

Цитология, 2020, T. 62, № 1, стр. 3-15
Транскрипционный фактор Zeb1: посттранскрипционная регуляция его активности в карциномах молочной железы человека
Д. Ю. Поздняков 1, О. Ю. Шувалов 1, Н. А. Барлев 1, А. Г. Миттенберг 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: a.mittenberg@gmail.com
Поступила в редакцию 03.07.2019
После доработки 30.09.2019
Принята к публикации 01.10.2019
Аннотация
В настоящем обзоре подробно описана модуляция экспрессии и функций Zeb1 – одного из транскрипционных факторов, регулирующих эпителиально-мезенхимный переход (ЭМП-ТФ), рассмотрены многочисленные и разнообразные посттранскрипционные и посттрансляционные механизмы регуляции активности Zeb1, а также рассматривается их роль в ангиогенезе, инвазии, метастазировании и формировании стволовых клеток опухоли. Ферменты, ответственные за посттрансляционные модификации ЭМП-ТФ, могут выступать в качестве удобных и эффективных терапевтических мишеней, однако подходить к данной проблеме следует с осторожностью, в силу возможного наличия у этих ферментов двойных функций – в качестве онкогенов или онкосупрессоров, в зависимости от клеточного контекста. Также в обзоре рассмотрена регуляция ЭМП-ТФ на уровне молекул РНК, при этом ключевую роль в сложной регуляторной цепи эпителиально-мезенхимного перехода играют многочисленные микро-РНК и длинные некодирующие РНК.
По данным мировой статистики, каждый год злокачественные новообразования молочной железы поражают более миллиона женщин во всем мире, из них в Российской Федерации регистрируется свыше 50 тыс. случаев. С каждым годом увеличивается и абсолютное число заболевших раком молочной железы (РМЖ) на 0.1–0.2%. РМЖ в России является ведущей онкологической патологией у женского населения (Гервас и др., 2019). В свою очередь, основной причиной смертности при злокачественных новообразованиях молочной железы является метастазирование. Метастазирующий РМЖ является одним из самых смертоносных видов РМЖ у женщин во всем мире с уровнем смертности более 2.1 на миллион случаев в год (Tevaarwerk et al., 2013). Метастазирование – многоэтапный процесс, в ходе которого раковые клетки распространяются по кровяному руслу и, в конечном итоге, приводят к образованию вторичных опухолевых узлов в отдаленных органах. Эпителиально-мезенхимальный переход (ЭМП) является ключевым процессом для перехода неинвазивного РМЖ в инвазивный и повышения устойчивости к традиционной химиотерапии, при котором поляризованные эпителиальные клетки теряют свои плотные межклеточные контакты, что приводит к повышению их миграционной способности, усилению инвазивных свойств и приобретению мезенхимного фенотипа. ЭМП характеризуется изменениями в экспрессии генов, в клеточной полярности, нарушением плотных контактов, значительным увеличением продукции компонентов внеклеточного матрикса, продукцией матриксных металлопротеиназ (MMP) и трансформирующего фактора роста-β (TGF-β), экспрессией маркеров раковых стволовых клеток, гипоксией, снижением экспрессии Е-кадгерина и другими молекулярно-биологическими событиями. Понимание функций маркеров ЭМП, связанных с метастазированием, а также путей их регуляции, имеет решающее значение для разработки потенциальных стратегий лечения пациенток с метастазирующим РМЖ. Недавние исследования показали, что ЭМП характеризуется потерей молекулы клеточной адгезии E-кадгерина, активацией таких мезенхимальных белков, как виментин, матриксные металлопротеиназы и N-кадгерин, и дерегуляцией сигнального пути Wnt, что приводит к проникновению эпителиальных клеток через базальную мембрану (см. обзор Поздняков и др., 2019).
