

Цитология, 2019, T. 61, № 8, стр. 634-648
Низкомолекулярные лиганды рецепторов лютеинизирующего и фолликулостимулирующего гормонов, как новое поколение регуляторов репродуктивных функций
А. О. Шпаков 1, *, Д. В. Дарьин 2, А. А. Бахтюков 1, К. В. Деркач 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
194223 Санкт-Петербург, Россия
2 Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
* E-mail: alex_shpakov@list.ru
Поступила в редакцию 25.04.2019
После доработки 17.05.2019
Принята к публикации 17.05.2019
Аннотация
Разработка новых регуляторов рецепторов лютеинизирующего (ЛГ) и фолликулостимулирующего (ФСГ) гормонов представляет собой одно из интенсивно развиваемых направлений молекулярной эндокринологии и репродуктологии, что обусловлено значительными проблемами при использовании препаратов гонадотропинов, причем как природных, так и рекомбинантных их форм. Наибольшие перспективы связывают с низкомолекулярными лигандами рецепторов ЛГ и ФСГ, которые по своей активности могут быть аллостерическими агонистами или антагонистами, положительными или негативными аллостерическими модуляторами или совмещать их функции. Основными преимуществами низкомолекулярных лигандов являются: 1) избирательность действия в отношении определенного внутриклеточного сигнального каскада, что особенно важно в условиях одновременной активации гонадотропинами сразу нескольких каскадов; 2) отсутствие конкуренции между гонадотропином и низкомолекулярными агонистами за связывание с рецептором, что обусловлено различиями в локализации ортостерического и аллостерического сайтов, с которыми они связываются; 3) возможность усиления эффекта гонадотропина при его совместном применении с низкомолекулярными аллостерическими агонистами или модуляторами, что обусловлено присущей им шапероноподобной активностью; 4) эффективность как при парентеральном, так и при пероральном введении. В настоящем обзоре суммированы и проанализированы данные литературы о дизайне и механизмах действия низкомолекулярных аллостерических лигандов рецепторов ЛГ и ФСГ, а также представлены недавние достижения авторов в области создания и изучения тиенопиримидиновых производных с активностью селективных агонистов рецептора ЛГ, влияющих на ЛГ-зависимые процессы в условиях in vitro и in vivo.
Гонадотропины – лютеинизирующий (ЛГ) и фолликулостимулирующий (ФСГ) гормоны и хорионический гонадотропин человека (ХГЧ) – являются важнейшими регуляторами функций репродуктивной системы у мужчин и женщин (Plant, 2015; Fournier, 2016; Шпаков, 2018). Основными направлениями их применения в клинике являются восстановление стероидогенеза, фолликулогенеза и сперматогенеза у пациентов с гипогонадотропным гипогонадизмом и с дефицитом половых стероидных гормонов, а также использование в качестве индукторов контролируемой овуляции во вспомогательных репродуктивных технологиях (De Leo et al., 2012; Ezcurra, Humaidan, 2014; Ulloa-Aguirre, Lira-Albarrán, 2016; Fournier, 2016; Шпаков, 2018).
Препараты гонадотропинов обычно выделяют из природных источников или получают генно-инженерным путем. Из мочи женщин, находящихся в постменопаузе, выделяют ФСГ, в то время как из мочи беременных женщин выделяют плацентарный ХГЧ. Наряду с этим в специальных клетках осуществляют синтез рекомбинантных форм всех трех гонадотропинов (De Leo et al., 2012; Levi Setti et al., 2015; Fournier, 2016). Однако как у природных, так и у рекомбинантных форм гонадотропинов имеются весьма существенные недостатки.
В случае мочевых форм ФСГ и ХГЧ основным недостатком является наличие в них биологически активных примесей, а также проблемы со стандартизацией препаратов, что приводит к варьированию их специфической биологической активности (Van Dorsselaer et al., 2011; Ezcurra, Humaidan, 2014). Необходимо также отметить, что выделяемый из мочи плацентарный ХГЧ отличается по структуре и функциональной активности от циркулирующих в крови мужчин и женщин ЛГ и сульфатированного ХГЧ, которые продуцируются гонадотрофами гипофиза (Cole, 2012; Ezcurra, Humaidan, 2014). При этом плацентарный ХГЧ вырабатывается только во время беременности, и его биологическая активность направлена в основном на регуляцию роста и развития эмбриона (Theofanakis et al., 2017). Мочевой ФСГ характеризуется высокой степенью N-гликозилирования, что снижает его сродство к рецептору ФСГ и ослабляет стимулирующие эффекты гонадотропина на клетки-мишени (Bousfield, Dias, 2011).
В свою очередь, рекомбинантные формы ЛГ, ХГЧ и ФСГ в значительной степени отличаются от природных форм гонадотропинов по посттрансляционным модификациям, в первую очередь по числу, степени разветвленности и заряду N-гликанов, что приводит к отчетливо выраженным изменениям их специфической активности и фармакологических характеристик (Шпаков, 2017). Более того, все препараты гонадотропинов характеризуются активностью агонистов, в то время как в клинической практике востребованы инверсионные агонисты и антагонисты, которые могут быть использованы для лечения гормонозависимых опухолей и контрацепции.
Все вышесказанное свидетельствует о необходимости разработки принципиально новых классов регуляторов рецепторов ЛГ и ФСГ. Наиболее перспективным подходом здесь является создание высокоселективных по отношению к рецепторам ЛГ и ФСГ низкомолекулярных аллостерических лигандов с активностью полных и инверсионных агонистов и нейтральных антагонистов, а также аллостерических модуляторов. Современному состоянию проблемы разработки таких лигандов и достижениям в изучении механизмов и мишеней их действия посвящен настоящий обзор.
СТРУКТУРНО-ФУНКЦИОНАЛЬНАЯ ОРГАНИЗАЦИЯ РЕЦЕПТОРОВ ГОНАДОТРОПИНОВ И МЕХАНИЗМЫ ИХ СВЯЗЫВАНИЯ С ЛИГАНДАМИ
Оба рецептора гонадотропинов относятся к группе δ родопсинового семейства рецепторов, сопряженных с гетеротримерными G-белками. Они содержат значительный по размеру внеклеточный N-концевой домен (эктодомен) и серпантинный домен, включающий семь гидрофобных, пронизывающих мембрану участков, которые соединены тремя внеклеточными и тремя цитоплазматическими петлями и образуют гептагеликальный трансмембранный канал. Гонадотропины с высоким сродством связываются с обогащенными остатками лейцина участками, формирующими основную часть эктодомена, в то время как аллостерический сайт, локализованный в полости трансмембранного канала, остается при этом свободным (Ji et al., 2004; Puett et al., 2007, 2010; Angelova et al., 2011; Ulloa-Aguirre et al., 2013; De Pascali et al., 2018). Необходимо отметить, что в большинстве рецепторов, сопряженных с G-белками, ситуация обратная – высокоаффинный (ортостерический) лигандсвязывающий сайт расположен внутри трансмембранного канала, а сравнительно небольшой N-концевой участок вместе с внеклеточными петлями формируют аллостерический сайт.
Все три гонадотропина представляют собой αβ-гетеродимеры, в которых α-субъединицы идентичны по первичной структуре, являясь продуктами одного и того же гена, в то время как β-субъединицы различаются и кодируются различными генами, определяя, тем самым, индивидуальность гонадотропина. Структура αβ-гетеродимерных комплексов гонадотропинов стабилизируется посредством цистиновых узлов, с помощью которых обеспечивается тесный контакт между центральными участками α- и β-субъединиц (Puett et al., 2007; De Pascali et al., 2018). За специфичность связывания с эктодоменом рецептора ответственна β-субъединица гонадотропина, в то время как α-субъединица необходима для физического взаимодействия между лиганд-активированным эктодоменом рецепторов ЛГ и ФСГ и их серпантинным доменом. После связывания гонадотропина с эктодоменом меняется конформация внеклеточных петель рецептора и его трансмембранного канала, что приводит к изменению функционального взаимодействия цитоплазматических петель рецептора как с различными типами гетеротримерных G-белков, так и с другими регуляторными белками, в первую очередь с β-аррестинами, что приводит к запуску сразу нескольких внутриклеточных сигнальных каскадов (Ulloa-Aguirre et al., 2013; Riccetti et al., 2017a, 2017b; De Pascali et al., 2018).
В настоящее время доказано сопряжение рецепторов ЛГ и ФСГ с тремя типами G-белков (Gs, Gq/11 и Gi/o), каждый из которых отвечаeт за активацию определенного сигнального пути. Через посредство Gs-белка гонадотропины стимулируют активность фермента аденилатциклазы (АЦ), что ведет к повышению внутриклеточного уровня цАМФ и стимуляции цАМФ-зависимых эффекторных белков – протеинкиназы А и факторов семейства Epac (Exchange Protein directly Activated by Cyclic AMP). Результатом стимуляции протеинкиназы А являются фосфорилирование и активация транскрипционного фактора CREB (cAMP-responsive element binding protein), контролирующего экспрессию большого числа цАМФ-зависимых генов, в то время как активация факторов семейства Epac приводит к стимуляции фосфатидилинозитол-3-киназы, митогенактивируемых протеинкиназ (МАПК) и ряда других эффекторных белков.
Активация цАМФ-зависимых путей является ключевым механизмом запуска гонадотропинами продукции половых стероидных гормонов (рис. 1). Необходимо отметить, что аденилатциклазная система играет решающую роль в зависимой от гонадотропинов регуляции физиологических и биохимических функций, поскольку экспрессия примерно двух третей генов, мишеней ЛГ, ХГЧ и ФСГ, контролируется цАМФ-зависимыми транскрипционными факторами (Ulloa-Aguirre et al., 2011).
