

Цитология, 2020, T. 62, № 1, стр. 16-23
Васкулярный компонент нейровоспаления при экспериментальной болезни Альцгеймера у мышей
А. В. Моргун 1, *, Е. Д. Осипова 1, Е. Б. Бойцова 1, О. Л. Лопатина 1, Я. В. Горина 1, Е. А. Пожиленкова 1, А. Б. Салмина 1
1 Красноярский государственный медицинский университет им. проф. В.Ф. Войно-Ясенецкого
Министерства здравоохранения РФ
660022 Красноярск, Россия
* E-mail: 441682@mail.ru
Поступила в редакцию 21.06.2019
После доработки 27.08.2019
Принята к публикации 03.09.2019
Аннотация
Исследованы новые RAGE- и CD147-опосредованные механизмы повреждения гиппокампа мышей за счет накопления бета-амилоида (Aβ), развития локального воспаления, нарушения метаболизма и повреждения гематоэнцефалического барьера на двух экспериментальных моделях болезни Альцгеймера in vivo. Изучены новые эффекты Aβ в ткани гиппокампа при хронической нейродегенерации альцгеймеровского типа, характеризующие нарушения нейропластичности, ангиогенеза, структурно-функциональную целостность гематоэнцефалического барьера, развитие локального нейровоспаления во взаимосвязи с особенностями экспрессии белков RAGE и CD147. Ранние нейродегенеративные изменения в гиппокампе, связанные с аккумуляцией Aβ, ассоциированы с интенсификацией неоангиогенеза и формированием аберрантных межклеточных контактов в слое эндотелия церебральных микрососудов в отдельных субрегионах гиппокампа и развитием локального нейровоспаления. По мере прогрессирования нейродегенерации неоангиогенез в гиппокампе подавляется.
Одним из наиболее распространенных в мире нейродегенеративных заболеваний является болезнь Альцгеймера (БА), характеризующаяся прогрессирующей потерей нейронов, в первую очередь гиппокампа, развитием когнитивных нарушений и других признаков деменции. В настоящее время заболевание остается неизлечимым, снижает продолжительность и качество жизни больных, что делает БА глобальной медицинской и социально-экономической проблемой (Graham et al., 2017).
Вопросам, посвященным этиологии и патогенезу БА, с момента открытия заболевания уделено много внимания. Известно, что биохимическими особенностями заболевания являются отложение амилоидных бляшек во внеклеточном пространстве и внутриклеточная аккумуляция гиперфосфорилированных нейрофибриллярных клубков. Однако вопрос о вкладе бета-амилоида (Aβ) и тау-белка в начало и развитие БА остается открытым (Armstrong, 2013). В последнее десятилетие обсуждается роль нарушений со стороны нейроваскулярной единицы (НВЕ) в патогенезе БА. НВЕ представляет совокупность клеток головного мозга (церебральные эндотелиоциты, перициты, периваскулярные астроциты и нейроны). Нарушения НВЕ, развивающиеся при БА, включают в себя изменения проницаемости гематоэнцефалического барьера (ГЭБ) и микроциркуляции вследствии нарушения глиоваскулярного контроля локального кровотока и нейрон-астроглиального метаболического сопряжения (van de Haar et al., 2016; Liu et al., 2018).
Патология церебральных микрососудов является одним из основных факторов риска развития деменции (Arvanitakis et al., 2016). В дополнении к этому, церебральная амилоидная ангиопатия, являясь важной причиной нарушения проницаемости ГЭБ и одним из патологических признаков БА, вызывает развитие сосудистых нарушений, что способствует снижению познавательной функции (Saito, Ihara, 2016). Таким образом, возникает вопрос о причинно-следственной связи между нейродегенерацией и сосудистым компонентом при развитии БА. Особенно это становится ясным, если учесть, что сосудистые нарушения развиваются достаточно рано, в том числе и до появления первых признаков нейродегенерации (Iturria-Medina et al., 2016), что обусловливает актуальность поиска новых молекулярных механизмов повреждения НВЕ и ГЭБ.
Повреждение кровеносных сосудов вызывает дисфункцию ГЭБ, что, в свою очередь, приводит к повреждению нейронов и накоплению Aβ в головном мозге (Salmina et al., 2016).
