress and inflammation in liver diseases. Oxid. Med. Cell Longev. 2016 : 4 234 061.
Lindquist J.A., Mertens P.R. 2018. Cold shock proteins: from cellular mechanisms to pathophysiology and disease. Cell Commun. Signal. 16 : 63.
Lynch L., Michelet X., Zhang S., Brennan P.J., Moseman A., Lester C. 2015. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg)-cells and macrophages in adipose tissue. Nat. Immunol. 16 : 85–95.
MacParland S.A., Liu J.C., Ma X.Z., Innes B.T., Bartczak A.M. et al. 2018. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9 : 4383.
Makarova-Rusher O.V., Medina-Echeverz J., Duffy A.G., Greten T.F. 2015. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 62 : 1420–1429.
Malato Y., Naqvi S., Schürmann N., Ng R., Wang B., Zape J. 2011. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest. 121 : 4850–4860.
Markose D., Kirkland P., Ramachandran P., Henderson N.C. 2018. Immune cell regulation of liver regeneration and repair. J. Immunol. Regener. Med. 2 : 1–10.
Michalopoulos G.K. 2007. Liver regeneration. J. Cell Physiol. 213 : 286–300.
Miura K., Yang L., van Rooijen N., Brenner D.A., Ohnishi H., Seki E. 2013. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 57 : 577–589.
Moroso V., Metselaar H.J., Mancham S., Tilanus H.W., Eissens D., van der Meer A. 2010. Liver grafts contain a unique subset of natural killer cells that are transferred into the recipient after liver transplantation. Liver Transpl. 16 : 895–908.
Mosher B., Dean R., Harkema J., Remick D., Palma J., Crockett E. 2001. Inhibition of Kupffer cells reduced CXC chemokine production and liver injury. J. Surg. Res. 99 : 201–210.
Nemeth E., Baird A.W., O’Farrelly C. 2009. Microanatomy of the liver immune system. Semin. Immunopathol. 31 : 333–343.
Norris S., Collins C., Doherty D.G., Smith F., McEntee G., Traynor O. 1998. Resident human hepatic lymphocytes are phenotypically different from circulating lymphocytes. J. Hepatol. 28 : 84–90.
O’Neill L.A.J., Pearce E.J. 2015. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213 : 15–23.
Pallett L.J., Gill U.S., Quaglia A., Sinclair L.V., Jover-Cobos M., Schurich A. 2015. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 21 : 591–600.
Park S.-H., Rehermann B. 2014. Immune responses to HCV and other hepatitis viruses. Immunity. 40 : 13–24.
Pellicoro A., Ramachandran P., Iredale J.P., Fallowfield J.A. 2014. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14 : 181–194.
Protzer U., Maini M.K., Knolle P.A. 2012. Living in the liver: hepatic infections. Nat. Rev. Immunol. 12 : 201–213.
Racanelli V., Sansonno D., Piccoli C., D’Amore F.P., Tucci F.A., Dammacco F. 2001. Molecular characterization of B cell clonal expansions in the liver of chronically hepatitis C virus-infected patients. J. Immunol. 167 : 21–29.
Radaeva S., Sun R., Jaruga B., Nguyen V.T., Tian Z., Gao B. 2006. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 130 : 435–452.
Rai R.M., Lee F.Y., Rosen A., Yang S.Q., Lin H.Z., Koteish A. 1998. Impaired liver regeneration in inducible nitric oxide synthase deficient mice. Proc. Natl. Acad. Sci. USA. 95 : 13 829–13 834.
Robinson M.W., Harmon C., O’Farrelly C. 2016. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 13 : 267–276.
Saeed S., Quintin J., Kerstens H.H.D., Rao N.A., Aghajanirefah A., Matarese F. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 345 : 1251086–1251086.
Sander L.E., Sackett S.D., Dierssen U., Beraza N., Linke R.P., Müller M. 2010. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207 : 1453–1464.
Sá-Pereira I., Roodselaar J., Couch Y., Consentino Kronka Sosthenes M., Evans M.C., Anthony D.C., Stolp H.B. 2018. Hepatic acute phase response protects the brain from focal inflammation during postnatal window of susceptibility. Brain Behav. Immun. 69 : 486–498.
Seki E., Brenner D. 2008. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 48 : 322–335.
Seki E., Schwabe R.F. 2015. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 61 : 1066–1079.
Selzner N., Selzner M., Odermatt B., Tian Y., Van Rooijen N., Clavien P.-A. 2003. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology. 124 : 692–700.