Рис. 1.
Сигнальные пути гонадотропинов с активностью лютеинизирующего гормона (ЛГ) и низкомолекулярных Gs-селективных агонистов. Гонадотропины, ЛГ и ХГЧ, специфически связываются с рецептором ЛГ и активируют различные типы G-белков (Gs и Gq/11) и β-аррестины, что приводит к запуску цАМФ-зависимых и фосфоинозитидных путей и каскада МАП-киназ (МАПК), контролирующих стероидогенез, рост, дифференцировку и апоптоз в клетках-мишенях. В свою очередь, Gs-селективные низкомолекулярные агонисты связываются с аллостерическим сайтом и через посредство Gs-белка стимулируют преимущественно цАМФ-зависимые пути, не оказывая заметного влияния на другие внутриклеточные каскады. Сокращения: АЦ – аденилатциклаза; И3Ф – инозитол-1,4,5-трифосфат; ПКА – протеинкиназа А; ПКС – протеинкиназа С; ФЛС – фосфолипаза С; цАМФ – циклический аденозинмонофосфат; ERK1/2 – киназы 1 и 2, регулируемые внеклеточными сигналами; Gq/11 – гетеротримерные G-белки q/11-семейства; Gs – гетеротримерные G-белки стимулирующего типа.
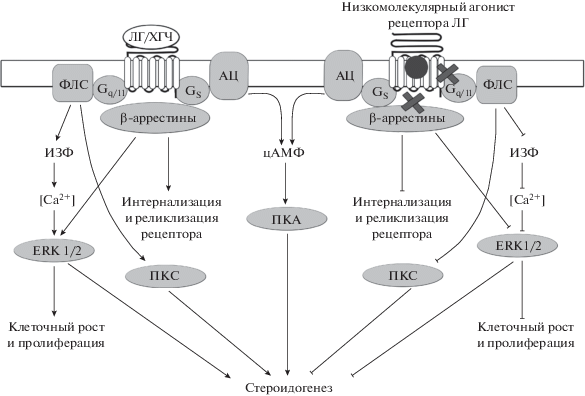
Активация гонадотропинами Gq/11-белков приводит к стимуляции фосфоинозитид-специфичной фосфолипазы Сβ (ФЛСβ), которая катализирует образование важнейших вторичных посредников – инозитол-3,4,5-трифосфата и диацилглицерина. Инозитол-3,4,5-трифосфат, активируя кальциевые каналы эндоплазматического ретикулума, обеспечивает повышение внутриклеточной концентрации кальция, что приводит к стимуляции Ca2+-зависимых эффекторных систем. В свою очередь, диацилглицерин повышает активность чувствительных к нему изоформ протеинкиназы С, что ведет к активации большого числа зависимых от этого фермента внутриклеточных систем (Ulloa-Aguirre et al., 2013; Riccetti et al., 2017a, 2017b; De Pascali et al., 2018; Landomiel et al., 2019).
Результатом гормональной активации рецепторов ФСГ и ЛГ является фосфорилирование их цитоплазматических участков, в первую очередь протяженного С-концевого домена. Этот процесс осуществляется серин/треониновыми протеинкиназами, которые относятся к семейству специфичных по отношению к G-белок-сопряженным рецепторам киназам. В фосфорилированном состоянии рецепторы приобретают способность специфично взаимодействовать с β-аррестинами, результатом чего является запуск ряда независимых от G-белков сигнальных путей, в том числе каскада МАПК (Kara et al., 2006; Reiter et al., 2012; Casarini et al., 2016; Riccetti et al., 2017b; Landomiel et al., 2019). β-Аррестиновые пути, как и цАМФ- и Ca2+-зависимые пути, вовлечены в регуляцию синтеза и секреции стероидных гормонов, контролируют рост и дифференцировку различных популяций клеток репродуктивной системы (рис. 1). Наряду с этим, β-аррестиновые пути играют исключительно важную роль в контроле интернализации, эндоцитоза и рециклизации комплексов гонадотропинов с рецепторами ЛГ и ФСГ, в значительной степени определяя судьбу рецепторных молекул после цикла их гормональной активации (Reiter et al., 2012; Riccetti et al., 2017b).
Специфическое связывание гонадотропина с рецептором индуцирует его переход в активную конформацию. Однако для активированных гормоном рецепторов ЛГ и ФСГ, как правило, характерна не одна, а несколько активных конформаций, каждая из которых отвечает за запуск определенного внутриклеточного сигнального каскада. Предпочтительность одной из активных конформаций обусловливает избирательную активацию гонадотропином определенного сигнального каскада, а соотношение между активными конформациями определяет паттерн таких каскадов. Стабильность и соотношение активных конформаций рецепторов ЛГ и ФСГ определяются типовой принадлежностью и структурными особенностями гонадотропина, в том числе степенью N-гликозилирования его α- и β-субъединиц, разветвленностью и зарядом N-гликанов, присутствием в α- и β-субъединицах мутаций, их способностью к олигомеризации и комплексообразованию с другими белками. Важную роль играют мутации и полиморфизмы в молекулах рецепторов, а также паттерн и функциональная активность нижележащих сигнальных белков, в том числе G-белков, β-аррестинов, рецепторных протеинкиназ (Cole, 2012; Fournier et al., 2015; Fournier, 2016; Nwabuobi et al., 2017; Landomiel et al., 2019).
Так, например, плацентарный ХГЧ с высокой эффективностью активирует аденилатциклазный сигнальный путь и повышает уровень цАМФ в клетках-мишенях, являясь мощным активатором стероидогенеза в клетках Лейдига семенников и в фолликулярных клетках яичников, но при этом в значительно меньшей степени влияет на Gq/11-зависимые каскады. В свою очередь, рекомбинантный ЛГ более эффективно стимулирует фосфолипазный и β-аррестиновый пути, но по способности стимулировать аденилатциклазу существенно уступает плацентарному ХГЧ (Riccetti et al., 2017b). Интересно отметить, что сульфатированная (гипофизарная) форма ХГЧ ближе по физиологическим эффектам и механизмам действия к ЛГ, хотя по первичной структуре идентична плацентарному ХГЧ. Различия в активности между сульфатированной и плацентарной формами ХГЧ обусловлены различиями в паттерне их N-гликозилирования соответственно в гонадотрофах гипофиза и клетках плаценты (Cole, 2012; Fournier et al., 2015; Шпаков, 2017, 2018; Nwabuobi et al., 2017).
В отличие от гонадотропинов, лиганды, которые взаимодействуют с аллостерическим сайтом рецепторов ЛГ и ФСГ, расположенным в их трансмембранном канале, более избирательны в плане регуляции внутриклеточных сигнальных каскадов. Это обусловлено тем, что, связываясь с аллостерическим сайтом, они стабилизируют, как правило, какую-то одну конформацию рецептора. В том случае, когда при взаимодействии с аллостерическим сайтом лиганды стабилизируют активную конформацию рецептора, они функционируют как полные аллостерические агонисты, положительные аллостерические модуляторы (ПАМ), усиливающие эффекты гонадотропинов, или, как аго-ПАМ, сочетающие эффекты полных агонистов и ПАМ. В том случае, когда результатом взаимодействия лигандов с аллостерическим сайтом является стабилизация неактивной конформации рецептора, их рассматривают как аллостерические антагонисты или негативные аллостерические модуляторы (НАМ), предотвращающие активацию рецептора гормоном (van der Westhuizen et al., 2015).
Поскольку узнающие и связывающие участки для гонадотропинов и низкомолекулярных аллостерических лигандов не перекрываются, то конкурентные взаимоотношения между ними отсутствуют. Соответственно, стимулирующие эффекты гонадотропинов и низкомолекулярных агонистов при их совместном применении могут характеризоваться аддитивностью, или, в случае наличия у аллостерического лиганда активности ПАМ или аго-ПАМ, их совместное введение с гонадотропином может приводить к потенцированию его эффекта. Ниже будут рассмотрены наиболее активные в условиях in vitro и in vivo низкомолекулярные лиганды аллостерического сайта рецепторов ЛГ и ФСГ, которые к настоящему времени разработаны нами и другими авторами.
НИЗКОМОЛЕКУЛЯРНЫЕ АГОНИСТЫ РЕЦЕПТОРА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА
Скрининг большого числа органических соединений с использованием библиотек химических структур позволил выявить несколько классов соединений, которые характеризовались активностью аллостерических регуляторов рецептора ЛГ. Среди них были производные терфенила и 1,3,5-замещенного пиразола, которые имели активность полных и инверсионных агонистов рецептора ЛГ (Jorand-Lebrun et al., 2007; Heitman, Ijzerman, 2008; Heitman et al., 2009). Производное терфенила LUF5771 с активностью инверсионного агониста ингибировало стимулирующие эффекты гонадотропинов с ЛГ-активностью и низкомолекулярных агонистов рецептора ЛГ (Heitman et al., 2009). В свою очередь, производное 1,3,5-замещенного пиразола (8-(1-(4-(трет-бутил)фенил)-3-(пиридин-3-ил)-1H-пиразол-5-ил)-2-(4-гидроксибензил)-4-оксооктанамид) проявляло активность полного агониста рецептора ЛГ, стимулируя активность АЦ и усиливая стероидогенез в клетках Лейдига, причем его действие выявлялось при наномолярных концентрациях вещества. Важно, что 8-(1-(4-(трет-бутил)фенил)-3-(пиридин-3-ил)-1H-пиразол-5-ил)-2-(4-гидроксибензил)-4-оксооктанамид был активным в условиях in vivo, повышая уровень тестостерона при пероральном введении самцам крыс (Jorand-Lebrun et al., 2007).