ЭМП представляет собой сложное, многоступенчатое явление, происходящее во время эмбрионального развития и прогрессирования опухоли, связанное с серьезным перепрограммированием экспрессии генов и приводящее к изменениям в судьбе и поведении клеток. Представляет значительный интерес обнаруженная в последние годы связь между ЭМП и раковыми стволовыми клетками, поскольку лишь незначительная популяция опухолевых клеток может инициировать и поддерживать развитие опухоли, и агрессивные опухолевые клетки обладают многими характеристиками эмбриональных клеток-предшественников. Обнаружено, что ЭМП приводит к возникновению у опухолевых клеток признаков стволовости, что обеспечивает молекулярный механизм опухолевого метастазирования и рецидивирования. Тем не менее, остается еще много вопросов, касающихся фундаментальных механизмов и регуляции ЭМП. Было показано, что многие эмбриональные транскрипционные факторы и пути, регулирующие ЭМП, активируются в моделях опухолей молочной железы и запускают ЭМП в контексте прогрессирования опухоли путем формирования комплексной сигнальной сети, в пределах которой ЭМП регулируется через интеграцию сигналов и контроль обратной связи. Несмотря на недавние достижения, многое остается неизвестным о программе ЭМП при прогрессировании рака и метастазировании, поскольку развитие злокачественной опухоли является сложным и многоступенчатым процессом, а ЭМП представляет собой лишь часть процесса инвазии опухолей и метастазирования. Часто трудно определить, является ли конкретная молекула или исследуемый путь специфическим для программы ЭМП или они работают параллельно с другими программами, такими как выживание и пролиферация клеток. В настоящее время ясно, что ЭМП не только запускается из программы внутри опухолевой клетки, но также зависит от сигналов микроокружения опухоли – внеклеточного матрикса, сосудистой системы, воспалительных клеток и фибробластов, так как специфические виды рака, несомненно, предпочитают конкретные сайты для метастазирования. В отличие от классического ЭМП, происходящего в эмбриональном развитии и представляющего собой относительно постоянное изменение в идентификации и судьбе клеток, маркеры и фенотип ЭМП часто не проявляются в отдаленных метастазах. Механизмы, лежащие в основе экспрессии этих маркеров ЭМП, пока установлены не до конца. Поэтому представляет значительный интерес выявление новых маркеров ЭМП и сигнальных путей, контролирующих процесс ЭМП в раковых заболеваниях человека, что приведет к лучшему пониманию прогрессии рака молочной железы и метастазирования и позволит разработать более эффективные терапевтические стратегии для профилактики метастазирования.
Ключевым фактором в обеспечении процессов ЭМП и последующей диссеминации раковых клеток является снижение уровня экспрессии Е-кадгерина. К настоящему времени известно значительное число транскрипционных репрессоров Е-кадгерина (TWIST1, SLUG, SNAIL1, ZEB1 (TCF8/dEF1), ZEB2 (SIP1), E2-2 (TCF4), E47 (TCF3), семейство FOX), которые часто используются в качестве биомаркеров ЭМП (Imani et al., 2016). Рассматриваемый в настоящем обзоре транскрипционный фактор Zeb1 активно экспрессируется в раковых клетках, расположенных на краю опухоли, – наиболее инвазивной области, и эта экспрессия ассоциирована с пониженным уровнем синтеза Е-кадгерина (Harb et al., 2018). Однако роль Zeb1 в процессах ЭМП не ограничивается лишь подавлением синтеза Е-кадгерина. Zeb1 способен также подавлять транскрипцию генов белков-регуляторов сплайсинга, что способствует, в частности, экспрессии сплайсированных изоформ рецептора фактора роста фибробластов (FGF), которые вызывают ЭМП клеток РМЖ в ответ на TGF-β (Horiguchi et al., 2012).
В сравнении с другими типами РМЖ, экспрессия Zeb1 значительно повышена в случае трижды негативного РМЖ (TN РМЖ) и в опухолях базального типа (Karihtala et al., 2013). Есть данные, что Zeb1 снижает экспрессию факторов полярности клеток, подавляет синтез базальной мембраны и активирует экспрессию матриксных металлопротеаз, таких как MMP-1, MMP-9 и MMP-14, тем самым способствуя ремоделированию базальной мембраны и стимуляции инвазии в окружающие ткани (Brabletz, Brabletz, 2010). В клеточных линиях рака толстой кишки, легкого и молочной железы человека принудительная экспрессия Zeb1 усиливала инвазивные и миграционные способности in vitro и метастазирование in vi-vo. Используя модель рака поджелудочной железы мыши, вызванную опосредованной Pdx1-cre активацией мутантного K-ras и мутантным p53, группа Браблеца недавно продемонстрировала, что Zeb1 является ключевым фактором локальной инвазии, способности к колонизации и отдаленного метастазирования (Krebs et al., 2017). Следует отметить, что подавление ЭМП-TФ Snai1 или Twist1 в той же клеточной модели не влияло на эти процессы, что указывает на важную роль Zeb1 (Zheng et al., 2015). Наконец, вовлечение ЭМП в каскад инвазии–метастазирования подчеркивает динамическую природу данного процесса, приобретение мезенхимальных признаков, усиливающих инвазивные и миграционные способности злокачественных клеток, в то время как для метастатической колонизации требуется мезенхимально-эпителиальный переход (МЭП) (Hugo et al., 2007; Nieto et al., 2016). Такая эпителиально-мезенхимная пластичность подтверждается обнаружением у пациентов с прогрессирующими метастазирующими опухолями циркулирующих опухолевых клеток с гибридным состоянием. Недавно микроскопическими методами была обнаружена пластичность клеток опухолей молочной железы мыши, что свидетельствует о важности приобретения временного мезенхимного состояния для миграции клеток (Beerling et al., 2016).