В целом, как показали клинические и экспериментальные исследования, цереброваскулярная дисфункция играет важную роль в развитии БА, а также церебральной амилоидной ангиопатии, с отложением амилоида в крупных сосудах и уменьшением клиренса амилоида из паренхимы мозга, в том числе из-за изменения транспорта амилоида через эндотелий и его неспособность к деградации (Carare et al., 2013; Steinberg et al., 2014; Keable et al., 2016).
Допускается, что избыточный амилоидогенез приводит к неоангиогенезу и формированию сети микрососудов с избыточной проницаемостью сосудистой стенки и усиленным транспортом амилоида из крови в мозг через поврежденный ГЭБ (Biron et al., 2011). В соответствии с этим предположением, при БА регистрируют повышенную экспрессию гиппокампальными эндотелиоцитами белков, транспортирующих Aβ из крови в ткань мозга (RAGE-белков) (Miller et al., 2008). Известно, что рецептор конечных продуктов гликирования белков (RAGE) опосредует многие физиологические функции – рост нейронов, выживание и регенерацию, играет важную роль в провоспалительных реакциях, индуцируя провоспалительные цитокины, является главным медиатором врожденного иммунного ответа и может участвовать в ряде патологических процессов, включая сахарный диабет, БА, системный амилоидоз (Kierdorf, Fritz, 2013; Ramasamy et al., 2016). Недавние исследования продемонстрировали участие RAGE в транспорте нейропептида окситоцина в ткань головного мозга (Yamamoto et al., 2019), что обеспечивает действие окситоцина в отношении социального поведения.
Интересна роль молекулы CD147 в патогенезе БА, так как есть доказательства важной роли CD147 в функционировании сердечно-сосудистой, нервной и иммунной систем (Agrawal et al., 2013; Kanyenda et al., 2014; Seizer et al., 2014). CD147 функционально сопряжен с транспортерами лактата (MCT1, MCT4), гамма-секретазой, участвующей в протеолиза белка-предшественника амилоида, а также регулирует экспрессию и активность матриксных металлопротеиназ (Успенская и др., 2018). CD147 экспрессируется эндотелиальными клетками и астроглией НВЕ/ГЭБ и может функционально интегрировать несколько сигнальных путей, активированных при реализации механизмов ангиогенеза и барьерогенеза.
Примечательно, что “классические” лиганды RAGE – конечные продукты гликирования – могут индуцировать экспрессию CD147 в некоторых клетках-эффекторах воспаления (Bao et al., 2010), тогда как лиганд CD147 – s100A9 – эффективно взаимодействует с RAGE, например, в участках тромбоза (von Ungern-Sternberg et al., 2018). Таким образом, логично предположить, что лиганды обоих рецепторов могут выступать в качестве т. н. аларминов, высвобождающихся из поврежденных клеток и инициирующих процессы локального воспаления и нарушения микроциркуляции. К категории аларминов также относятся белки группы HMGB1, которые инициируют многие механизмы, лежащие в основе т. н. вторичной альтерации при воспалении при остром нарушении мозгового кровообращения (Kim et al., 2018), черепно-мозговой травме и эпилепсии (Paudel et al., 2018). Известно, что HMGB1 выступают в качестве лигандов RAGE, в том числе в головном мозге при болезни Альцгеймера (Deane, 2012; Meneghini et al., 2013), будучи ответственными за некоторые механизмы повреждения ГЭБ (Festoff et al., 2016).
В связи с вышеизложенным, а также с учетом роли локального нейровоспаления в прогрессирование нейродегенерации альцгеймеровского типа, CD147 и RAGE могут рассматриваться в качестве потенциальных молекул-мишеней, изменение экспрессии которых способно изменить характер течения цереброваскулярной патологии при БА.
Целью настоящего исследований стало изучение молекулярных механизмов нарушения церебрального неоангиогенеза и структурно-функциональной целостности ГЭБ при экспериментальном моделировании БА in vivo, оценка роли Aβ в регуляции метаболизма клеток эндотелия церебральных микрососудов, процессов церебрального ангиогенеза и поддержания структурно-функциональной целостности ГЭБ в норме и при развитии церебральной амилоидной ангиопатии.