Однако наибольшие успехи достигнуты в разработке агонистов рецептора ЛГ на основе производных тиенопиримидина. В 2002 г. голландскими учеными были открыты первые тиенопиримидиновые производные с активностью агонистов рецептора ЛГ, и наиболее эффективными среди них были соединения Org41841 [N-трет-бутил-5-амино-4-(3-метоксифенил)-2-(метилтио)тиено[2,3-d]пиримидин-6-карбоксамид] и Org43553 [5-амино-N-(трет-бутил)-2-(метилтио)-4-(3-(2-морфолинацетамидо)фенил) тиено[2,3-d]пиримидин-6-карбоксамид] (Van Straten et al., 2002). В дальнейшем соединение Org43553 зарекомендовало себя, как высокоселективный и высокоэффективный аллостерический агонист рецептора ЛГ. Оно не только активировало ЛГ-зависимые сигнальные пути в условиях in vitro, но и было эффективным стимулятором стероидогенеза в условиях in vivo (Heitman et al., 2008; van Koppen et al., 2008; van de Lagemaat et al., 2009, 2011; van Straten, Timmers, 2009). Основываясь на структуре соединения Org43553, нами были сконструированы, синтезированы и в дальнейшем обстоятельно изучены тиенопиримидиновые производные TP01 [5-амино-N-(трет-бутил)-4-(3-(изоникотинамидо)фенил)-2-(метилтио)тиено[2,3-d]пиримидин-6-карбоксамид], TP03 [5-амино-N-трет-бутил-2-(метилсульфанил)-4-(3-(никотинамидо)фенил)тиено[2,3-d]пиримидин-6-карбоксамид] и TP23 [5-амино-N-(трет-бутил)-4-(3-(2-хлороникотинамидо)фенил)-2-(метилтио)тиено[2,3-d]пиримидин-6-карбоксамид] с активностью полных агонистов рецептора ЛГ, которые, так же как и соединение Org43553, были активны в условиях in vitro и in vivo (Деркач и др., 2014, 2016а, 2016б; Шпаков и др., 2014а, 2014б; Бахтюков и др., 2017, 2019а).
С использованием молекулярного докинга соединений Org41 841 и Org43 553 в аллостерический сайт рецептора ЛГ и соотношений “структура–активность” для этих производных и их аналогов была осуществлена реконструкция трансмембранного канала рецептора ЛГ и родственного ему рецептора тиреотропного гормона (ТТГ). Это позволило идентифицировать те аминокислотные остатки и структурные элементы в серпантинных доменах рецепторов ЛГ и ТТГ, которые ответственны за их переход в активную конформацию и за передачу сигнала от связанного с гормоном эктодомена к цитоплазматическим сайтам рецепторов, ответственным за взаимодействие с G-белками и β-аррестинами (Jaschke et al., 2006; Moore et al., 2006).
Показано, что замены аминокислотных остатков, локализованных во второй внеклеточной петле и в пятом и шестом трансмембранных участках рецептора ТТГ, на соответствующие остатки рецептора ЛГ приближают аллостерический сайт рецептора ТТГ к таковому рецептора ЛГ. Мутантные рецепторы ТТГ с одиночной заменой Leu570Phe или с двойными заменами Leu570Phe/Phe585Thr и Leu570Phe/Tyr643Phe во второй внеклеточной петле приобретали способность связываться с соединением Org41841 (агонистом рецептора ЛГ) со значениями EC50, равными 800–1000 нМ. Эти данные указывают на участие остатков Lуг570Phe, Phe585Thr и Tyr643Phe в формировании связывающей поверхности аллостерического сайта в рецепторе ЛГ (Jaschke et al., 2006).
Одновременная замена девяти аминокислотных остатков (Ile560Val и Leu570Phe во второй внеклеточной петле, Prp577Thr, Ala579Ser, Leu580Gln, Ala581Val и Phe585Thr в пятом трансмембранном участке, Tyr643Phe и Ile648Ala в шестом трансмембранном участке), формирующих аллостерический сайт рецептора ТТГ, на соответствующие им остатки в рецепторе ЛГ, обеспечивала связывание соединения Org41841 с мутантным рецептором ТТГ с тем же сродством, что и с рецептором ЛГ. Максимальное стимулирующее действие Org41841 на активность АЦ при его связывании с мутантным рецептором ТТГ было близко таковому ЛГ, что указывает на сходство механизмов трансдукции сигналов через серпантинные домены рецепторов ЛГ и ТТГ (Jaschke et al., 2006). Установлено, что отрицательно заряженный остаток Glu506, локализованный в третьем трансмембранном участке рецепторов ЛГ и ТТГ, также участвует в специфическом связывании с Org41841. Его замена на остаток аланина полностью блокировала связывание низкомолекулярного агониста с рецептором ЛГ и предотвращала стимуляцию АЦ (Jaschke et al., 2006; Moore et al., 2006).
Важной особенностью низкомолекулярных аллостерических агонистов является то, что они способны стабилизировать какую-то одну активную конформацию рецептора и, тем самым, селективно стимулируют определенный сигнальный каскад. Так, соединение Org43 553 даже в сравнительно высоких концентрациях (от 1 до 10 мкМ) стимулировало активность фосфоинозитид-специфичной ФЛСβ только на 33–37%, что составляет менее 5% от соответствующего эффекта гонадотропина (van Koppen et al., 2008). При этом вещество Org43553, как в высоких, так и низких концентрациях, эффективно стимулировало активность АЦ и цАМФ-зависимых транскрипционных факторов. Это указывает на избирательность стимулирующего влияния Org43 553 в отношении аденилатциклазной системы, реализуемого через Gs-белки, и об отсутствии его значимого действия на активность Gq/11-белков и ФЛСβ (van Koppen et al., 2008).
Нами с помощью техники АДФ-рибозилирования и пептидной стратегии показана избирательность тиенопиримидинового производного TP03 с активностью агониста рецептора ЛГ по отношению к аденилатциклазной системе в тестикулярных и овариальных мембранах крыс (Деркач и др., 2017). В мембранах, обработанных холерным токсином, который гиперактивирует Gs-белки и выключает их из сигнальной трансдукции, стимулирующие эффекты TP03 в отношении активности АЦ и ГТФ-связывания Gs-белков полностью подавлялись. В то же время обработка мембран коклюшным токсином, инактивирующим Gi-белки, или их инкубация с пептидом 349–359 Gαq/11-субъединицы, нарушающим взаимодействие рецептора с Gq/11-белками, существенного влияния на регуляторные эффекты TP03 не оказывала. Только при высокой концентрации TP03 (10–4 М) в небольшой степени влиял на ГТФ-связывание Gq/11-белков. Необходимо отметить, что регуляторные эффекты ХГЧ в тех же условиях не были специфичными в отношении определенного типа G-белков (Деркач и др., 2017). Следует отметить, что значительная часть синтезированных и изученных нами тиенопиримидиновых производных и других аллостерических лигандов рецептора ЛГ не показала значимого влияния на активность аденилатциклазной системы и стероидогенную активность клеток-мишеней и была исключена из дальнейшего анализа. Однако эти соединения могут активировать независимые от Gs-белков сигнальные каскады, включающие Gq/11-белки и β-аррестины, вследствие чего могут представлять интерес как для изучения молекулярных механизмов действия аллостерических регуляторов рецептора ЛГ, так и для практического их использования в медицине.
Избирательность сигнальной трансдукции и отсутствие заметного влияния на аррестиновые сигнальные пути, ответственные за даун-регуляцию и рециклизацию рецепторов ЛГ, играет важную роль в сохранении чувствительности тканей-мишеней к гонадотропинам при длительной обработке тиенопиримидиновыми производными, что было нами продемонстрировано для TP03 (Бахтюков и др., 2019а). Показано, что семисуточная обработка самцов крыс с помощью ХГЧ и TP03 повышала у них уровень тестостерона, но динамика стимулирующего эффекта этих препаратов была различной. Так при обработке ХГЧ в первые сут прирост концентрации тестостерона был максимальным и затем снижался, в то время как стероидогенный эффект TP03, напротив, повышался, достигая максимума на седьмые сут обработки. Если в первые сут прирост концентрации тестостерона под действием TP03 был в 4 раза ниже такового для ХГЧ, то на седьмые сут эксперимента стероидогенные эффекты ХГЧ и TP03 выравнивались (Бахтюков и др., 2019а). Причинами этого являются изменение чувствительности рецептора ЛГ к ХГЧ и тиенопиримидинам и различные механизмы действия этих агонистов рецептора ЛГ на систему стероидогенеза в клетках Лейдига.
Не только длительное, но и однократное введение ХГЧ вызывает статистически значимое снижение экспрессии гена Lhr, кодирующего рецептор ЛГ, в семенниках самцов крыс. В то же время при длительном введении животным TP03 экспрессия гена Lhr сохраняется и даже повышается, что является важным фактором сохранения чувствительности клеток Лейдига к эндогенным гонадотропинам (Бахтюков и др., 2019а). Необходимо отметить, что развитие резистентности репродуктивных тканей к гонадотропинам является одной из наиболее острых проблем при их использовании для лечения репродуктивных дисфункций и во вспомогательных репродуктивных технологиях (Banker, Garcia-Velasco, 2015; Riccetti et al., 2017a).
