

Цитология, 2019, T. 61, № 3, стр. 208-217
Регуляция функций белка р53 в ответ на тепловой стресс
С. Е. Парфеньев 1, А. Н. Смотрова 1, М. А. Шкляева 1, Н. А. Барлев 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: nick.a.barlev@gmail.com
Поступила в редакцию 06.10.2018
После доработки 10.12.2018
Принята к публикации 11.12.2018
Аннотация
В процессе супрессии опухолей человека одну из важнейших функций выполняет белок p53, кодируемый геном TP53. Мутации в ДНК-связывающем домене р53 приводят к изменению его конформации, способствуя образованию аберрантных внутриклеточных белковых комплексов, включающих в себя белки теплового шока (Hsp70). Это может провоцировать возникновение агрессивных типов опухолей, включая рак молочной железы. В связи с этим, изучение механизмов регуляции мутантных форм p53 в составе таких стабильных комплексов представляется крайне актуальным. Целью настоящей работы являлось изучение регуляции мутантного по R175H белка р53 (mutр53-R175H) при тепловом стрессе в клетках MDA-MB-231 рака молочной железы in vitro. Было установлено, что тепловой стресс вызывает резкое снижение уровня белков wtp53 (p53 дикого типа) и mutp53-R175H, который постепенно восстанавливается после снятия стресса. Также мы обнаружили, что mutp53-R175H повышает внутриклеточный уровень Hsp70 в норме и снижает его уровень после теплового стресса. При этом сам белок mutp53-R175H меняет свою внутриклеточную локализацию, как в нормальных условиях, так и в ответ на тепловой шок, находясь в составе Hsp70-содержащих белковых комплексов. Таким образом, поведение wtр53 и mutp53-R175H в ответ на тепловой шок отличаются друг от друга, по-видимому, за счет различного взаимодействия с белковыми комплексами, которые регулируют их стабильность и внутриклеточную локализацию.
Белок p53 является транскрипционным фактором, который способен активировать или подавлять работу большого числа генов, ответственных за остановку клеточного цикла, старение и апоптоз (Reinhardt, Schumacher, 2012). Помимо кодирующих генов, р53 также регулирует транскрипцию генов, не кодирующих РНК, которые в свою очередь контролируют репрессию сотен других генов (Barlev et al., 2010). При раковых заболеваниях человека очень часто выявляются мутации генa TP53, отвечающего за синтез белка р53 (Petitjean et al., 2007).
Известно, что более 80% мутаций TP53, встречающихся в опухолях человека, локализовано в ДНК-связывающем домене (Olivier et al., 2010). Мутации в ДНК-связывающем домене р53, так называемые мутации приобретения функции (gain-of-function mutation; GOF), вызывают локальную денатурацию его трехмерной структуры таким образом, что меняется спектр белков-партнеров белка р53 и сила взаимодействия с ними, приводя к усилению ракового потенциала клеток. Ярким примером является мутация R175H (замена аргинина на гистидин в 175 положении) – одна из наиболее часто встречающихся мутаций при карциноме молочной железы (Olivier et al., 2006). Было показано, что мутантный по R175H белок р53 (mutp53-R175H) способен образовывать стабильные комплексы с шаперонными белками теплового шока (БТШ) Hsp40, Hsp70, Hop и Hsp90 (King et al., 2001; Zylicz et al., 2001). Важно отметить, что повышение уровня экспрессии БТШ наблюдается во многих типах опухолей (Calderwood et al., 2006). Например, при раке груди повышение уровня Hsp70 коррелирует с неблагоприятным прогнозом для больного (Steiner et al., 2006). Поэтому изучение характера взаимодействия mutр53-R175H с БТШ является важной задачей, решение которой позволит глубже понять механизмы образования раковых клеток при мутациях белка р53 в ДНК-связывающем домене, в целом, и при мутации R175H, в частности, а также разработать новые подходы для борьбы с онкологическими заболеваниями, обусловленными данной мутацией.
В норме р53 имеет короткий период полураспада благодаря существованию ряда убиквитин-лигаз (Pirh2, COP-1, WWP1 и др.), которые присоединяют молекулы убиквитина к белку р53, что приводит к его дальнейшему узнаванию 19S-субъединичным комплексом и разрушению в 20S-субъединичном комплексе протеасом (Momand et al., 1992; Mittenberg et al., 2008; Дакс и др., 2013). Ключевую роль в данном процессе играет E3-убиквитин-лигаза Mdm2 (Marouco et al., 2013). Важно отметить, что p53 является транскрипционным активатором для гена MDM2, таким образом, возникает негативная регуляторная обратная петля взаимодействия p53 и Mdm2 (Haupt et al., 1997), характер которого может меняться вследствие ряда пост-трансляционных модификаций р53. При клеточном стрессе происходит стабилизация p53 за счет его фосфорилирования протеинкиназами по остаткам серина в положениях 15 и 20, нарушающего взаимодействие р53 с Mdm2 (Meek, Anderson, 2009), вследствие чего р53 накапливается в клетке, где выполняет координацию множества сигнальных путей в ответ на повреждения ДНК (Vogelstein et al., 2000).