МАТЕРИАЛ И МЕТОДИКА
Моделирование нейродегенерации. Использовали две модели БА – трансгенную модель (сформировавшаяся нейродегенерация) и модель с интрагиппокампальным введением амилоида (ранние стадии заболевания и реализация нейротоксического действия Aβ). Животных содержали в клетках со свободным доступом к воде и корму при постоянной температуре 20 ± 2°С и фиксированном режиме освещения (12 ч света и 12 ч темноты).
В экспериментах использовали 3 группы животных. Контрольная группа – мыши линии C57BL/6, самцы в возрасте 4 мес. (n = 5) – ложно-оперированные животные после введения растворителя Aβ – фосфатно-солевого буферного раствора (PBS). Генетическая модель БА – мыши линии B6SLJ –Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas, сам-цы в возрасте 9 мес. (n = 5), были получены из лаборатории The Jackson Laboratory (США).
Экспериментальная БА – мыши линии C57BL/6, самцы в возрасте 4 мес. (n = 5). Моделирование БА осуществляли интрагиппокампальным введением 1 мкл препарата агрегированного Aβ 1-42 (Sigma-Aldrich, США) по стереотаксическим координатам мозга в зону СА1 гиппокампа. Препарат Aβ растворяли в PBS до концентрации 50 мкM с последующей агрегацией в термостате при 37°С в течение 7 сут (Epelbaum et al., 2015). Оценку признаков БА с 7 сут после оперативного вмешательства проводили с использованием нейроповеденческого тестирования “Распознавание нового объекта” (Sipos et al., 2007; Miedel et al., 2017). Верификацию экспериментальной модели БА осуществляли окрашиванием 0.015%-ным раствором Тиофлавина S (Sigma-Aldrich, США). После введения амилоида в ткани головного мозга наблюдали флуоресцирующие амилоидные бляшки с характерным спектром испускания.
Иммуногистохимия. На 2 сут после операции осуществляли транскардиальную перфузию 4%-ным параформальдегидом с последующим забором головного мозга. Мозг фиксировали в 10%-ном нейтральном забуференном формалине, после чего погружали в 20%-ный раствор сахарозы. С помощью микротома Thermo Scientific Microm HM 650 (Thermo Fisher Scientific, США) изготавливали срезы толщиной 50 мкм. Методом непрямой иммуногистохимии для свободно плавающих срезов проводили регистрацию целевых маркеров (Encinas, Enikolopov, 2008).
После промывки в PBS, срезы блокировали 3%-ным козьим сывороточным альбумином в PBS и 1%-ным тритоном X-100 в течение 1 ч при комнатной температуре с последующим инкубированием в течение ночи с первичными антителами к RAGE (ab3611; Abcam, Великобритания), HMGB1 (ab77302; Abcam, Великобритания), к CD147 (ab108317; Abcam, Великобритания), JAM1 (ab52647; Abcam, Великобритания) и к CD31 (PECAM-1)-FITC (F8402; Sigma Aldrich, США). Все антитела использзовали в разведении 1 : 1000. После инкубации с первичными антителами, срезы промывали и инкубировали со вторичными антителами, конъюгированными с Alexa (Abcam, Великобритания) в разведении 1 : 1000 в течение 2 ч при комнатной температуре. Ядра клеток окрашивали DAPI (Thermo Fisher Scientific, США).
Изображения получали с помощью конфокального микроскопа Olympus FV 10i (Olympus, Япония). В срезах головного мозга подсчитывали количество клеток, экспрессирующих целевые маркеры, не менее чем в пяти полях зрения.
Производили подсчет доли клеток, экспрессирующих целевые молекулы (от общего числа клеток в области сосудов в поле зрения, рассчитанного по ядрам DAPI-позитивных клеток, локализующихся в области сосудов), в не менее 5 полях зрения. Солоколизацию целевых меток оценивали по коэффициенту корреляции Пирсона с использованием специализированного програмного обеспечения для микроскопа Olympus FV 10i (Zinchuk V. et al., 2007).
Статистическую обработку полученных результатов проводили методами непараметрической статистики. Для сравнения показателей в независимых выборках применяли критерий Манна−Уитни, сравнение зависимых выборок осуществляли с помощью критерия Уилкоксона. Различия принимали значимыми при P < 0.05. Результаты представлены в виде средних значений и их ошибки.