Zeb1 контролирует экспрессию многих онкогенных и онкосупрессорных микроРНК, в том числе miR-34a. Zeb1 подавляет экспрессию miR-34a для запуска ремоделирования актинового цитоскелета и обеспечения инвазии и метастазирования (Ahn et al., 2012).
Было обнаружено, что Zeb1 и Zeb2 совместно обеспечивают инвазию и миграцию клеток опухоли. В экспериментах in vitro и in vivo показано, что в снижении агрессивности рецидивирующей глиомы более эффективен нокдаун и Zeb1, и Zeb2, нежели только одного из факторов (Suzuki et al., 2018).
Участие Zeb1 в регуляции процессов метастазирования зафиксировано и в случае других злокачественных новообразований. Так, Zeb1 при РМЖ облегчал образование метастазов в костях, запуская экспрессию внеклеточных факторов noggin, follistatin и chordin-like 1, которые инактивируют лиганды активина и костные морфогенетические белки семейства TGF-β (Mock et al., 2015). В свою очередь, при метастатических поражениях лимфатических узлов при плоскоклеточной карциноме матки была отмечена пониженная экспрессия Zeb1, чего не обнаруживалось в первичных опухолях, а у части опухолевых клеток в лимфатических узлах наблюдалось повышение мембранной экспрессии Е-кадгерина (Ma et al., 2015).
Центральный механизм регуляции ЭМП-ТФ основан на петлях отрицательной обратной связи с участием нескольких видов микроРНК (miR) (Bracken et al., 2008; Burk et al., 2008; Jang et al., 2014; Wu et al., 2017; Bryzgunova et al., 2018). Особое значение имеет регуляторный механизм, основанный на взаимной репрессии членов семейства miR-200 и белков Zeb. Zeb1 и Zeb2 регулируют пять членов семейства miR-200, которые образуют два кластера −miR-200b/miR-200a/miR-429 и miR-200c/miR-141, экспрессирующиеся в виде двух полицистронных транскриптов. Экспрессия членов семейства miR-200 приводит к подавлению их мишеней, Zeb1 и Zeb2, что является достаточным условием для восстановления экспрессии гена E-кадгерина, отвечающего за межклеточные контакты. При этом, промоторы, регулирующие транскрипцию кластеров miR-200, в свою очередь, непосредственно подавляются Zeb1 и Zeb2, тем самым формируя петлю двойной обратной связи. Аналогичный механизм работает и для контроля программы ЭМП, управляемой ЭМП-ТФ SNAIL1. SNAIL1 непосредственно подавляется еще одной микроРНК, miR-34, чья собственная транскрипция подавляется SNAIL1 (Siemens et al., 2011).
В предыдущей работе (Поздняков и др., 2019) нами были проанализированы пути регуляции активности фактора Zeb1 на уровне транскрипции, однако посттранскрипционная регуляция была незаслуженно обойдена вниманием; в настоящем обзоре мы восстановим полноту картины.