Основываясь на фармакофорной гипотезе взаимодействия р53 с Mdm2, происходящего с участием гидрофобных аминокислот Phe19, Trp23 и Leu26, был разработан ряд ингибиторов межбелкового взаимодействия между р53 и Mdm2 (Davidovich et al., 2015; Fedorova et al., 2018). В их число входит нутлин (Vassilev et al., 2004), производные которого планируется использовать для борьбы с различными формами онкологии, связанными со сверхэкспрессией Mdm2, и в настоящее время проходят клинические испытания (Fedorova et al., 2018).
В более ранних исследованиях было показано, что инактивация Mdm2 происходит в результате его захвата в тримерный комплекс Mdm2-Hsp90-mutp53 (mutp53 – мутантный р53), при этом связывание с Hsp90 ингибирует Е3-убиквитин-лигазную функцию Mdm2 (Peng et al., 2001). Вероятно, Hsp90 действует аналогично белку Arf, который за счет связывания с Mdm2 подавляет его убиквитин-лигазную активность (Pomerantz, et al., 1998). В раковых клетках mutp53 вступает в устойчивые взаимодействия с Hsp90, который эффективно ингибирует не только Mdm2, но и другую убиквитин-лигазу, CHIP, что приводит к аберрантной стабилизации mutp53 (Li et al., 2011). В норме, шаперон-зависимая E3-лигаза CHIP связывается с белком Hsp70, являющимся резидентной частью комплекса Hsp90 и обычно способствует деградации своих белков-клиентов, однако убиквитинирующая способность CHIP может быть нарушена в опухолевых клетках (Li et al., 2011). В экспериментах с нокдауном Hsf1 (транскрипционного фактора, отвечающего за экспрессию многих БТШ) в клетках, экспрессирующих mutp53, было продемонстрировано, что в результате резкого снижения уровней Hsp70 и Hsp90 в клетке происходит быстрая дестабилизация mutp53 и уменьшение периода его жизни (Li et al., 2011). Это еще раз подтверждает предположение об участии данных шаперонов в процессе стабилизации mutp53.
В настоящей работе осуществляется исследование динамики взаимодействия белка mutр53-R175H с Hsp70, который активизируется при тепловом шоке, а также внутриклеточной локализации mutр53-R175H.
МАТЕРИАЛ И МЕТОДИКА
В работе использовали клеточную линию рака молочной железы человека MDA-MB-231, характеризующуюся мутацией mutр53-R175H, на основе которой были получены три линии (MDA-MB-231-mutp53-R175H-GFP, MDA-MB-231-wtp53-GFP, MDA-MB-231-GFP) путем трансфицирования лентивирусными векторами, кодирующими mutp53-R175H-GFP, wtp53-GFP и GFP. Линия MDA-MB-231-GFP служила контрольной. Также были задействованы клетки рака молочной железы человека MCF7 с повышенной экспрессией Hsp70. MDA-MB-231 и MCF7 получены из коллекции биоматериалов ATCC (American Type Culture Collection, США). В работе использовали бактериальные клетки E. coli (штамм BL21), которые подвергли трансформации экспрессионными плазмидами. В результате были созданы бактериальные клетки с оверэкспрессией GST, wtp53-GST, mutp53-GST; причем бактерии E. coli, экспрессирующие GST, применяли в качестве контроля. Исходные клетки E. coli были приобретены в компании ЗАО Евроген (Россия).
Для культивирования клеток эукариот использовали среду DMEM (Gibco, США), содержащую 10% фетальной бычьей сыворотки (FBS) (HyClone, США), смесь антибиотиков пенициллина и стрептомицина (Биолот, Россия) и L-глутамин (Биолот, Россия). Кроме того, в работе использовали фосфатно-солевой буфер (PBS, конечная концентрация 10 мкг/мл), содержащий 10 мМ Na2HPO4, 1.76 мМ KH2PO4, 137 мМ NaCl и 2.7 мМ KCl, pH 7.4 и 0.25%-ный раствор трипсина (Gibco, США) в PBS.
Трансдукция. Стабильные клеточные линии MDA-MB-231-mutp53-R175H-GFP, MDA-MB-231-wtp53-GFP, MDA-MB-231-GFP получали с использованием нескольких циклов трансдукции лентивирусными частицами. Клетки HEK293-T, полученные из коллекции биоматериалов ATCC (США), трансфицировали плазмидной смесью (15 мкг экспрессирующего вектора, несущего одну из вставок: mutp53-R175H-GFP, wtp53-GFP или GFP, 9.35 мкг psPAX2 и 5.32 мкг pMD2.G) с применением Turbofect (ThermoFischer, США) в соответствии с рекомендациями изготовителя. Среду меняли через 16 ч после трансфекции, а затем лентивирусные частицы собирали каждые 24 ч. Собранные частицы концентрировали путем ультрацентрифугирования в градиенте сахарозы в течение 2 ч при 72 000 g. Для заражения вирусными частицами высевали по 50 000 клеток на лунку 24-луночного планшета, используя среду RPMI 1640 (Gibco, Великобритания). Спустя 4 ч в лунки добавляли лентивирусные частицы и полибрен до конечной концентрации 8 мкг/мл. Клетки с вирусами инкубировали в течение 24 ч. Через 4 сут долю трансформированных клеток оценивали с помощью проточного цитометра Guava EasyCyte 8 (EMD Millipore, MA, США), после чего производили сортировку трансдуцированных клеток на основе флуоресценции GFP с использованием прибора BD FACSAria III (BD, США).