РЕЗУЛЬТАТЫ
Интрагиппокампальное введение Aβ вызвало развитие нейродегенерации альцгеймеровского типа у экспериментальных животных, которое сопровождалось увеличением количества HMGB1-иммунопозитивных клеток в зубчатой извилине гиппокампа – 4.6 ± 0.6% по сравнению с контрольной группой (1.8 ± 0.4%) (Р = 0.0014) (рис. 1а).
Рис. 1.
Относительное количество клеток, иммунопозитивных на белки группы HMGB1 (а), RAGE (б), CD147 (в) и CD31 (г) у контрольных мышей и мышей с моделью болезни Альцгеймера. Иммуногистохимия с использованием соответствующих антител. Контроль – ложно-оперированные животные с введением PBS; БА – болезнь Альцгеймера смоделирована посредством интрагиппокампального введения препарата бета-амилоида; B6SLJ – мыши с генетической моделью болезни Альцгеймера. СА1, СА2, СА3, DG – зоны гиппокампа.
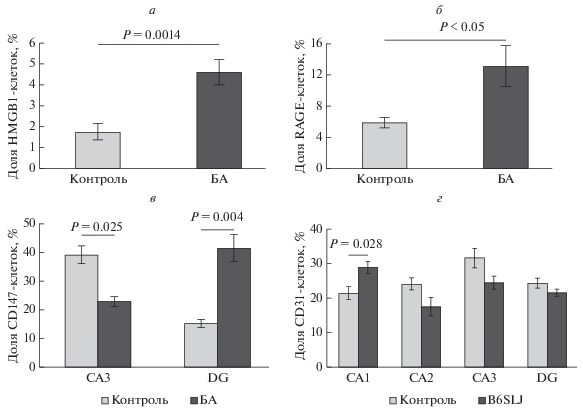
Однонаправленные изменения обнаружили при регистрации RAGE-иммунопозитивных клеток – увеличение количества гиппокампальных клеток, экспрессирующих RAGE, в экспериментальной группе до 13.2 ± 2.6%, по сравнению с ложно-оперированными животными (5.9 ± 0.7%) (Р < 0.05) (рис. 1б).
Учитывая, что Aβ и HMGB1 являются эндогенными лигандами RAGE белков, повышение экспрессии RAGE в эндотелиальных клетках гиппокампа животных с одновременным увеличением экспрессии HMGB1 и аккумуляцией Aβ при экспериментальной БА свидетельствует о реализации RAGE-опосредованных механизмов локального воспаления и эндотелиальной дисфункции.
Следующим этапом мы оценили некоторые особенности ангиогенеза, реализуемые посредством молекулы CD147 при экспериментальной БА. У животных с генетической моделью БА в области гиппокампа СА3 количество эндотелиоцитов, экспрессирующих CD147 (23 ± 1.6%) было меньше (Р = 0.025), чем в контрольной группе (39.1 ± 3.1%) (рис. 1в). Одновременно с этим в зубчатой извилине гиппокампа зафиксировали повышенное (Р = 0.004) количество CD147-иммунопозитивных клеток (41.4 ± 4.7%) по сравнению с ложно-оперированными животными (15.3 ± 1.4%) (рис. 1в).
В субрегионах гиппокампа CA1 и CA2 значимых различий в экспрессии CD147 не наблюдали.
Учитывая выявленные нарушения ангиогенеза у животных с экспериментальной моделью БА, мы оценили особенности экспрессии маркера эндотелиальной дисфункции (CD31) и белка адгезивных контактов (JAM1) в гиппокампе. В экспериментальной группе доля CD31-иммунопозитивных эндотелиоцитов была в два раза больше, чем в контрольной группе животных, и составляла 5.5 ± 0.9% против 2.4 ± 0.4% соответственно (Р = 0.0053).
Однако при моделировании БА в эндотелиальных клетках произошло снижение индекса солокализации CD31 с молекулой адгезионных контактов – JAM1, который составил 21 ± 7%. В контрольной группе индекс солокализации достигал 92.7 ± 5.2% (Р < 0.0001).
Обнаруженное увеличение количества CD31-иммунопозитивных эндотелиоцитов у животных с моделью ранних нарушений вызвало интерес к оценке характера экспрессии CD31 в отдельных субрегионах гиппокампа у животных с уже сформировавшимися нейродегенеративными изменениями (генетическая модель БА).