Посттранскрипционные этапы регуляции Zeb1, альтернативный сплайсинг, микроРНК и длинные некодирующие РНК
Процессы ЭМП регулируются на посттранскрипционном уровне несколькими механизмами, через контроль над сплайсингом и стабильностью молекул РНК. Например, в первичных опухолях и модели клеток РМЖ, наблюдали альтернативный сплайсинг, ассоциированный с ЭМП и опосредованный действием различных классов факторов сплайсинга – RBFOX, MBNL, CELF, hnRNP, ESRP (Shapiro et al., 2011). Изменения экспрессии ЭМП-ТФ, в том числе Zeb1, оказывали значительное влияние на альтернативный сплайсинг в ходе ЕМТ через регуляцию ряда факторов (ESPR, RBM47, QKI) и обеспечение реализации специфической эпителиальной или мезенхимной программы сплайсинга (Yang et al., 2016). Недавние исследования (Li et al., 2018b) показали функциональное значение некоторых из таких альтернативно сплайсированных изоформ. Эктопическая экспрессия Zeb1 в клетках HLMER индуцировала экспрессию РНК-связывающих белков RBFOX1 и QKI, которые способствуют приобретению мезенхимного фенотипа посредством регуляции актин-связывающего белка филамина B (FL-NB) (Li et al., 2018b). Следует отметить, что такие изменения в пути альтернативного сплайсинга имеют существенное значение для опухолевых клеток, поскольку альтернативная изоформа филамина B способна индуцировать ЭМП, высвобождая ТФ FOXC1 из ингибирующего комплекса (Li et al., 2018b).
Механизмы создания разнообразия белков могут также затрагивать регуляцию времени жизни белка через стабилизацию 3'-нетранслируемой области его мРНК. В регуляции стабильности мРНК Zeb1 участвуют различные РНК-связывающие белки, в их числе hnRNP D и PTBP3, которые, как следует из результатов недавних исследований, повышают стабильность транскрипта Zeb1 (Al-Khalaf, Aboussekhra, 2014; Li et al., 2016a; Hou et al., 2018). Экспрессия и функции РНК-связывающих белков могут регулироваться несколькими способами, оказывая влияние на экспрессию мРНК фактора Zeb1 и индукцию ЕМТ. Например, AUF1 (hnRNP D) является мишенью beclin1 и двух опухолевых супрессорных микроРНК, miR-141 и miR-146b-5p (Al-Khalaf, Aboussekhra, 2014; Li et al., 2016a).
Для процессов ЭМП характерны нарушения экспрессии микроРНК (Riaz et al., 2013), а специфический набор микроРНК связан с различными эпителиальными и мезенхимными признаками опухолевых клеточных моделей (Zhang, Ma, 2012). Современные подходы к секвенированию микроРНК привели к открытию новых молекул, регулирующих ЭМП как in vitro, так и in vivo. Одной из них является miR-1199-5p (Diepenbruck et al., 2017), для которой показана способность к дестабилизации транскрипта Zeb1 через связывание со специфическими сайтами на его 3'-НТР, что приводит, тем самым, к снижению экспрессии Zeb1. Еще одним примером микроРНК, участвующей в регуляции экспрессии Zeb, является miR-448. Уровень ее экспрессии в клетках РМЖ снижен, но искусственное его повышение заметно подавляло пролиферацию, миграцию и инвазию раковых клеток. Выяснилось, что miR-448 способна снижать уровень Zeb1/2, напрямую взаимодействуя с 3'-НТР мРНК данных транскрипционных факторов (Ma et al., 2018).
В последнее время в качестве эпигенетического репрессора функций Zeb1 используется ингибитор HDAC1, моцетиностат, запускающий экспрессию miR-203, что возобновляет чувствительность к химиотерапии и может стать эффективной стратегией борьбы с раком (Meidhof et al., 2015).
Как уже упоминалось выше, в основе регуляции ЭМП-ТФ лежат петли отрицательной обратной связи, в которых задействован ряд микро-РНК (Bracken et al., 2008; Burk et al., 2008; Jang et al., 2014; Wu et al., 2017; Bryzgunova et al., 2018), под контролем сигнального пути TGF-β и ряда других внешних стимулов (Lu et al., 2013; Tian et al., 2013). Были предложены и экспериментально подтверждены две различные модели взаимного регулирования. Так, было показано, что молекулы miR-34 и miR-200 функционируют совместно для управления ЭМП (Tian et al., 2014). Для описываемой петли обратной связи возможны три стабильных состояния с разным соотношением Zeb1/miR-200: с эпителиальным (высокий miR-200, низкий Zeb), мезенхимным (низкий miR-200, высокий Zeb) и гибридным (средний miR-200, средний Zeb) фенотипами (Lu et al., 2013).
В ответ на внешние стимулы, например, TGF-β, или под воздействием другой микроРНК, может происходить изменение соотношения miR-200/Zeb1 и статуса ЭМП. Например, в опухолях молочной железы miR-22 индуцирует ЭМП и способствует приобретению черт стволовости, путем подавления экспрессии miR-200a и miR-200c, что приводит к повышению уровня Zeb1. Модуляция соотношения mi-R-200/Zeb1, miR22, вносит существенный вклад в метастазирование опухоли, а оверэкспрессия miR22 коррелирует с плохим клиническим исходом у пациентов (Song et al., 2013).