Иммуноблотинг. Клетки лизировали по стандартному протоколу (Laemmli, 1970). Для этого к охлажденным клеткам, находящимся в чашках и предварительно промытых PBS, добавляли 5-кратный буфер Laemmli и инкубировали 1 мин при комнатной температуре. Далее с чашек собирали лизат и выдерживали 5 мин при 96°C. Разделение белков проводили в 13%-ном полиакриламидном геле в денатурирующих условиях. Затем переносили на PVDF-мембрану, которую инкубировали с соответствующими антителами. Для выявления белков использовали первичные антитела (в конечной концентрации в растворе 1 мкг/мл) к белкам: β-актину (Sigma Aldrich, США), p53 (Millipore, Германия), Hsp70 (Santa Cruz, США) и Mdm2 (Millipore, Германия), а также вторичные антитела (Thermo Scientific, США) в конечной концентрации ~0.2 мкг/мл. Для детекции и визуализации результатов электрофореза, гели окрашивали раствором InstantBlue Protein Stain (Expedeon, США) в течение 1 ч. Получение изображений ПААГ и мембран, окрашенных антителами, проводили при помощи системы для визуализации изображений ChemiDoc Touch Imaging System (Bio-Rad, США).
Тепловой шок и подготовка препаратов к флуоресцентной микроскопии. Клетки на покровных стеклах культивировали до 70%-ной конфлюэнтности и выдерживали в сухом СО2-инкубаторе при температуре 43°C в течение 40 мин. Далее часть стекол с клетками в чашках Петри дважды промывали PBS объемом 1 мл и фиксировали 5%-ным формалином в течение 20 мин, промывали три раза по 1 мл PBS в течение 5 мин. Другую часть клеток на стеклах инкубировали при 37°C и подвергали фиксации спустя еще 1 или 4 ч. После фиксации удаляли PBS, добавляли раствор детергента тритона-Х100. Фиксированные клетки окрашивали с помощью 500 мкл раствора DAPI в PBS в течение 3 мин и промывали PBS дважды по 5 мин. Затем покровные стекла с клетками переворачивали “вниз лицом” и переносили на предметные, на которые были заранее нанесены капля глицерина. Визуализацию проводили с использованием флуоресцентного микроскопа LSM 5PASCAL (Zeiss, Германия) на длинах волн 470 и 365 нм для визуализации флуоресценции белка GFP и красителя DAPI.
Трансформация бактериальных клеток экспрессионными плазмидными векторами, индукция экспрессии генов и очистка рекомбинантного белка. Пробирки с клетками E. coli размораживали на льду и добавляли по 10 нг экспрессионной плазмиды pGEX-5X-1, содержащей GST-таг, или слитый с wtр53 GST-таг, или слитый с mutр53 GST-таг. Инкубировали на льду 15 мин. Затем подвергали тепловому шоку в течение 1 мин при температуре 42°C. Далее пробирки с клетками помещали в лед на 5 мин. Затем добавляли по 1 мл среды LB (Amresco, США), инкубировали 1 ч при 37°C и постоянном перемешивании. После этого пробирки с клетками бактерий центрифугировали при 4000 g и 4°C в течение 30 с. Осадок, содержащий бактериальные клетки, ресуспендировали, наносили на чашки Петри с LB-агаром с ампициллином (1 : 1000). Чашки Петри помещали на 1 сут в термостат при температуре 37°C. Прошедшие на чашках селекцию бактерии перемещали в пробирки с жидкой средой LB с ампициллином (1 : 1000) и наращивали в шейкере в течение 12 ч при 37°C. Далее культуру разводили средой LB (1 : 100) и оставляли при 37°C, наращивали клетки до значений оптической плотности OD600 = 0.7–0.8. Добавляли ИПТГ (изопропил-β-D-1-тиогалактопиранозид) до финальной концентрации 0.4 мМ/л и проводили индукцию экспрессии таргетных генов в течение 10 ч при температуре 16°C (понижение температуры помогает увеличить стабильность белка р53) до OD600 = 1.5–2.0. Далее суспензию бактерий центрифугировали при 3000 g в течение 5 мин при 4°C, затем ресуспендировали в среде LB и добавляли лизирующий буфер (1 : 50). Пробы соницировали на льду 4 раза по 15 с с перерывами по 30 с. Затем образцы центрифугировали 15 мин при 3000 g при 4°C и отбирали супернатант.