В наших экспериментах, ХГЧ и TP03 повышали экспрессию гена Star, кодирующего белок StAR (steroidogenic acute regulatory protein), который осуществляет транспорт холестерина из внутриклеточных депо в митохондрии. Этот процесс является скорость-лимитирующей стадией стероидогенеза. При этом стимулирующий эффект гонадотропина на экспрессию гена для StAR существенно превосходил таковой TP03 (Бахтюков и др., 2019а). Выявлена закономерность, согласно которой степень повышения экспрессии гена Star тем больше, чем больше степень снижения экспрессии гена Lhr и стероидогенного эффекта агониста рецептора ЛГ. Это может указывать на компенсаторный характер повышения экспрессии гена Star при нарушении передачи ЛГ-сигнала в клетках Лейдига, приводящего к снижению активности протеинкиназы А. В условиях длительного воздействия ХГЧ существенно повышалась экспрессия гена Cyp11a1, кодирующего цитохром Р450scc (C27 cholesterol side-chain cleavage cytochrome P450), который превращает холестерин в прегненолон, а на седьмые сут эксперимента отмечали также повышение экспрессии гена Hsd3b, кодирующего дегидрогеназу 3β-HSD, превращающую прегненолон в прогестерон. Примечательно, что при использовании TP03 существенных изменений в экспрессии генов, кодирующих цитохром Р450scc и дегидрогеназу 3β-HSD, выявлено не было, что свидетельствует об умеренном, в отличие от ХГЧ, влиянии этого низкомолекулярного агониста на систему стероидогенеза в условиях длительной обработки им животных (Бахтюков и др., 2019а).
Наряду с избирательностью сигнальной трансдукции, что характеризует тиенопиримидиновые производные, как селективные bias-агонисты рецептора ЛГ, определенный вклад в стабильность их стероидогенного эффекта при длительном применении вносят отличия фармакокинетических характеристик низкомолекулярных агонистов от таковых гонадотропинов. При введении крысам соединения Org43 553 период его полувыведения составил около 3 ч и был в среднем в два раза меньше, чем соответствующий показатель для ХГЧ, что является свидетельством более быстрой деградации и выведения низкомолекулярного агониста (van de Lagemaat et al., 2009). Снижение времени полувыведения имеет большое практическое значение, поскольку не только предотвращает резистентность тканей к эндогенным гонадотропинам, особенно при длительном введении низкомолекулярного агониста, но и снижает риск развития синдрома гиперстимуляции яичников, который является опасным осложнением при контролируемой индукции овуляции с помощью ХГЧ и ЛГ.
В отличие от гонадотропинов, ни однократная, ни многократная обработка самок крыс соединением Org43553 не вызывала у них повышения размеров яичников и проницаемости сосудов и не провоцировала синдром гиперстимуляции яичников (van de Lagemaat et al., 2011). Следует отметить, что триггером этого синдрома является мощный всплеск экспрессии и активности фактора роста эндотелия сосудов. Показано, что гонадотропины приводят к резкому повышению содержания фактора роста эндотелия сосудов в яичниках, в то время как тиенопиримидиновые производные таким действием не обладают. Признаки синдрома гиперстимуляции яичников отсутствовали и у женщин, которые для индукции овуляции перорально получали соединение Org43 553. При этом введение Org43 553 не только индуцировало овуляцию, но и позволяло получать ооциты высокого качества, что в дальнейшем обеспечивало нормальное развитие эмбрионов (Gerrits et al., 2013).
Поскольку ортостерический и аллостерический сайты в рецепторе ЛГ пространственно разделены, то низкомолекулярные агонисты рецептора ЛГ не оказывают ингибирующего влияния на связывание ЛГ и ХГЧ (Jorand-Lebrun et al., 2007; Heitman et al., 2008; van Koppen et al., 2008). Стимулирующие активность АЦ эффекты тиенопиримидиновых производных и гонадотропинов характеризуются аддитивностью, по крайней мере, при их концентрациях ниже значений EC50 (Heitman et al., 2008; van Koppen et al., 2008). Аддитивность эффектов тиенопиримидинов и ХГЧ была продемонстрирована нами в экспериментах in vitro и in vivo. Преинкубация тестикулярных мембран с TP03 приводила к повышению стимулирующего действия ХГЧ (10–10 М) на активность АЦ (Шпаков и др., 2014б; Деркач и др., 2016а). При введении ХГЧ самцам крыс, которых предварительно обрабатывали TP03, было выявлено потенцирование стероидогенного эффекта гонадотропина, причем потенцирующее действие TP03 в наибольшей степени выявлялось при низких дозах ХГЧ (Шпаков и др., 2019).
Способность тиенопиримидиновых производных усиливать эффекты гонадотропинов может быть обусловлена их шапероноподобным действием, которое впервые было описано при изучении восстанавливающего влияния соединения Org42 599 (трифторацетатной соли Org43553) на активность мутантных рецепторов ЛГ с заменами Ala593Pro и Ser616Tyr (Newton et al., 2011). Присутствие Org42 599 в культуре клеток с экспрессированными в них мутантными рецепторами не только стимулировалo экспрессию мутантного рецептора, но также нормализовалo его фолдинг и повышалo плотность рецепторов ЛГ на поверхности клеток. Шапероноподобный эффект Org42 599 обусловлен способностью этого соединения проникать внутрь клетки и там связываться с аллостерическим сайтом рецепторов ЛГ, локализованных в мембране эндоплазматического ретикулума (рис. 2). Это способствует правильной укладке полипептидной цепи рецепторных молекул, образованию ими олигомерных комплексов и транслокации в плазматическую мембрану (Newton et al., 2011). Необходимо подчеркнуть, что рецепторы ЛГ с мутациями Ala593Pro и Ser616Tyr в их трансмембранных участках в отсутствие шаперонов не способны встраиваться в плазматическую мембрану и, соответственно, не могут активироваться гонадотропинами. Такие мутантные формы рецепторов ЛГ выявлены у пациентов с гипоплазией клеток Лейдига и вызванным этим бесплодием (Kremer et al., 1995; Latronico et al., 1996; Mizrachi, Segaloff, 2004; Ulloa-Aguirre et al., 2014).
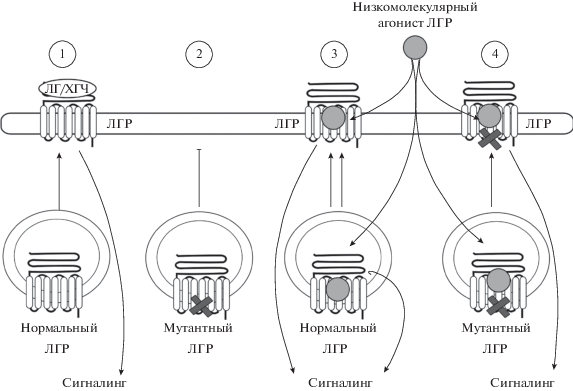
Рис. 2.
Аллостерические агонисты рецепторов гонадотропинов, как низкомолекулярные шапероны. 1 – транслокация в мембрану нормальной формы рецептора ЛГ (ЛГР) в условиях активации гонадотропином; 2 – отсутствие транслокации мутантного ЛГР; 3 – транслокация в мембрану нормальной формы ЛГР после его внутриклеточного связывания с низкомолекулярным агонистом; 4 – транслокация мутантного ЛГР после его связывания с низкомолекулярным агонистом, что обеспечивает способность ЛГР активироваться эндогенными гонадотропинами. Будучи гидрофобными соединениями, низкомолекулярные агонисты проникают внутрь клетки и взаимодействуют там с рецепторами гонадотропинов. Это могут быть синтезированные de novo или транспортированные внутрь клетки в результате эндоцитоза рецепторы гонадотропинов, или мутантные формы рецепторов с нарушенным фолдингом, неспособные эффективно транслоцироваться в плазматическую мембрану. Связывание рецепторов с аллостерическими агонистами стабилизирует их структуру и способствует транслокации в плазматическую мембрану, повышая чувствительность клеток к эндогенным гонадотропинам.
Аллостерические сайты даже близкородственных рецепторов характеризуются высокой вариабельностью первичной структуры и пространственной организации, что делает лиганды аллостерических сайтов более специфичными к рецепторам в сравнении с лигандами высокоаффинных (ортостерических) сайтов (van der Westhuizen et al., 2015; Lindsley et al., 2016). Однако в случае низкомолекулярных аллостерических регуляторов рецепторов ЛГ, ФСГ и ТТГ специфичность не столь высока, и достаточно часто возникают ситуации, когда лиганд аллостерического сайта одного рецептора способен (хотя и с существенно меньшей эффективностью) влиять на функциональную активность другого рецептора, что показано для ряда высокоактивных низкомолекулярных аллостерических агонистов рецептора ЛГ. Причиной этого, как можно полагать, является инверсия местоположения аллостерического и ортостерического сайтов в рецепторах гипофизарных гликопротеиновых гормонов (van der Westhuizen et al., 2015). Вследствие этого мониторинг кросс-реактивности низкомолекулярных аллостерических агонистов рецептора ЛГ по отношению к рецепторам ТТГ и ФСГ является важной составляющей при их фармакологической характеристике (van Koppen et al., 2008; Бахтюков и др., 2019б). В то же время, анализ синтезированных нами соединений TP01, TP03 и TP23 в условиях in vitro и in vivo показал, что они слабо или вовсе не влияют на активность рецепторов ТТГ и ФСГ. При обработке ими крыс не было выявлено изменений как базальных уровней тиреоидных гормонов тироксина и трийодтиронина, так и стимулирующего действия на них тиролиберина (Бахтюков и др., 2019б).