У животных с генетической моделью БА мы выявили тенденцию (Р = 0.223) к снижению количества эндотелиоцитов, экспрессирующих CD31, в зубчатой извилине гиппокампа (21.5 ± 1%) по сравнению с контролем (24.3 ± 1.4%). Аналогичная ситуация наблюдается в зонах СА2 и СА3 гиппокампа (рис. 1г). Однако в субрегионе гиппокампа СА1 доля CD31-иммунопозитивных клеток оказалась выше в экспериментальной группе (28.9 ± 1.7%) по сравнению с контрольной (21.4 ± 1.8%) (Р = 0.028).
Таким образом, можно предположить, что начальные этапы нейродегенерации альцгеймеровского типа сопровождаются интенсификацией процессов неоангиогенеза в гиппокампе, однако по мере прогрессирования заболевания неоангиогенез подавляется. Исключение составляет субрегион гиппокампа CA1, в котором высокий уровень экспрессии CD31-иммунопозитивных клеток сохраняется.
ОБСУЖДЕНИЕ
Нами получены новые данные о фундаментальных механизмах сопряжения процессов нейропластичности в норме и при хронической нейродегенерации, а также механизмах развития амилоидной ангиопатии. В ходе работы реализован экспериментальный подход с использованием двух экспериментальных моделей БА in vivo для подтверждения возможного участия в патогенезе CD147- и RAGE-опосредованных механизмов в развитии эндотелиальной дисфункции, ремоделировании сосудистой сети и нарушениях структурной целостности гематоэнцефалического барьера. Изучены новые эффекты Aβ в ткани гиппокампа (на ранних этапах и при сформированной нейродегенерации альцгеймеровского типа), характеризующие нарушения ангиогенеза (экспрессия CD31 в различных областях гиппокампа), структурно-функциональной целостности ГЭБ (экспрессия белка JAM1), развития локального нейровоспаления (экспрессия HMGB1) во взаимосвязи с особенностями экспрессии CD147 и RAGE белков.
Ранние нейродегенеративные изменения в гиппокампе, связанные с аккумуляцией Aβ, сопровождаются интенсификацией неоангиогенеза, нарушением формирования межэндотелиальных контактов, наростанием эндотелиальной дисфункции в гиппокампе и развитием локального нейровоспаления, с последующим угнетением неоангиогенеза по мере прогрессирования заболевания.
Обнаруженное увеличение экспрессии CD31 на фоне редуцирования экспрессии JAM1 в клетках эндотелия микрососудов гиппокампа максимально в CA1 субрегионе. Вероятнее всего, аккумуляция Aβ в сосудистой стенке вызывает интенсификацию ангиогенеза и формирование новых сосудов с аберрантной экспрессией JAM1, что одновременно ассоциировано с увеличением экспрессии CD147 как молекулы, регулирующей процессы ангиогенеза и внеклеточного метаболизма амилоида. Считается, что уменьшение количества CD31-иммунопозитивных клеток характерно для нейровоспаления и нейродегенеративных заболеваний. В целом, уменьшение экспрессии CD31 в ткани головного мозга характерно для эндотелиальной дисфункции и/или гибели клеток эндотелия. В частности, снижение уровня экспрессии CD31 отмечали у пациентов с БА, что позволяет предположить, что во время прогрессирования заболевания наблюдается обширная дегенерация эндотелия (Grammas, 2011; Magaki et al., 2018).
Обнаруженные нами наиболее ранние изменения в гиппокампе, которые характеризуются увеличением количества CD31-иммунопозитивных эндотелиоцитов и четырехкратным уменьшением коэффициента солокализации JAM1 и CD31 в этих клетках, позволяют предположить, что аккумуляция Aβ вызывает интенсификацию ангиогенеза и формирование новых сосудов, но с аберрантной экспрессией белка адгезивных контактов JAM1. В свою очередь, нарушение экспрессии белков адгезивных контактов, обеспечивающих целостность эндотелиального монослоя, при интенсивном неоангиогенезе сопровождается формированием функционально некомпетентного ГЭБ на ранних стадиях нейродегенерации альцгеймерского типа.