GST-pull-down-анализ. К пробам, содержащим очищенный рекомбинантный белок бактерий, добавляли по 300 мкл суспензии, состоящей из гранул глутатион-сефарозы (100 мкл гранул на пробу) и PBS, включающий в себя 0.6% детергента NP40 (Нонидет P-40). Пробы перемешивали в течение 1 ч при 4°C и центрифугировали 1 мин при 1000 g и комнатной температуре. Осадки трижды промывали PBS, содержащим 1 мМ PMSF и 0.6% NP40, и центрифугировали при тех же условиях. Далее осадки снова промывали в PBS с PMSF и добавляли по 1 мл PBS с PMSF, тщательно ресуспендировали. Затем отбирали по 200 мкл и центрифугировали в течение 10 с при 1000 g. Осадки промывали в 1 мл буфера С следующего состава (мМ): 20 TrisCl, 420NaCl, 1.5 MgCl2, 0.2 EDTA, 1 PMSF и 25% глицерина от общего объема; pH буфера доводили до 8.0 с помощью NaOH и добавляли в осадки по 620 мкл экстракта MCF7, предварительно отцентрифугировав их при 1000 g при комнатной температуре в течение 10 с. Далее получившиеся суспензии инкубировали при постоянном перемешивании в течение 1.5 ч при 4°C и центрифугировали 10 с при 1000 g, отбирали супернатант с несвязавшимися белками. Осадок с гранулами и провзаимодействовашими с ними белками трижды промывали буфером С, центрифугируя при 1000 g в течение 10 с при комнатной температуре. После этого гранулы, связавшиеся с белками ресуспендировали в 20 мкл PBS.
Статистическую обработку результатов проводили с использованием программ ImageJ 1.50i и GraphPad Prism 6.01. Программу ImageJ применяли для определения яркости свечения белка GFP в ядерной области клеток после проведения теплового шока. Полученные значения анализировали в GraphPad Prism путем сравнения групп данных по U-критерию Манна–Уитни при уровне значимости 0.05.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Р53 – зависимое повышение уровня Hsp70. В работе использовали клеточную линию рака молочной железы человека MDA-MB-231, характеризующуюся наработкой mutp53-R175H в результате мутации гена ТР53, присутствующую одновременно в обеих аллелях. Эта линия была стабильно трансфицирована лентивирусными векторами, в результате чего были получены 3 вариации линии: MDA-MB-231-mutp53-R175H-GFP, MDA-MB-231-wtp53-GFP и MDA-MB-231-GFP. После проведения иммуноблотинга с использованием антител, специфичных к р53 и Hsp70, нами установлено, что в клеточных линиях, содержащих экзогенный р53 (особенно mutp53-R175H), увеличено содержание белка Hsp70 (рис. 1). Повышение уровня Hsp70 при усилении экспрессии эктопического TP53 может свидетельствовать о существовании механизма регуляции транскрипционной активности гена HSPA1 за счет p53. Другим объяснением этого феномена может служить их взаимная стабилизация на белковом уровне.
Рис. 1.
Сравнение уровней белков р53 и Hsp70 в клетках линий MDA-MB-231-GFP (дорожка 1), MDA-MB-231-wtp53-GFP (2), MDA-MB-231-mutp53-R175H-GFP (3), полученных путем трансфицирования лентивирусными векторами, кодирующими mutp53-R175H-GFP, wtp53-GFP и GFP, из линии рака молочной железы человека MDA-MB-231 с мутацией R175H в гене р53 (mutр53-R175H). p53-GFP включает в себя р53 дикого типа, сшитый GFP (wtp53-GFP) и mutр53-R175H, сшитый с GFP (mutр53-R175H-GFP). Данные получены с помощью Вестерн-блот-анализа.

Анализ динамики локализации mutp53-R175H и wtp53, сшитых с зеленым флуоресцентным белком (GFP). Известно, что активация экспрессии HSPA1 и функций Hsp70 происходит при тепловом стрессе. В связи с этим, опухолевые клетки линий MDA-MB-231-GFP, MDA-MB-231-wtp53-GFP и MDA-MB-231-mutp53-R175H-GFP подвергали воздействию теплового шока.
Белок wtp53 до теплового шока распределялся в клетке диффузно (рис. 2а, A), а сразу после стресса концентрировался в ядре (рис. 2а, Д). Спустя 4 ч он снова равномерно распределялся во внутриклеточном пространстве (рис. 2а, З). Белок mutp53-R175H вел себя противоположным образом: до теплового шока он в основном выявлялся в ядре (рис. 2а, В), после теплового шока его количество в ядре и цитоплазме выравнивалось (рис. 2а, Е), а спустя 4 ч снова наблюдали его повышенное содержание в ядре (рис. 2а, И). Статистический расчет яркости свечения GFP в составе wр53-GFP и mutp53-R175H-GFP в анализируемых трех клеточных линиях, полученных в результате трансфекции различными векторами (рис. 2б) показывает статистически достоверное повышение уровня wр53-GFP в клетках линии MDA-MB-231-wtp53-GFP и понижение уровня mutp53-R175H-GFP в клетках линии MDA-MB-231-mutp53-R175H-GFP после теплового шока (Р < 0.0001). Через 4 ч после теплового шока происходит неполное восстановление уровней wtp53 (P = 0.0145) и mutp53-R175H (P = 0.0035). Это вероятно связано с тем, что для восстановления прежних значений уровней р53 клеткам требуется больше времени.
Рис. 2.
Иммуноцитохимическая окраска клеток линий MDA-MB-231-GFP, MDA-MB-231-wtp53-GFP, MDA-MB-231-mutp53-R175H-GFP, не подвергавшихся тепловому шоку (К – контроль), сразу после теплового шока (ТШ) и через 4 ч после него (а), а также интенсивность флюоресценции (ИФ) белка GFP ядерной области этих же клеток в тех же условиях (б). б: данные представлены в виде медиан и квартилей; значение Р < 0.001 показано четырьмя звездочками.