НИЗКОМОЛЕКУЛЯРНЫЕ АГОНИСТЫ И АНТАГОНИСТЫ РЕЦЕПТОРА ФОЛЛИКУЛОСТИМУЛИРУЮЩЕГО ГОРМОНА
Первый низкомолекулярный агонист рецептора ФСГ был разработан еще в 2001 г. и представлял собой пиперидинкарбоксамид, который в наномолярных концентрациях стимулировал активность АЦ в CHO-клетках с экспрессированным в них рецептором ФСГ. Однако будучи активным в условиях in vi-tro, пиперидинкарбоксамид при введении животным не оказывал никакого влияния на зависимые от ФСГ физиологические процессы и был исключен из дальнейших исследований (El Tayer et al., 2001). Дальнейший поиск низкомолекулярных регуляторов рецептора ФСГ позволил выявить большое число различающихся по структуре органических соединений с активностью аллостерических агонистов, антагонистов и модуляторов рецептора ФСГ (van Straten, Timmers, 2009; Nataraja et al., 2015; Anderson et al., 2018, 2019). Среди них производные тиазолидинов (Maclean et al., 2004; Wrobel et al., 2006; Yanofsky et al., 2006; Arey, 2008), тетрагидрохинолинов (Van Straten et al., 2005), гексагидрохинолинов (Grima Poveda et al., 2006; van Koppen et al., 2013), тиенопиримидинов (Hanssen, Timmers, 2003), бензамидов (van Koppen et al., 2013), дикетопиперазинов (Guo et al., 2004a, 2004b), а также замещенные γ-лактамы (Pelletier et al., 2005) и N-алкилированные сульфанилпиперазины (Magar et al., 2002).
Наибольший интерес представляют производные тиазолидинов, тетра- и гексагидрохинолинов и бензамидов, среди которых имеются полные и инверсионные агонисты и нейтральные антагонисты рецептора ФСГ. Так, в 2004 г. было разработано соединение 4-(5-(2-((2-(1H-индол-3-ил)этил)амино)-2-оксоэтил)-2-(4-(бензилокси)фенил)-4-оксотиазолидин-3-ил)-2-хлоробензамид, которое является производным тиазолидинов. Оно в наномолярных концентрациях подавляло стимулирующий эффект ФСГ и по своему фармакологическому профилю было отнесено к инверсионным агонистам (Maclean et al., 2004). В дальнейшем на его основе были разработаны тиазолидины с активностью полных агонистов (Pelletier et al., 2005; Wrobel et al., 2006; Yanofsky et al., 2006), в том числе соединение 3-(2S,5R)-5-(2-((3-этокси-4-метоксифенетил)амино)-2-оксоэтил)-4-оксо-2-(4-(фенилэтенил)фенил)тиазолидин-3-ил)бензамид. Оно с высокой эффективностью стимулировало цАМФ-зависимые сигнальные каскады и стероидогенез в клетках-мишенях, а в условиях in vivo индуцировало развитие преовуляторных фолликулов и овуляцию у неполовозрелых самок крыс (Yanofsky et al., 2006; Sriraman et al., 2014).
Наряду с собственным стимулирующим действием, это соединение потенцирует стимулирующие эффекты низких концентраций ФСГ, выступая здесь в качестве ПАМ для рецептора ФСГ. Дальнейшая модификация структуры тиазолидинов с активностью полных агонистов привела к созданию соединения 3-(2S,5R)-5-(2-((2-(1H-индол-3-ил)этил)амино)-2-оксоэтил)-4-оксо-2-(5-(фенилэтенил)тиофен-2-ил)тиазолидин-3-ил)бензамид, которое в низких концентрациях активировало Gs-белки и цАМФ-зависимые каскады и, тем самым, было отнесено к полным агонистам рецептора ФСГ. Важно отметить, что в высоких концентрациях оно ингибировало индуцированную гонадотропином активацию Gs-белков и стимулировало Gi-белки, через которые осуществляется ингибирование активности АЦ, функционируя как НАМ для рецептора ФСГ (Arey, 2008; Zoenen et al., 2012).
В 2006 г. появилось сообщение о разработке соединения Org214444-0, производного 4-фенил-5-оксо-l,4,5,6,7,8-гексагидрохинолина, которое в отсутствие ФСГ со значением EC50 около 1 нМ вызывало активацию АЦ в культуре СНО-клеток с экспрессированным в них рецептором ФСГ, а также стимулировало стероидогенез в клетках гранулезы человека и крысы (Grima Poveda et al., 2006; van Koppen et al., 2013). Наряду с этим, соединение Org214444-0 повышало сродство ФСГ к рецептору и эффективность стимуляции АЦ гонадотропином, что свидетельствует о присущей ему активности аго-ПАМ. Пероральное введение Org214444-0 вызывало стимуляцию фолликулогенеза и индуцировало овуляцию у зрелых самок крыс, что указывает на его устойчивость и хорошую всасываемость в желудочно-кишечном тракте (van Koppen et al., 2013).
Среди производных тетрагидрохинолина наибольший интерес представляет соединение 10, которое с высокой эффективностью ингибировало индуцированный ФСГ рост фолликулов и овуляцию у мышей (Van Straten et al., 2005). Изучение механизмов его действия показало, что соединение N-(1-ацетил-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-[1,1'-бифенил]-4-карбоксамид нарушает функциональное взаимодействие между эктодоменом и серпантинным доменом рецептора ФСГ, предотвращая передачу гормонального сигнала с эктодомена на цитоплазматические участки рецептора, активирующие Gs-белок, что типично для НАМ (Van Straten et al., 2005). Активность НАМ была присуща и 7-{4-[бис-(2-карбамоил-этил)-амино]-6-хлор-(1,3,5)-триазин-2-иламино)-4-гидрокси-3-(4-метокси-фенилазо)-нафтален}-2-сульфоновой кислоте, которая снижала специфическое связывание ФСГ с рецептором, подавляла стимулирующее действие гонадотропина на АЦ и систему стероидогенеза, а в условиях in vivo предотвращало индуцированную гонадотропином овуляцию у крыс (Arey et al., 2002; Wrobel et al., 2002).
Среди производных бензамида наиболее активными были соединения ADX61623, ADX68692 и ADX68693, которые демонстрировали активность аллостерических антагонистов рецептора ФСГ (Dias et al., 2011, 2014). Необходимо отметить, что все они имели необычный фармакологический профиль, что, как можно полагать, обусловлено сложным характером их влияния на регулируемые ФСГ сигнальные пути в клетках-мишенях. В условиях in vitro, соединение ADX61623 при низких концентрациях ингибировало индуцированную ФСГ продукцию цАМФ и прогестерона в фолликулярных клетках, а при высоких концентрациях повышало выработку эстрадиола, предшественником которого является, в том числе, и прогестерон (Dias et al., 2011). Из трех производных бензамида только соединение ADX68692 было активным в условиях in vivo. При пероральном и подкожном введении оно нарушало цикличность у зрелых самок крыс и уменьшало число извлеченных качественных ооцитов (Dias et al., 2014). Однако отметим, что соединение ADX68692 влияло, хотя и в меньшей степени, на рецептор ЛГ, усиливая продукцию прогестерона и снижая синтез тестостерона при обработке им первичной культуры клеток Лейдига крысы (Ayoub et al., 2016).
Подводя итог, следует отметить, что, несмотря на большое число разработанных аллостерических лигандов рецептора ФСГ, они до сих пор не используются в клинике, что обусловлено проблемами с их биодоступностью и деградацией, а также мало изученными побочными эффектами (Anderson et al., 2019). В то же время серьезные проблемы и ограничения, связанные с применением коммерческих препаратов ФСГ в клинике, а также необходимость разработки селективных ингибиторов функциональной активности рецептора ФСГ, которые в настоящее время отсутствуют, являются весомым аргументом в пользу дальнейшего изучения механизмов действия низкомолекулярных лигандов рецептора ФСГ и их внедрения в клиническую практику.
ЗАКЛЮЧЕНИЕ
Обобщая полученные к настоящему времени результаты в области изучения низкомолекулярных аллостерических лигандов рецепторов ЛГ и ФСГ, можно отметить следующие их особенности и преимущества, которые важны как для молекулярной эндокринологии и биохимии гормональных сигнальных систем, так и для клинической репродуктологии.
1) Низкомолекулярные аллостерические агонисты рецепторов ЛГ и ФСГ не конкурируют с гонадотропинами за места связывания и, тем самым, не подавляют собственные эффекты ЛГ, ХГЧ и ФСГ, а в ряде случаев даже их усиливают, действуя как ПАМ или аго-ПАМ. Ингибирование стимулирующего эффекта гонадотропинов низкомолекулярными лигандами с активностью НАМ осуществляется не вследствие конкуренции между ними за связывание с рецептором, а в результате аллостерического влияния НАМ на процесс трансдукции гонадотропинового сигнала.
2) Низкомолекулярные лиганды рецепторов гонадотропинов характеризуются селективностью в отношении определенных внутриклеточных сигнальных каскадов, функционируя как bias-лиганды, что позволяет предсказывать функциональный ответ клетки на их действие и предотвращает некоторые побочные эффекты, характерные для гонадотропинов (рис. 1).