Shen K., Zheng S.-S., Park O., Wang H., Sun Z., Gao B. 2008. Activation of innate immunity (NK/IFN-gamma) in rat allogeneic liver transplantation: contribution to liver injury and suppression of hepatocyte proliferation. Amer. J. Physiol. Gastrointest. Liver Physiol. 294 : G1070–G1077.
Simpson N., Cho Y.W., Cicciarelli J.C., Selby R.R., Fong T.-L. 2006. Comparison of renal allograft outcomes in combined liver-kidney transplantation versus subsequent kidney transplantation in liver transplant recipients: analysis of UNOS database. Transplantation. 82 : 1298–1303.
Strey C.W., Markiewski M., Mastellos D., Tudoran R., Spruce L.A., Greenbaum L.E. 2003. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J. Exp. Med. 198 : 913–923.
Su G.L., Klein R.D., Aminlari A., Zhang H.Y., Steinstraesser L., Alarcon W.H. 2000. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 31 : 932–936.
Sun R., Gao B. 2004. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma). Gastroenterology. 127 : 1525–1539.
Tacke F., Luedde T., Trautwein C. 2009. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 36 : 4–12.
Takeuchi O., Akira S. 2010. Pattern recognition receptors and inflammation. Cell. 140 : 805–820.
Tall A.R., Yvan-Charvet L. 2015. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15 : 104–116.
Tannahill G.M., Curtis A.M., Adamik J., Palsson-McDermott E.M., McGettrick A.F., Goel G. 2013. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 496 : 238–242.
Thomson A.W., Knolle P.A. 2010. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 10 : 753–766.
Thouas G.A., Dominguez F., Green M.P., Vilella F., Simon C., Gardner D.K. 2015. Soluble ligands and their receptors in human embryo development and implantation. Endocrin. Rev. 36 : 92–130.
Tilg H., Moschen A.R. 2010. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 52 : 1836–1846.
Tokita D., Sumpter T.L., Raimondi G., Zahorchak A.F., Wang Z., Nakao A. 2008. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J. Hepatol. 49 : 1008–1018.
Tu Z., Bozorgzadeh A., Crispe I.N., Orloff M.S. 2007. The activation state of human intrahepatic lymphocytes. Clin. Exp. Immunol. 149 : 186–193.
Tu Z., Bozorgzadeh A., Pierce R.H., Kurtis J., Crispe I.N., Orloff M.S. 2008. TLR-dependent cross talk between human Kupffer cells and NK cells. J. Exp. Med. 205 : 233–244.
van Egmond M., van Garderen E., van Spriel A.B., Damen C.A., van Amersfoort E.S., van Zandbergen G., van Hattum J., Kuiper J., van de Winkel J.G. 2000. FcalphaRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat. Med. 6 : 680–685.
Wang J., Knaut H. 2014. Chemokine signaling in development and disease. Development. 141 : 4199–4205.
Wieland A., Frank D.N., Harnke B., Bambha K. 2015. Systematic review: microbial dysbiosis and nonalcoholic fatty liver disease. Aliment Pharmacol. Ther. 42 : 1051–1063.
Wilson G.K., Tennant D.A., McKeating J.A. 2014. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J. Hepatol. 61 : 1397–1406.
Wu J., Meng Z., Jiang M., Zhang E., Trippler M., Broering R. 2010. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 129 : 363–374.
Wuensch S.A., Spahn J., Crispe I.N. 2010. Direct, help-independent priming of CD8+ T-cells by adeno-associated virus-transduced hepatocytes. Hepatology. 52 : 1068–1077.
Yanagisawa K., Yue S., van der Vliet H.J., Wang R., Alatrakchi N., Golden-Mason L. 2013. Ex vivo analysis of resident hepatic pro-inflammatory CD1d-reactive T-cells and hepatocyte surface CD1d expression in hepatitis C. J. Viral Hepat. 20 : 556–565.
Yang L., Jhaveri R., Huang J., Qi Y., Diehl A.M. 2007. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab. Invest. 87 : 927–937.
Yu J., Hu Y., Gao Y., Li Q., Zeng Z., Li Y., Chen H. 2018. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell Death Discov. 5 : 34.
Zhou Z., Xu M.-J., Gao B. 2016. Hepatocytes: a key cell type for innate immunity. Cell. Mol. Immunol. 13 : 301–315.
Zimmermann H.W., Bruns T., Weston C.J., Curbishley S.M., Liaskou E., Li K.‑K. 2015. Bidirectional transendothelial migration of monocytes across hepatic sinusoidal endothelium shapes monocyte differentiation and regulates the balance between immunity and tolerance in liver. Hepatology. 63 : 233–246.
Дополнительные материалы отсутствуют.