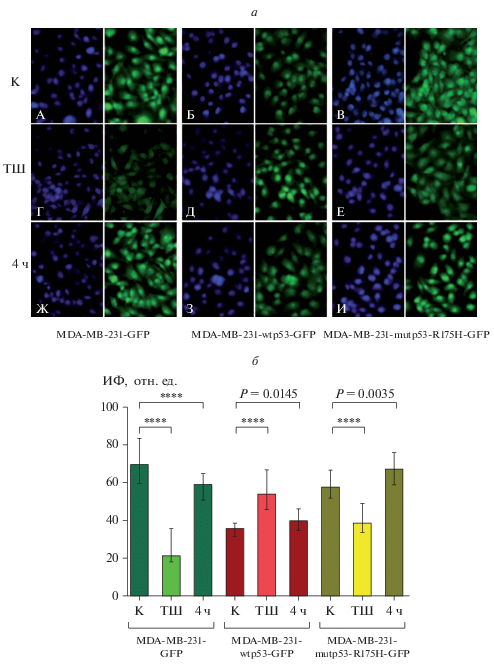
Рис. 3.
Сравнение уровней белков mutр53-R175H, р53-GFP (wtр53-GFP, mutр53-R175H-GFP) и Hsp70 в клетках линий MDA-MB-231-GFP (дорожки 1), MDA-MB-231-wtp53-GFP (2), MDA-MB-231-mutp53-R175H-GFP (3), не подвергавшихся тепловому шоку (К – контроль), сразу после теплового шока (ТШ), через 1 ч (верхняя панель) и через 4 ч (нижняя панель) после ТШ. Данные получены с помощью Вестерн-блот-анализа.

В норме, белок Mdm2 связывается с p53 и обеспечивает присоединение убиквитина к его С-концу и способствует деградации p53, в результате чего происходит снижение концентрации p53, и, как следствие, уменьшение транскрипционной активности гена MDM2. Снижение уровня белка Mdm2 в клетке приводит к восстановлению содержания р53. Таким образом, осуществляется взаимная регуляция p53 и Mdm2 (Vogelstein et al., 2000). Этим объясняется снижение уровня р53 через 4 ч после теплового шока (рис. 2а, З), в то время, когда, по-видимому, осуществляется усиление работы гена MDM2, вызванное увеличением уровня wtp53.
Повышение концентрации mutp53-R175H в ядре в нормальных условиях, вероятно, связано с его усиленным импортом в ядро, обусловленным повышением сродства к белкам-переносчикам, вследствие нарушения третичной структуры mutp53-R175H. Скорее всего, в данном процессе принимает участие Hsp70, поскольку известно, что Hsp70 способен обеспечивать транспорт p53 из цитоплазмы в ядро (Narayanan et al., 2006). Согласно литературным данным по изучению внутриклеточной локализации p53, в отсутствие Mdm2 и Hsp70, белок mutp53 распределяется в клетке диффузно, а при повышенном уровне Hsp70 белок mutp53 распределяется преимущественно в цитоплазме; при этом сверх-экспрессия MDM2 приводит к тому, что большая часть mutp53 перемещается в ядро (Wiech et al., 2012).
Отсутствие деградации mutp53-R175H внутри ядра может быть следствием того, что Hsp70 защищает mutp53-R175H от воздействия Mdm2. Это косвенно подтверждается тем, что инактивация Mdm2 происходит в результате образования тримерного комплекса Mdm2-Hsp90-mutp53, при этом связывание с Hsp90 ингибирует Е3-убиквитин-лигазные функции Mdm2 (Peng et al., 2001). Комплекс Hsp90 – сложный многокомпонентный шаперонный аппарат клетки, включающий Hsp70; и есть все основания предполагать, что Hsp70 в составе комплекса Mdm2-Hsp70-mutp53 играет аналогичную роль, что и в комплексе Mdm2-Hsp90-mutp53 относительно взаимодействия р53 и Mdm2.
При изучении действия ингибиторов Hsp90 на регуляцию стабильности комплексов с mutp53 было показано, что ингибирование шаперонных функций Hsp90 таким агентом, как 17AAG (17-N-аллиламино-17-деметоксигельданамицин) приводит к быстрой деградации mutp53 в результате его высвобождения из комплексов, включающих в себя Hsp90, и реактивации убиквитин-лигаз, входящих в состав таких комплексов (Li et al., 2011). По-видимому, в этом процессе участвуют, как Mdm2, так и убиквитин-лигаза, CHIP (carboxy-terminus of Hsc70 interacting protein). Доказательством этому служит тот факт, что нокдаун Mdm2 или CHIP вызывает значительное снижение активности процесса деградации mutp53, а нокдаун по обеим убиквитин-лигазам практически прекращает этот процесс (Li et al., 2011).