3) Действие низкомолекулярных аллостерических агонистов избирательно и, в отличие от гонадотропинов, существенно не затрагивает аррестиновые пути, ответственные за даун-регуляцию рецепторов ЛГ и ФСГ. Вследствие этого в условиях обработки такими агонистами чувствительность тканей к гонадотропинам сохраняется, что позволяет использовать длительные курсы низкомолекулярных агонистов для регуляции зависимых от гонадотропинов физиологических функций, а также использовать их совместно с гонадотропинами
4) Поскольку низкомолекулярные аллостерические агонисты способны проявлять шапероноподобные свойства в отношении рецепторов ЛГ и ФСГ, предотвращая их внутриклеточную деградацию и стимулируя транслокацию в мембрану, их можно использовать для усиления ответа клеток на гонадотропины (рис. 2). Использовать можно как в случае мутантных форм рецепторов ЛГ и ФСГ, имеющих сниженную способность к транслокации в мембрану, так и в условиях метаболических нарушений, воспалительных и аутоиммунных процессов, которые ведут к нарушению посттрансляционного процессинга и деградации рецепторов ЛГ и ФСГ, а также при старении (Бахтюков и др., 2018).
5) Низкомолекулярные аллостерические лиганды рецепторов ЛГ и ФСГ могут быть активны не только при парентеральных способах введения, но и при пероральном способе доставки, поскольку они в большинстве своем стабильны в желудочно-кишечном тракте и хорошо всасываются клетками кишечника.
Возникает закономерный вопрос, почему при столь очевидных преимуществах перед гонадотропинами аллостерические лиганды рецепторов ЛГ и ФСГ до сих пор не применяются в медицине. Одними из основных причин этого являются недостаточная изученность их фармакокинетики и распределения в организме, а также проблемы с созданием лекарственных форм на их основе, поскольку в большинстве своем они являются высоко гидрофобными веществами, которые растворяются, как правило, только в ДМСО и других слабо полярных растворителях. Попытки снизить гидрофобность аллостерических лигандов с помощью модификации их структуры путем введения гидрофильных групп приводят к частичной или полной потере ими биологической активности, что обусловлено нарушением их проникновения в трансмембранный канал рецепторов гонадотропинов (Grima Poveda et al., 2006). Наибольшие надежды здесь связывают с использованием солюбилизирующих агентов, которые способны повысить растворимость этих соединений, не изменяя их структуру.
Список литературы
Бахтюков А.А., Деркач К.В., Дарьин Д.В., Шарова Т.С., Шпаков А.О. 2018. Ослабление базальной и стимулированной агонистами рецептора лютеинизирующего гормона продукции тестостерона у стареющих самцов крыс. Успехи геронтологии. 31(5) : 654–661. (Bakhtyukov A.A., Derkach K.V., Dar’in D.V., Sharova T.S., Shpakov A.O. 2018. The weakening of the basal and luteinizing hormone receptor agonist-stimulated testosterone production in aging male rats. Adv. Gerontol. 31(5) : 654–661.)
Бахтюков А.А., Деркач К.В., Дарьин Д.В., Шпаков А.О. 2019а. Стероидогенный эффект низкомолекулярного агониста рецептора лютеинизирующего гормона при его введении самцам крыс. Докл. Акад. наук. 484(6) : 103–106. (Bakhtyukov A.A., Derkach K.V., Dar’in D.V., Shpakov A.O. 2019a. Conservation of steroidogenic effect of the low-molecular-weight agonist of luteinizing hormone receptor in the course of its long-term administration to male rats. Dokl. Biochem. Biophys. 484 : 78–81.)
Бахтюков А.А., Деркач К.В., Дарьин Д.В., Шпаков А.О. 2019б. Тиенопиримидиновые производные специфично активируют стероидогенез в семенниках, но не влияют на функции щитовидной железы. Журн. эвол. биохим. физиол. 55(1): 28–36. (Bakhtyukov A.A., Derkach K.V., Dar’in D.V., Shpakov A.O. 2019b. Thienopyrimidine derivatives specifically activate steroidogenesis in the testes, but do not affect the function of the thyroid gland. Zhurn. Evol. Biokhim. Fiziol. 55(1): 28–36.)
Бахтюков А.А., Соколова Т.В., Дарьин Д.В., Деркач К.В., Шпаков А.О. 2017. Сравнительное изучение стимулирующего эффекта низкомолекулярного агониста рецептора лютеинизирующего гормона и хорионического гонадотропина на стероидогенез в клетках Лейдига крысы. Рос. Физиол. журн. им. И.М. Сеченова. 103(10) : 1181–1192. (Bakhtyukov A.A., Sokolova T.V., Dar’in D.V., Derkach K.V., Shpakov A.O. 2017. A comparative study of the stimulating effect of a low molecular weight agonist of the luteinizing hormone receptor and chorionic gonadotropin on steroidogenesis in rat Leydig cells. Ros. Fiziol. Zhurn. Im. I.M. Sechenova. 103(10) : 1181–1192.)
Деркач К.В., Бахтюков А.А., Шпаков А.А., Дарьин Д.В., Шпаков А.О. 2017. Особенности регуляции гетеротримерных G-белков хорионическим гонадотропином и низкомолекулярным агонистом рецептора лютеинизирующего гормона. Цитология. 59(7) : 474–481. (Derkach K.V., Bakhtyukov A.A., Shpakov A.A., Dar’in D.V., Shpakov A.O. 2017a. Specificity of heterotrimeric G protein regulation by human chorionic gonadotropin and low-molecular agonist of luteinizing hormone receptor. Cell Tissue Biol. 11(6) : 475–482.)
Деркач К.В., Дарьин Д.В., Лобанов П.С., Шпаков А.О. 2014. Тиенопиримидиновые производные повышают уровень тестостерона при их интратестикулярном, внутрибрюшинном и пероральном введении самцам крыс. Докл. Акад. наук. 459(3) : 382–385. (Derkach K.V., Dar’in D.V., Lobanov P.S., Shpakov A.O. 2014. Intratesticular, intraperitoneal, and oral administration of thienopyrimidine derivatives increases the testosterone level in male rats. Dokl. Biol. Sci. 459(1) : 326–329.)
Деркач К.В., Дарьин Д.В., Бахтюков А.А., Лобанов П.С., Шпаков А.О. 2016а. Изучение функциональной активности новых низкомолекулярных агонистов рецептора лютеинизирующего гормона in vitro и in vivo. Биол. мембраны. 33(4) : 263–271. (Derkach K.V., Dar’in D.V., Bakhtyukov A.A., Lobanov P.S., Shpakov A.O. 2016. In vitro and in vivo studies of functional activity of new low molecular weight agonists of the luteinizing hormone receptor. Biochemistry (Moscow). Suppl. Ser. A: Memb. Cell Biol. 10(4) : 294–300.)
Деркач К.В., Легкодух А.С., Дарьин Д.В., Шпаков А.О. 2016б. Стимулирующее влияние тиенопиримидинов, структурных аналогов Org43553, на активность аденилатциклазы в семенниках и на продукцию тестостерона у самцов крыс. Цитология. 58(8) : 602–609. (Derkach K.V., Legkodukh A.S., Dar’in D.V., Shpakov A.O. 2017. The stimulating effect of thienopyrimidines structurally similar to Org43553 on adenylate cyclase activity in the testes and on testosterone production in male rats. Cell Tiss. Biol. 11(1) : 73–80.)
Шпаков А.О. 2017. Гликозилирование гонадотропинов, как важнейший механизм регуляции их активности. Рос. Физиол. журн. им. И.М. Сеченова. 103(9) : 1004–1021. (Shpakov A.O. 2017. Glycosylation of gonadotropins, as the most important mechanism of regulation of their activity. Ros. Fiziol. Zhurn. Im. I. M. Sechenova. 103(9) : 1004–1021.)
Шпаков А.О. 2018. Гонадотропины – от теории к клинической практике. СПб: ПОЛИТЕХ-ПРЕСС. 347 c. (Shpakov A.O. 2018. Gonadotropins - from theory to clinical practice. SPb: POLITECH-PRESS. 347 pp.)
Шпаков А.О., Бахтюков А.А., Деркач К.В., Дарьин Д.В. 2019. Усиление стероидогенного эффекта хорионического гонадотропина при его совместном введении с низкомолекулярным агонистом рецептора лютеинизирующего гормона. В кн.: Рецепторы и внутриклеточная сигнализация. Междунар. конф. 20–24 мая 2019 г., Пущино. 862–867. (Shpakov A.O., Bakhtyukov A.A., Derkach K.V., Dar’in D.V. 2019. Enhancing the steroidogenic effect of chorionic gonadotropin when it is co-administered with low molecular weight agonist of luteinizing hormone receptor. Proceedings of the International Conference “Receptors and Intracellular Signaling”. May 20–24, 2019, Pushchino. 862–867.)
Шпаков А.О., Дарьин Д.В., Деркач К.В., Лобанов П.С. 2014а. Стимулирующее влияние тиенопиримидиновых производных на аденилатциклазную сигнальную систему в семенниках крыс. Докл. Акад. наук. 456(4) : 494–498. (Shpakov A.O., Dar’in D.V., Derkach K.V., Lobanov P.S. 2014a. The stimulating influence of thienopyrimidine compounds on the adenylyl cyclase systems in the rat testes. Dokl. Biochem. Biophys. 456 : 104–107.)
Шпаков А.О., Деркач К.В., Дарьин Д.В., Лобанов П.С. 2014б. Активация аденилатциклазы тиенопиримидиновыми производными в семенниках и яичниках крыс. Цитология. 56(5) : 346–352. (Shpakov A.O., Derkach K.V., Dar’in D.V., Lobanov P.S. 2014b. Activation of adenylyl cyclase by thienopyrimidine derivatives in rat testes and ovaries. Cell Tiss. Biol. 8(5) : 400–406.)