Указанные изменения регистрируются на фоне развития локального воспаления, в том числе за счет белка HMGB1, высвобождающегося из поврежденных клеток. Увеличение экспрессии RAGE белков на клетках церебрального эндотелия обеспечивает действие HMGB1 в отношении этих клеток, что может являться причиной развития повреждения и дисфункции эндотелия. Известно, что HMGB1 является маркером нейровоспаления и играет существенную роль в патогенезе БА – индуцирует дегенерацию клеток, модифицирует процессы агрегации Aβ и является независимым медиатором действия Aβ (Fujita et al., 2016). С другой стороны, HMGB1 является лигандом RAGE в различных тканях, и их взаимодействие индуцирует процессы воспаления (Weber et al., 2015). А взаимодействие Aβ с RAGE-позитивными эндотелиоцитами, нейронами и микроглией инициирует окислительный стресс, образование липидных пероксидов (Chen et al., 2018).
В совокупности с накоплением Aβ в сосудистой стенке, что стимулирует активацию провоспалительных цитокинов и активных форм кислорода, все указанные процессы вызывают повреждение нейронов, гладкомышечных клеток и перицитов, эндотелиальную дисфункцию, способствуя повреждению ГЭБ (Erickson, Banks, 2013). Помимо прочего, RAGE рассматривают как транспортер Aβ в головной мозг из периферической крови, а RAGE-рецепторы опосредуют транслокацию Aβ из внеклеточного пространства во внутриклеточное (Andrade et al., 2018; Reich et al., 2018).
Аналогичным образом, в гиппокампе возрастает экспрессия CD147 (преимущественно в зубчатой извилине), что может носить характер “ответной” реакции на аккумуляцию Аβ и интенсификацию ремоделирования микрососудов.
CD147 является молекулой, экспрессия которой в клетках НВЕ регулирует процессы протеолиза белка-предшественника амилоида, секреции матриксных металлопротеиназ, транспорта лактата, а также ангиогенез (Vetrivel et al., 2008; Muramatsu, 2016). Важно отметить, что при повреждении головного мозга экспрессия CD147 увеличивается (Wei et al., 2014).
Интересно, что у животных контрольной группы зубчатая извилина гиппокампа демонстрирует минимальный (по сравнению с другими субрегионами гиппокампа) уровень экспрессии CD147.
Известно, что уровень экспрессии CD147 обратно пропорционален локальной продукции Аβ вследствие того, что CD147 негативно контролирует активность ассоциированной с ним в составе мультипротеинового комплекса гамма-секретазы и стимулирует внеклеточную деградацию Aβ. Логично предположить, что высокий уровень экспрессии CD147 в зубчатой извилине гиппокампа, а также зарегистрированные нами высокие уровни экспрессии HMGB1 и RAGE в этом регионе совокупно маркируют интенсивность процессов воспаления, ремоделирования микрососудов и метаболизма амилоида.
Список литературы
Успенская Ю.А., Комлева Ю.К., Горина Я.В., Пожиленкова Е.А., Белова О.А., Салмина А.Б. 2018. Полифункциональность CD147 и новые возможности для диагностики и терапии. Сибирское медицинское обозрение. № 4. С. 22. (Uspenskaya Yu.A., Komleva Yu.K., Gorina Ya.V., Pozhilenkova EA, Belova OA, Salmina A.B. 2018. Multifunctionality of CD147 and new opportunities for diagnosis and therapy. Siberian Medical Review. № 4. P. 22.)
Agrawal S.M. Williamson J., Sharma R., Kebir H., Patel K., Prat A., Yong V.W. 2013. Extracellular matrix metalloproteinase inducer shows active perivascular cuffs in multiple sclerosis. Brain. V. 6. P. 1760.
Andrade S., Ramalho M.J., Pereira M.D.C., Loureiro J.A. 2018. Resveratrol brain delivery for neurological disorders prevention and treatment. Front. Pharmacol. V. 9. P. 1261.
Armstrong R.A. 2013. What causes alzheimer’s disease? Folia neuropathologica. V. 3. P. 169.
Arvanitakis Z., Capuano A.W., Leurgans S.E., Bennett D.A., Schneider J.A. (2016). Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. V. 9. P. 934.
Bao W., Min D., Twigg S.M., Shackel N.A., Warner F.J., Yue D.K., McLennan S.V. 2010. Monocyte CD147 is induced by advanced glycation end products and high glucose concentration: possible role in diabetic complications. Am. J. Physiology-Cell Physiology. V. 5. P. C1212.
Biron K.E., Dickstein D.L., Gopaul R., Jefferies W.A. 2011. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PloS One. V. 8: e23789.