Ранее в экспериментах с эктопической экспрессией CHIP было показано возможное участие CHIP, как альтернативной E3-лигазы, в процессах убиквитинирования и деградации mutp53 (Lukashchuk, Vousden, 2007; Muller et al., 2008). Кроме того, относительно недавно было установлено, что триоксид мышьяка, используемый при лечении лейкемии, может взаимодействовать с ингибиторами Hsp90 и способствовать запуску деградации mutp53 E3-убиквитин-лигазой Pirh2 (Yan et al., 2014). Таким образом, согласно данным из литературы, в настоящее время можно говорить о возможном участии уже трех убиквитин-лигаз: Mdm2, CHIP и Pirh2 в образовании комплексов с белками теплового шока и mutp53, а также в быстрой деградации mutp53 при ингибировании Hsp90. По-видимому, в дальнейшем этот список может расширяться. Дальнейшие исследования должны определить правомочность наших предположений.
Выход mutp53-R175H из ядра, в отличие от wtp53, сразу после воздействия тепловым шоком (рис. 2a, Д, Е) можно объяснить тем, что в ядре активизируется де-убиквитиназа HAUSP (Brooks et al., 2007) и замещает собой Mdm2 в комплексе Mdm2-Hsp70-mutp53-R175H. При этом, Mdm2, высвобождаясь из комплекса, осуществляет автоубиквитинирование и деградирует в протеосомах (Dai, Gu, 2010). Можно предположить, что в отсутствие Mdm2 комплекс Hsp70-mutp53-R175H (HAUSP-Hsp70-mutp53-R175H) по каким-то причинам покидает ядро. Поэтому при замещении Mdm2 на HAUSP вновь образовавшийся комплекс HAUSP-Hsp70-mutp53-R175H не является стабильным и теряет HAUSP, что приводит к выходу Hsp70-R175H из ядра. В качестве альтернативного объяснения можно выдвинуть предположение о том, что mutp53-R175H накапливается в ядре из-за пост-трансляционных модификаций Mdm2 в результате стресса. Ковалентные модификации ослабляют взаимодействие между Mdm2 и mutp53-R175H, и, как следствие, mutp53-R175H остается в ядре. Это предположение подтверждается тем фактом, что фосфорилированный в условиях стресса с помощью АТМ киназы белок Mdm2 хуже справляется с экспортом р53 из ядра (Maya et al., 2001).
Изменение уровней р53 и Hsp70 после теплого стресса. На следующем этапе исследования мы изучили динамику изменения уровней р53 и Hsp70 в клетках с помощью вестерн-блот-анализа белков проб, полученных из клеток тех же линий (MDA-MB-231-GFP, MDA-MB-231-wtp53-GFP и MDA-MB-231-mutp53-R175H-GFP), не подвергавшихся тепловому шоку и на разных сроках после теплового шока (сразу, через 1 и 4 ч).
Сразу после теплового шока было отмечено резкое повышение содержания Hsp70 в клеточных линиях MDA-MB-231-GFP и MDA-MB-231-wtp53-GFP, которое оставалось на том же уровне как минимум в течение 4-х ч, что отражает нормальную реакцию клетки в ответ на тепловой стресс (рис. 3). В отличие от вышеперечисленных клеток, в клетках MDA-MB-231-mutp53-R175H-GFP через 1 ч после теплового шока происходило заметное снижение уровня Hsp70 и его неполное восстановление спустя 4 ч. Подобное изменение уровня Hsp70 может являться следствием сложных процессов, затрагивающих различные межбелковые взаимодействия между mutp53-R175H с другими белками. Мы предполагаем, что в данных условиях может происходить активация убиквитин-лигаз, входящих в состав комплексов с mutp53-R175H и обусловленная этим протеасомная деградация Hsp70. В частности, для Hsp-ассоциированной Е3-убиквитин-лигазы CHIP показана такая способность к Hsp70-независимой активации под действием теплового стресса (Younger et al., 2004).
Сразу после теплового шока количество mutp53-R175H, а также mutp53-R175H-GFP и wtp53-GFP в клетках снижалось, затем постепенно восстанавливалось почти до прежнего уровня. Объяснение этому факту можно найти в работе, в которой на клетках рака печени человека было показано, что в условиях стресса высокоспецифичный шаперон gp96, являющийся гомологом Hsp90, повышает убиквитин-лигазную активность Mdm2 и уменьшает эффективность протеосомного ингибитора MG132 (Wu et al., 2015). Таким образом, мы предполагаем, что при тепловом стрессе деградация р53, происходит в результате его убиквитинирования и последующей протеосомной деградации, опосредованной воздействием шаперона gp96.