Arey B.J. 2008. Allosteric modulators of glycoprotein hormone receptors: discovery and therapeutic potential. Endocrine. 34 : 1–10.
Arey B.J., Deecher D.C., Shen E.S., Stevis P.E., Meade E.H., Wrobel J., Frail D.E., Lopez F.J. 2002. Identification and characterization of a selective, nonpeptide follicle-stimulating hormone receptor antagonist. Endocrinol. 143 : 3822–3829.
Anderson R.C., Newton C.L., Anderson R.A., Millar R.P. 2018. Gonadotropins and Their Analogs: Current and Potential Clinical Applications. Endocr. Rev. 39 : 911–937.
Anderson R.C., Newton C.L., Millar R.P. 2019. Small Molecule Follicle-Stimulating Hormone Receptor Agonists and Antagonists. Front. Endocrinol. (Lausanne). 9: 757. https://doi.org/10.3389/fendo.2018.00757
Angelova K., Felline A., Lee M., Patel M., Puett D., Fanelli F. 2011. Conserved amino acids participate in the structure networks deputed to intramolecular communication in the lutropin receptor. Cell Mol. Life Sci. 68 : 1227-1239.
Ayoub M.A., Yvinec R., Jégot G., Dias J.A., Poli S.M., Poupon A., Crépieux P., Reiter E. 2016. Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implications in steroidogenesis. Mol. Cell. Endocrinol. 436 : 10–22.
Banker M., Garcia-Velasco J.A. 2015. Revisiting ovarian hyper stimulation syndrome: Towards OHSS free clinic. J. Hum. Reprod. Sci. 8 : 13–17.
Bousfield G.R., Dias J.A. 2011. Synthesis and secretion of gonadotropins including structure-function correlates. Rev. Endocr. Metab. Disord. 12 : 289–302.
Casarini L., Reiter E., Simoni M. 2016. β-Arrestins regulate gonadotropin receptor-mediated cell proliferation and apoptosis by controlling different FSHR or LHCGR intracellular signaling in the hGL5 cell line. Mol. Cell. Endocrinol. 437 : 11–21.
Cole L.A. 2012. hCG, the wonder of today’s science. Reprod. Biol. Endocrinol. 10: 24. https://doi.org/10.1186/1477-7827-10-24
De Leo V., Musacchio M.C., Di Sabatino A., Tosti C., Morgante G., Petraglia F. 2012. Present and future of recombinant gonadotropins in reproductive medicine. Curr. Pharm. Biotechnol. 13 : 379–391.
De Pascali F., Tréfier A., Landomiel F., Bozon V., Bruneau G., Yvinec R., Poupon A., Crépieux P., Reiter E. 2018. Follicle-Stimulating Hormone Receptor: Advances and Remaining Challenges. Int. Rev. Cell. Mol. Biol. 338 : 1–58.
Dias J.A., Bonnet B., Weaver B.A., Watts J., Kluetzman K., Thomas R.M., Poli S., Mutel V., Campo B. 2011. A negative allosteric modulator demonstrates biased antagonism of the follicle stimulating hormone receptor. Mol. Cell. Endocrinol. 333 : 143–150.
Dias J.A., Campo B., Weaver B.A., Watts J., Kluetzman K., Thomas R.M., Bonnet B., Mutel V., Poli S.M. 2014. Inhibition of follicle-stimulating hormone-induced preovulatory follicles in rats treated with a nonsteroidal negative allosteric modulator of follicle-stimulating hormone receptor. Biol. Reprod. 90 : 19. https://doi.org/10.1095/biolreprod.113.109397
El Tayer N., Reddy A., Buckler D. 2001. Applied Research Systems ARS Holding N.A., assignee FSH Mimetics for the Treatment of Infertility. Unites States patent US 6,235,755.
Ezcurra D., Humaidan P. 2014. A review of luteinising hormone and human chorionic gonadotropin when used in assisted reproductive technology. Reprod. Biol. Endocrinol. 12: 95.https://doi.org/10.1186/1477-7827-12-95
Fournier T. 2016. Human chorionic gonadotropin: Different glycoforms and biological activity depending on its source of production. Ann. Endocrinol. (Paris). 77 : 75–81.
Fournier T., Guibourdenche J., Evain-Brion D. 2015. Review: hCGs: Different sources of production, different glycoforms and functions. Placenta. 36 (Suppl. 1): S60–S65.
Gerrits M., Mannaerts B., Kramer H., Addo S., Hanssen R. 2013. First evidence of ovulation induced by oral LH agonists in healthy female volunteers of reproductive age. J. Clin. Endocrinol. Metab. 98 : 1558–1566.
Grima Poveda P.M., Karstens Willem F.J., Timmers C.M. 2006. Inventors, N.V. Organon, assignee 4-Phenyl-5-Oxo-1,4,5,6,7,8-Hexahydroquinoline Derivatives for the Treatment of Infertility. United States patent US 8,022,218.
Guo T., Adang A.E., Dolle R.E., Dong G., Fitzpatrick D., Geng P., Ho K.K., Kultgen S.G., Liu R., McDonald E., McGuinness B.F., Saionz K.W., Valenzano K.J., van Straten N.C., Xie D., Webb M.L. 2004a. Small molecule biaryl FSH receptor agonists. Part 1: Lead discovery via encoded combinatorial synthesis. Bioorg. Med. Chem. Lett. 14 : 1713–1716.
Guo T., Adang A.E., Dong G., Fitzpatrick D., Geng P., Ho K.K., Jibilian C.H., Kultgen S.G., Liu R., McDonald E., Saionz K.W., Valenzano K.J., van Straten N.C., Xie D., Webb M.L. 2004b. Small molecule biaryl FSH receptor agonists. Part 2: Lead optimization via parallel synthesis. Bioorg. Med. Chem. Lett. 14 : 1717–1720.
Hanssen R.G.J.M., Timmers C.M. 2003. Inventors Preparation of Thienopyrimidines with Combined FSH and LH Activity. Patent US WO/2003/020726.
Heitman L.H., Ijzerman A.P. 2008. G protein-coupled receptors of the hypothalamic-pituitary-gonadal axis: A case for Gnrh, LH, FSH, and GPR54 receptor ligands. Med. Res. Rev. 28 : 975–1011.
Heitman L.H., Narlawar R., de Vries H., Willemsen M.N., Wolfram D., Brussee J., Ijzerman A.P. 2009. Substituted terphenyl compounds as the first class of low molecular weight allosteric inhibitors of the luteinizing hormone receptor. J. Med. Chem. 52 : 2036–2042.
Heitman L.H., Oosterom J., Bonger K.M., Timmers C.M., Wiegerinck P.H.G., Ijzerman A.P. 2008. [3H]Org 43553, the first low-molecular-weight agonistic and allosteric radioligand for the human luteinizing hormone receptor. Mol. Pharmacol. 73 : 518–524.
Jäschke H., Neumann S., Moore S., Thomas C.J., Colson A.O., Costanzi S., Kleinau G., Jiang J.K., Paschke R., Raaka B.M., Krause G., Gershengorn M.C. 2006. A low molecular weight agonist signals by binding to the transmembrane domain of thyroid-stimulating hormone receptor (TSHR) and luteinizing hormone/chorionic gonadotropin receptor (LHCGR). J. Biol. Chem. 281 : 9841–9844.
Ji I., Lee C., Jeoung M., Koo Y., Sievert G.A., Ji T.H. 2004. Trans-activation of mutant follicle-stimulating hormone receptors selectively generates only one of two hormone signals. Mol. Endocrinol. 18 : 968–978.
Jorand-Lebrun C., Brondyk B., Lin J., Magar S., Murray R., Reddy A., Schroff H., Wands G., Weiser W., Xu Q., McKenna S., Brugger N. 2007. Identification, synthesis, and biological evaluation of novel pyrazoles as low molecular weight luteinizing hormone receptor agonists. Bioorg. Med. Chem. Lett. 17 : 2080–2085.
Kara E., Crépieux P., Gauthier C., Martinat N., Piketty V., Guillou F., Reiter E. 2006. A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol. Endocrinol. 20 : 3014–3026.
Kremer H., Kraaij R., Toledo S.P., Post M., Fridman J.B., Hayashida C.Y., van Reen M., Milgrom E., Ropers H.H., Mariman E., et al. 1995. Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat. Genet. 9 : 160–164.
Landomiel F., De Pascali F., Raynaud P., Jean-Alphonse F., Yvinec R., Pellissier L.P., Bozon V., Bruneau G., Crépieux P., Poupon A., Reiter E. 2019. Biased Signaling and Allosteric Modulation at the FSHR. Front. Endocrinol. (Lausanne). 10: 148. https://doi.org/10.3389/fendo.2019.00148
Latronico A.C., Anasti J., Arnhold I.J., Rapaport R., Mendonca B.B., Bloise W., Castro M., Tsigos C., Chrousos G.P. 1996. Brief report: Testicular and ovarian resistance to luteinizing hormone caused by inactivating mutations of the luteinizing hormone-receptor gene. N. Engl. J. Med. 334 : 507–512.
Levi Setti P.E., Alviggi C., Colombo G.L., Pisanelli C., Ripellino C., Longobardi S., Canonico P.L., De Placido G. 2015. Human recombinant follicle stimulating hormone (rFSH) compared to urinary human menopausal gonadotropin (HMG) for ovarian stimulation in assisted reproduction: A literature review and cost evaluation. J. Endocrinol. Invest. 38 : 497–503.