Carare R.O. Hawkes C.A., Jeffrey M., Kalaria R.N., Weller R.O. 2013. Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol. Appl. Neurobiol. V. 6. P. 593.
Chen F., Ghosh A., Hu M., Long Y., Sun H., Kong L., Hong H., Tang S. 2018. RAGE-NF-κB-PPARγ signaling is involved in AGEs-induced upregulation of amyloid-β influx transport in an in vitro BBB model. Neurotox. Res. V. 2. P. 284.
Deane R.J. 2012. Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med. Chem. V. 7. P. 915.
Encinas J.M., Enikolopov G. 2008. Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell Biol. V. 85. P. 243.
Epelbaum S., Youssef I., Lacor P.N., Chaurand P., Duplus E., Brugg B., Duyckaerts C., Delatour B. 2015. Acute amnestic encephalopathy in amyloid-β oligomer–injected mice is due to their widespread diffusion in vivo. Neurobiol. Aging. V. 6. P. 2043.
Erickson M.A., Banks W.A. 2013. Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cerebral Blood Flow Metabolism. V. 10. P. 1500.
Festoff B.W., Sajja R.K., van Dreden P., Cucullo L. 2016. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflam. V. 1. P. 194.
Fujita K., Motoki K., Tagawa K., Chen X., Hama H., Nakajima K., Homma H., Tamura T., Watanabe H., Katsuno M., Matsumi C., Kajikawa M., Saito T., Saido T., Sobue G., Miyawaki A., Okazawa H. 2016. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Reports. V. 6 : 31895. https://doi.org/10.1038/srep31895
Graham W.V., Bonito-Oliva A., Sakmar T.P. 2017. Update on Alzheimer’s disease therapy and prevention strategies. Ann. Rev. Med. V. 1. P. 413.
Grammas P. 2011. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation. V. 8 : 26. https://doi.org/10.1186/1742-2094-8-26
Haar H.J. van de, Jansen J.F.A., van Osch M.J.P., van Buchem M.A., Muller M., Wong S.M., Hofman P.A.M., Burgmans S., Verhey F.R.J., Backes W.H. 2016. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol. Aging. V. 45. P. 190.
Iturria-Medina Y. Sotero R.C., Toussaint P.J., Mateos-Pérez J.M., Evans A.C. 2016. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. V. 21 : 11934. https://doi.org/10.1038/ncomms11934
Kanyenda L.J., Verdile G., Martins R., Meloni B.P., Chieng J., Mastaglia F., Laws S.M., Anderton R.S., Boulos S. 2014. Is cholesterol and amyloid-β stress induced CD147 expression a protective response? Evidence that extracellular cyclophilin a mediated neuroprotection is reliant on CD147. J. Alzheimer’s Disease. V. 3. P. 545.
Keable A., Fenna K., Yuen H.M., Johnston D.A., Smyth N.R., Smith C., Rustam Al-Shahi Salman, Samarasekera N., James N.A.R., Attems J., Kalaria R.N., Wellera R.O., Carare R.O. 2016. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta. V. 5. P. 1037.
Kierdorf K., Fritz G. 2013. RAGE regulation and signaling in inflammation and beyond. J. Leukocyte Biol. V. 1. P. 55.
Kim I.D., Lee H., Kim S.W., Lee H.K., Choi J., Han P.L., Lee J.K. 2018. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Disease. V. 4. P. 426.
Liu X., Hou D., Lin F., Luo J., Xie J., Wang Y., Tian Y. 2018. The role of neurovascular unit damage in the occurrence and development of Alzheimer’s disease. Rev. Neurosci. https://doi.org/10.1515/revneuro-2018-0056
Magaki S., Tang Z., Tung S., Williams C.K., Lo D., Yong W.H., Khanlou N., Vinters H.V. 2018. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging. V. 70. P. 70.
Meneghini V., Bortolotto V., Francese M.T., Dellarole A., Carraro L., Terzieva S., Grilli M. 2013. High-mobility group box-1 protein and β-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-kb axis: relevance for Alzheimer’s disease. J. Neurosci. V. 14. P. 6047.