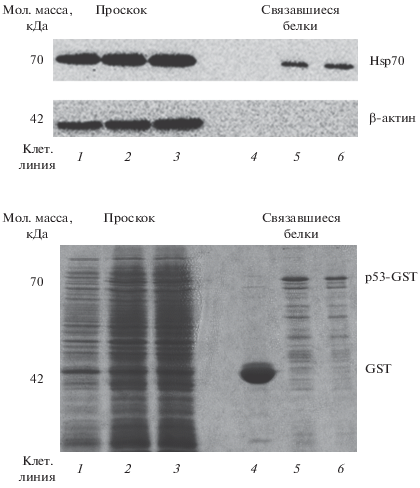
Взаимодействие mutp53-R175H и Hsp70. Для оценки специфичности этого взаимодействия мы провели GST-pull-down-анализ. Экспрессированные в E. сoli рекомбинантные белки GST, сшитые с wtp53, или с mutp53-R175H, очищали методом аффинной хроматографии на глутатион-сефарозе (субстрат для фермента GST) и затем смешивали с экстрактом клеток линии MCF7, которые характеризуются повышенной экспрессией HSPA1. Связавшиеся с GST-химерами белки анализировали с помощью вестерн-блотинга. Анализ показал, что Hsp70 связывается с химерными белками wtp53-GST и mutp53-R175H-GST примерно с равной эффективностью (рис. 4) При этом связывание было специфичным, поскольку Hsp70 не связывался с контрольным белком GST. Из этого следует, что мутация R175H в белке p53 не влияет на силу взаимодействия mutp53-R175H с Hsp70. Таким образом, преимущественная локализация mutp53-R175H в ядре при нормальных условиях, как было показано нами в данной работе скорее всего обусловлена не усилением сродства mutp53-R175H к Hsp70, а повышением экспрессии HSPA1 и, как следствие, увеличением вероятности образования комплекса mutp53-Hsp70 и перемещением комплекса в ядро. Возможно, что mutp53-R175H также повышает сродство Mdm2 к комплексу mutp53-Hsp70.
Рис. 4.
Результаты GST-pull-down-анализа для сравнения силы взаимодействия Hsp70 с wtp53 и mutp53-R175H. Вверху: выявление белка Hsp70, наработанного в клетках MCF7, с помощью Вестерн-блот-анализа экстрактов верхней фазы (1–3) и осадка (4–6) клеток E. coli, сверхэкспрессирующих GST (дорожки 1, 4), wtp53-GST (2, 5), mutp53-R175H-GST (3, 6) на втором этапе GST-пулдаун-анализа. Β-аctin – контроль нагрузки. Внизу: электрофореграмма оставшихся в растворе (дорожки 1–3) и связавшихся с гранулами глутатион-сефарозы (4–6) белков после проведения первого этапа GST-пулдаун- анализа. Гель окрашен красителем Instant blue. Белки выделены из экстрактов клеток E. coli с оверэкспрессией GST (дорожки 1, 4), wtp53-GST (2, 5), mutp53-R175H-GST (3, 6).
Суммируя все вышеописанные результаты, полученные в ходе настоящей работы, нам удалось установить, что: mutp53-R175H, в отличие от р53 дикого типа, вызывает повышение уровня Hsp70 скорее всего за счет усиления экспрессии гена HSPA1, но при тепловом шоке наблюдается падение уровня Hsp70 в клетках линии MDA-MB-231-mutp53-R175H-GFP. Также нами было показано, что mutp53-R175H в отличие от wtp53, в нормальных условиях локализуется преимущественно в ядре, а при тепловом стрессе перемещается в цитоплазму, причем после снятия стрессового воздействия постепенно возвращается в ядро. Этот процесс не связан напрямую с повышенным сродством mutp53-R175H к Hsp70, а вероятно происходит из-за взаимодействия с другими белками, принимающими участие в образовании комплексов, включающих в себя p53 и Hsp70. Наконец, нами обнаружено резкое уменьшение уровня белков wtp53 и mutp53-R175H в ответ на тепловой шок, а также постепенное их восстановление после теплового стресса.
В итоге можно сделать вывод о том, что поведение wtр53 и mutp53-R175H в ответ на тепловой шок различается между собой: mutp53-R175H усиливает экспрессию гена HSPA1 в нормальных условиях, но вызывает усиленную деградацию Hsp70 при тепловом шоке, меняя при этом собственную внутриклеточную локализацию. В связи с этим представляется актуальным изучение белкового интерактома mutp53-R175H при тепловом шоке, поскольку интерактанты, регулирующие стабильность и внутриклеточную локализацию mutp53-R175H, будут a priori являться новыми мишенями для таргетной противораковой терапии. Одним из таких направлений может рассматриваться поиск ингибиторов HSPA1 или Hsp70 для предотвращения образования стабилизирующих комплексов типа Mdm2-Hsp70-mutp53-R175H в раковых клетках.
Работа была выполнена при финансовой поддержке Российского научного фонда (проект № 14-50-00068).
Список литературы
Дакс А.А., Мелино Д., Барлев Н.А. 2013. Роль различных Е3-убиквитинлигаз в регуляции активности онкосупрессора р53. Цитология. 55 (10) : 673–687. (Daks A.A., Melino D., Barlev N.A. 2013. The role of different E3 ubiquitin ligases in regulation of the P53 tumor suppressor protein. Tsitologiya. 55 (10) : 673–687).
Barlev N.A., Sayan B.S., Candi E., Okorokov A.L. 2010. The microRNA and p53 families join forces against cancer. Cell Death Differ. 17 : 373–375.
Brooks C.L., Li M., Hu M., Shi Y., Gu W. 2007. The p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene. 26 : 7262–7266.
Calderwood S.K., Khaleque M.A., Sawyer D.B., Ciocca D.R. 2006. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem. Sci. 31 : 164–172.
Dai C., Gu W. 2010. p53 post-translational modification: deregulated in tumorigenesis. Nat. Rev. Cancer. 16 : 528–536.