Lindsley C.W., Emmitte K.A., Hopkins C.R., Bridges T.M., Gregory K.J., Niswender C.M., Conn P.J. 2016. Practical Strategies and Concepts in GPCR Allosteric Modulator Discovery: Recent Advances with Metabotropic Glutamate Receptors. Chem. Rev. 116 : 6707–6741.
Maclean D., Holden F., Davis A.M., Scheuerman R.A., Yanofsky S., Holmes C.P., Fitch W.L., Tsutsui K., Barrett R.W., Gallop M.A. 2004. Agonists of the follicle stimulating hormone receptor from an encoded thiazolidinone library. J. Comb. Chem. 6 : 196–206.
Magar S., Goutopoulos A., Liao Y., Schwarz M., Russell T.J. 2002. Piperazine Derivatives and Methods of Use. Patent WO2004031182A1.
Mizrachi D., Segaloff D.L. 2004. Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol. Endocrinol. 18 : 1768–1777.
Moore S., Jaeschke H., Kleinau G., Neumann S., Costanzi S., Jiang J.K., Childress J., Raaka B.M., Colson A., Paschke R., Krause G., Thomas C.J., Gershengorn M.C. 2006. Evaluation of small-molecule modulators of the luteinizing hormone/choriogonadotropin and thyroid stimulating hormone receptors: Structure-activity relationships and selective binding patterns. J. Med. Chem. 49 : 3888–3896.
Nataraja S.G., Yu H.N., Palmer S.S. 2015. Discovery and development of small molecule allosteric modulators of glycoprotein hormone receptors. Front. Endocrinol. (Lausanne). 6 : 142. https://doi.org/10.3389/fendo.2015.00142
Newton C.L., Whay A.M., McArdle C.A., Zhang M., van Koppen C.J., van de Lagemaat R., Segaloff D.L., Millar R.P. 2011. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA. 108 : 7172–7176.
Nwabuobi C., Arlier S., Schatz F., Guzeloglu-Kayisli O., Lockwood C.J., Kayisli U.A. 2017. hCG: Biological Functions and Clinical Applications. Int. J. Mol. Sci. 18 : pii : E2037. https://doi.org/10.3390/ijms18102037
Pelletier J.C., Rogers J., Wrobel J., Perez M.C., Shen E.S. 2005. Preparation of highly substituted gamma-lactam follicle stimulating hormone receptor agonists. Bioorg. Med. Chem. 13 : 5986–5995.
Plant T.M. 2015. 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-pituitary-gonadal axis. J. Endocrinol. 226 : T41–T54.
Puett D., Angelova K., da Costa M.R., Warrenfeltz S.W., Fanelli F. 2010. The luteinizing hormone receptor: insights into structure-function relationships and hormone-receptor-mediated changes in gene expression in ovarian cancer cells. Mol. Cell. Endocrinol. 329 : 47–55.
Puett D., Li Y., DeMars G., Angelova K., Fanelli F. 2007. A functional transmembrane complex: The luteinizing hormone receptor with bound ligand and G protein. Mol. Cell. Endocrinol. 260–262 : 126–136.
Reiter E., Ahn S., Shukla A.K., Lefkowitz R.J. 2012. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52 : 179–197.
Riccetti L., De Pascali F., Gilioli L., Potì F., Giva L.B., Marino M., Tagliavini S., Trenti T., Fanelli F., Mezzullo M., Pagotto U., Simoni M., Casarini L. 2017a. Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod. Biol. Endocrinol. 15 : 2. https://doi.org/10.1186/s12958-016-0224-3
Riccetti L., Yvinec R., Klett D., Gallay N., Combarnous Y., Reiter E., Simoni M., Casarini L., Ayoub M.A. 2017b. Human luteinizing hormone and chorionic gonadotropin display biased agonism at the LH and LH/CG receptors. Sci. Rep. 7 : 940. https://doi.org/10.1038/s41598-017-01078-8
Sriraman V., Denis D., de Matos D., Yu H., Palmer S., Nataraja S. 2014. Investigation of a thiazolidinone derivative as an allosteric modulator of follicle stimulating hormone receptor: evidence for its ability to support follicular development and ovulation. Biochem. Pharmacol. 89 : 266–275.
Theofanakis C., Drakakis P., Besharat A., Loutradis D. 2017. Human Chorionic Gonadotropin: The Pregnancy Hormone and More. Int. J. Mol. Sci. 18 : pii: E1059. https://doi.org/10.3390/ijms18051059
Ulloa-Aguirre A., Crepieux P., Poupon A., Maurel M.C., Reiter E. 2011. Novel pathways in gonadotropin receptor signaling and biased agonism. Rev. Endocr. Metab. Dis. 12 : 259–274.
Ulloa-Aguirre A., Dias J.A., Bousfield G., Huhtaniemi I., Reiter E. 2013. Trafficking of the follitropin receptor. Methods Enzymol. 521 : 17–45.
Ulloa-Aguirre A, Lira-Albarrán S. 2016. Clinical Applications of Gonadotropins in the Male. Prog. Mol. Biol. Transl. Sci. 143 : 121–174.
Ulloa-Aguirre A., Zariñán T., Dias J.A., Conn P.M. 2014. Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function. Mol. Cell. Endocrinol. 382 : 411–423.
van de Lagemaat R., Raafs B.C., van Koppen C., Timmers C.M., Mulders S.M., Hanssen R.G. 2011. Prevention of the onset of ovarian hyperstimulation syndrome (OHSS) in the rat after ovulation induction with a low molecular weight agonist of the LH receptor compared with hCG and rec-LH. Endocrinology. 152 : 4350–4357.
van de Lagemaat R., Timmers C.M., Kelder J., van Koppen C., Mosselman S., Hanssen R.G. 2009. Induction of ovulation by a potent, orally active, low molecular weight agonist (Org 43553) of the luteinizing hormone receptor. Hum. Reprod. 24 : 640–648.
van der Westhuizen E.T., Valant C., Sexton P.M., Christopoulos A. 2015. Endogenous allosteric modulators of G protein-coupled receptors. J. Pharmacol. Exp. Ther. 353 : 246–260.
Van Dorsselaer A., Carapito C., Delalande F., Schaeffer-Reiss C., Thierse D., Diemer H., McNair D.S., Krewski D., Cashman N.R. 2011. Detection of prion protein in urine-derived injectable fertility products by a targeted proteomic approach. PLoS ONE. 6: e17815. https://doi.org/10.1371/journal.pone.0017815
van Koppen C.J., Verbost P.M., van de Lagemaat R., Karstens W.J., Loozen H.J., van Achterberg T.A., van Amstel M.G., Brands J.H., van Doornmalen E.J., Wat J., Mulder S.J., Raafs B.C., Verkaik S., Hanssen R.G., Timmers C.M. 2013. Signaling of an allosteric, nanomolar potent, low molecular weight agonist for the follicle-stimulating hormone receptor. Biochem. Pharmacol. 85 : 1162–1170.
van Koppen C.J., Zaman G.J., Timmers C.M., Kelder J., Mosselman S., van de Lagemaat R., Smit M.J., Hanssen R.G. 2008. A signaling-selective, nanomolar potent allosteric low molecular weight agonist for the human luteinizing hormone receptor. Naunyn Schmiedebergs Arch. Pharmacol. 378 : 503–514.
van Straten N.C., Schoonus-Gerritsma G.G., van Someren R.G., Draaijer J., Adang A.E., Timmers C.M., Hanssen R.G., van Boeckel C.A. 2002. The first orally active low molecular weight agonists for the LH receptor: Thienopyr(im)idines with therapeutic potential for ovulation induction. ChemBioChem. 3 : 1023–1026.
van Straten N.C., Timmers C.M. 2009. Non-Peptide ligands for the gonadotropin receptors. Annu. Rep. Med. Chem. 44 : 171–188.
Van Straten N.C., van Berkel T.H., Demont D.R., Karstens W.J., Merkx R., Oosterom J., Schulz J., van Someren R.G., Timmers C.M., van Zandvoort P.M. 2005. Identification of substituted 6-amino-4-phenyltetrahydroquinoline derivatives: potent antagonists for the follicle-stimulating hormone receptor. J. Med. Chem. 48 : 1697–1700.
Wrobel J., Green D., Jetter J., Kao W., Rogers J., Pérez M.C., Hardenburg J., Deecher D.C., López F.J., Arey B.J., Shen E.S. 2002. Synthesis of (bis)sulfonic acid, (bis)benzamides as follicle-stimulating hormone (FSH) antagonists. Bioorg. Med. Chem. 10 : 639–656.
Wrobel J., Jetter J., Kao W., Rogers J., Di L., Chi J., Peréz M.C., Chen G.C., Shen E.S. 2006. 5-alkylated thiazolidinones as follicle-stimulating hormone (FSH) receptor agonists. Bioorg. Med. Chem. 14 : 5729–5741.
Yanofsky S.D., Shen E.S., Holden F., Whitehorn E., Aguilar B., Tate E., Holmes C.P., Scheuerman R., MacLean D., Wu M.M., Frail D.E., López F.J., Winneker R., Arey B.J., Barrett R.W. 2006. Allosteric activation of the follicle-stimulating hormone (FSH) receptor by selective, nonpeptide agonists. J. Biol. Chem. 281 : 13226–13233.
Zoenen M., Urizar E., Swillens S., Vassart G., Costagliola S. 2012. Evidence for activity-regulated hormone-binding cooperativity across glycoprotein hormone receptor homomers. Nat. Commun. 3 : 1007. https://doi.org/10.1038/ncomms1991
Дополнительные материалы отсутствуют.