Miedel C.J., Patton J.M., Miedel A.N., Miedel E.S., Levenson J.M. 2017. Assessment of spontaneous alternation, novel object recognition and limb clasping in transgenic mouse models of amyloid-β and tau neuropathology. J. Vis. Exp. V. 123. https://doi.org/10.3791/55523
Miller M.C., Tavares R., Johanson C.E., Hovanesian V., Donahue J.E., Gonzalez L., Silverberg G.D., Stopa E.G. 2008. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. V. 1230. P. 273.
Muramatsu T. 2016. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. V. 5. P. 481.
Paudel Y.N., Shaikh M.F., Chakraborti A., Kumari Y., Aledo-Serrano Á., Aleksovska K., Alvim M.K.M., Othman I. 2018. HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front. Neurosci. V. 12. P. 628.
Ramasamy R., Shekhtman A., Schmidt A.M. 2016. The multiple faces of RAGE–opportunities for therapeutic intervention in aging and chronic disease. Expert Opinion Therap. Targets. V. 4. P. 431.
Reich D., Gallucci G., Tong M., de la Monte S.M. 2018. Therapeutic advantages of dual targeting of PPAR-δ and PPAR-γ in an experimental model of sporadic Alzheimer’s disease. J. Parkinson’s Dis. Alzheimer’s Dis. V. 1. P. 1.
Saito S., Ihara M. 2016. Interaction between cerebrovascular disease and Alzheimer pathology. Current Opinion Psychiatry. V. 2. P. 168.
Salmina A.B., Pozhilenkova E.A., Morgun A.V., Kuvacheva N.V., Shuvaev A.N., Lopatina O.L., Boitsova E.B., Taranushenko T.E. 2016. Glial dysfunction and blood-brain barrier impairment in the developing brain. Adv. Neuroimmune Biol. V. 2. P. 69.
Seizer P., Gawaz M., May A.E. 2014. Cyclophilin A and EMMPRIN (CD147) in cardiovascular diseases. Cardiovasc. Res. V. 1. P. 17.
Sipos E., Kurunczi A., Kasza A., Horváth J., Felszeghy K., Laroche S., Toldi J., Párducz A., Penke B., Penke Z. 2007. Beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neurosci. V. 1. P. 28.
Steinberg M., Hess K., Corcoran C., Mielke M.M., Norton M., Breitner J., Green R., Leoutsakos J., Welsh-Bohmer K., Lyketsos C., Tschanz J. 2014. Vascular risk factors and neuropsychiatric symptoms in Alzheimer’s disease: The cache county study. Int. J. Geriatric Psychiatry. V. 2. P. 153.
Ungern-Sternberg S.N.I., von Zernecke A., Seizer P. 2018. Extracellular matrix metalloproteinase inducer EMMPRIN (CD147) in cardiovascular disease. Int. J. Mol. Sci. V. 19 : pii: E507. https://doi.org/10.3390/ijms19020507
Vetrivel K.S., Zhang X., Meckler X., Cheng H., Lee S., Gong P., Lopes K.O., Chen Y., Iwata N., Yin K.J., Lee J.M., Parent A.T., Saido T.C., Li Y.M., Sisodia S.S., Thinakaran G. 2008. Evidence that CD147 modulation of beta-amyloid (Abeta) levels is mediated by extracellular degradation of secreted Abeta. J. Biol. Chem. V. 28. P. 19489.
Weber D.J., Allette Y.M., Wilkes D.S., White F.A. 2015. The HMGB1-RAGE inflammatory pathway: implications for brain injury-induced pulmonary dysfunction. Antiox. Red. Signaling. V. 17. P. 1316.
Wei M., Li H., Shang Y., Zhou Z., Zhang J. 2014. Increased CD147 (EMMPRIN) expression in the rat brain following traumatic brain injury. Brain Res. V. 1585. P. 150.
Yamamoto Y., Liang M., Munesue S., Deguchi K., Harashima A., Furuhara K., Yuhi T., Zhong J., Akther S., Goto H., Eguchi Y., Kitao Y., Hori O., Shiraishi Y., Ozaki N., et al. 2019. Vascular RAGE transports oxytocin into the brain to elicit its maternal bonding behaviour in mice. Commun. Biol. V. 1. P. 76.
Zinchuk V., Zinchuk O, Okada T. 2007. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: Pushing pixels to explore biological phenomena. Acta Histochem Cytochem. V. 40. P. 101.
Дополнительные материалы отсутствуют.