Davidovich P., Aksenova V., Petrova V., Tentler D., Orlova D., Smirnov S., Gurzhiy V., Okorokov A.L., Garabadzhiu A., Melino G., Barlev N., Tribulovich V. 2015. Discovery of novel isatin-based p53 inducers. Med. Chem. Lett. 6 : 856–860.
Fedorova O., Daks A., Petrova V., Petukhov A., Lezina L., Shuvalov O., Davidovich P., Kriger D., Lomert E., Tentler D., Kartsev V., Uyanik B., Tribulovich V., Demidov O., Melino G. Barlev N.A. 2018. Novel isatin-derived molecules activate p53 via interference with Mdm2 to promote apoptosis. Cell Cycle. 17 : 1917–1930.
Haupt Y., Maya R., Kazaz A., Oren M. 1997. Mdm2 promotes the rapid degradation of p53. Nature. 387 : 296–299.
King F.W., Wawrzynow A., Hohfeld J., Zylicz M. 2001. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 20 : 6297–6305.
Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227 : 680–685.
Li D., Marchenko N.D., Schulz R., Fischer V., Velasco-Hernandez T., Talos F., Moll U.M. 2011. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mo. Cancer Res. 9 : 577–588.
Lukashchuk N., Vousden K.H. 2007. Ubiquitination and degradation of mutant p53. Mol. Cell Biol. 27: 8284–8295.
Marouco D., Garabadgiu A.V., Melino G., Barlev N.A. 2013. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 4 : 1556–1571.
Maya R., Balass M., Kim S.-T., Shkedy D., Leal J.-F.M., Shifman O., Moas M., Buschmann T., Ronai Z., Shiloh Y., Kastan M., Katzir E., Oren M. 2001. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Devel. 15 : 1067–1077.
Meek D.W., Anderson C.W. 2009. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. doi 10.1101/cshperspect.a000950
Mittenberg A.G., Moiseeva T.N., Barlev N.A. 2008. Role of proteasomes in transcription and their regulation by covalent modifications. Front. Biosci. 13 : 7184–7192.
Momand J., Zambetti G.P., Olson D.C., George D., Levine A.J. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 69 : 1237–1245.
Muller P., Hrstka R., Coomber D., Lane D.P., Vojtesek B. 2008. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene. 27 : 3371–3383.
Narayanan. N.K., Narayanan B.A., Bosland M., Condon M.S., Nargi D. 2006. Docosahexaenoic acid in combination with celecoxib modulates HSP70 and p53 proteins in prostate cancer cells. Int. J. Cancer. 119 : 1586–1598.
Olivier M., Hollstein M., Hainaut P. 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. doi 10.1101/cshperspect.a001008
Olivier M., Langer A., Carrieri P., Bergh J., Klaar S., Eyfjord J., Theillet C., Rodriguez C., Lidereau R., Bieche I., Varley J., Bignon Y., Uhrhammer N., Winqvist R., Jukkola-Vuorinen A., Niederacher D., Kato S., Ishioka C., Hainaut P., Borresen-Dale A.-L. 2006. The clinical value of somatic TP53 gene mutations in 1.794 patients with breast cancer. Clin. Cancer Res. 12 : 1157–1167.
Peng Y.H., Chen L.H., Li C.G., Lu W.G., Chen J.D. 2001. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 276 : 40583–40590.
Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M. 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat. 28 : 622–629.
Pomerantz J., Schreiber-Agus N., Liegeois N.J., Silverman A., Alland L., Chin L., Potes J., Chen K., Orlow I., Lee H.W., Cordon-Cardo C., dePinho R.A. 1998. The Ink4a tumor suppressor gene product, p19 Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 92 : 713–723.
Reinhardt H.C., Schumacher B. 2012. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends genet. 28 : 128–136.
Steiner K., Graf M., Hecht K., Reif S., Rossbacher L., Pfister K., Kolb H.-J., Schmetzer H.M., Multhoff G. 2006. High HSP70-membrane expression on leukemic cells from patients with acute myeloid leukemia is associated with a worse prognosis. Leukemia. 20 : 2076–2079.
Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E.A. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 303 : 844–848.
Vogelstein B., Lane D., Levine AJ. 2000. Surfing the p53 network. Nature. 408 : 307–310.
Wiech M., Olszewski M.B., Tracz-Gaszewska Z., Wawrzynow B., Zylicz M., Zylicz A. 2012. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One. doi 10.1371/journal.pone.0051426
Wu B., Chu X., Feng C., Hou J., Fan H., Liu N., Li C., Kong X., Ye X., Meng S. 2015. Heat shock protein gp96 decreases p53 stability by regulating Mdm2 E3 ligase activity in liver cancer. Cancer Letters. 359 : 325–334.
Yan W., Jung Y.S., Zhang Y., Chen X. 2014. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PloS one. doi 10.1371/journal.pone.0103497
Younger J.M., Ren H.Y., Chen L., Fan C.Y., Fields A., Patterson C., Cyr D.M. 2004. A foldable CFTRΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70–CHIP E3 ubiquitin ligase. J. Cell Biol. 167 : 1075–1085.
Zylicz M., King F.W., Wawrzynow A. 2001. Hsp70 interactions with the p53 tumour suppressor protein. EMBO J. 20 : 4634–4638.
Дополнительные материалы отсутствуют.