Представители семейства молекул miR-200 способны связываться с 3'-НТР мРНК Zeb1, образуя двойную петлю отрицательной обратной связи, которая регулирует экспрессию miR-200 и Zeb1 (Nishijima et al., 2016).
Еще одним примером регуляции Zeb1 через miR-200 является редактирование РНК аденозиндеаминазой ADAR. Так, было обнаружено, что отредактированная микро-РНК miR-200b стимулирует клеточную инвазию и миграцию, главным образом, благодаря своей ослабленной способности к подавлению Zeb1/Zeb2, вместо этого негативно воздействуя на другую группу мишеней – рецептор ингибиторующего фактора лейкемии (LIFR) (Wang et al., 2017b).
Важнейшая роль Zeb1 в контроле пролиферации и выживания клеток реализуется путем взаимной конкуренции между Zeb1 и белками семейства р53. Zeb1 вовлечен в регуляцию транскрипции изоформ p63 и p73, в то время как р53 контролирует уровень данного ЭМП-ТФ путем активации транскрипции кластеров miR-200c/141 и miR-200a/miR200b/429 (Fontemaggi et al., 2005). Было показано, что уровни экспрессии таких ЭМП-ТФ, как SNAI1, SNAI2, TGF-B1, и TGF-B2 не изменялись при нокауте гена TP53, в отличие от Zeb1/2, уровень экспрессии которого значительно возрастал при нокдауне или делеции р53. Одновременное подавление и Zeb1, и Zeb2 значительно снижало мезенхимальные черты и способность к диссеминации раковых клеток, однако при подавлении только одного из них, серьезных изменений не наблюдалось. Таким образом, предполагается, что оба ЭМП-ТФ, и Zeb1, и Zeb2, играют ключевую роль в р53-зависимой регуляции ЭМП (Kim et al., 2011).
В то время как p53 дикого типа регулирует ЭМП с помощью микроРНК, подавляющих экспрессию Zeb1 и Zeb2 (Kim et al., 2011), на модели клеток рака эндометрия было показано, что мутантный p53 непосредственно связывается с геном miR-130b, которая является специфичным ингибитором фактора Zeb1, и предотвращает его активацию, что приводит к повышению уровня Zeb1 и последующей активации ингибиторов экспрессии E-кадгерина – Snail и BMI-1 (Dong et al., 2013).
Еще одним примером негативной регуляции Zeb1 является miR-33b, однако и тут имеет место петля обратной связи: было показано, что Zeb1 подавляет экспрессию miR-33b для обеспечения процессов инвазии и метастазирования в клетках рака легкого (Qu et al., 2015). К числу негативных регуляторов Zeb1 следует отнести и miR-101, инактивация транскрипции которой запускала MEK1/2-ERK2-Zeb1 сигнальный путь, активируемый ЭФР, что способствовало прохождению ЭМП и миграции клеток (Chandra Mangalhara et al., 2017).
Также недавно было обнаружено, что лизофосфатидная кислота 1 (LPA1) запускает в клетках РМЖ базального типа экспрессию онко-miR-21 через активацию PI3K/Zeb1 сигнального пути. Высокий уровень экспрессии miR-21 обеспечивает пролиферацию и инвазию многих типов раковых клеток (Sahay et al., 2015). В Т-клеточной лимфоме кожи Zeb1 не способен взаимодействовать с собственным сайтом связывания в промоторе гена интерлейкина 15 (IL-15) из-за чрезмерного его метилирования, что приводило к повышенной транскрипции IL-15, и избыточная его секреция в Т-клетках подавляла работу отрицательной ауторегуляторной петли, опосредованной HDAC1. Затем происходила активация HDAC1 и HDAC6 с последующей индукцией транскрипции onco-miR-21 (Mishra et al., 2016).
В другом исследовании было показано, что высокий уровень miR-27a подавляет экспрессию компонента Е3 убиквитин-лигазного комплекса Fbxo45, путем связывания с 3'-НТР его мРНК, тем самым нарушая формирование Skp1-Pam-Fbxo45 атипичного E3 убиквитин-лигазного комплекса, что в конечном счете приводило к стабилизации Zeb1 (Xu et al., 2015).
Как следует из результатов недавних исследований, в качестве механизма, позволяющего избежать регуляции Zeb1 c помощью микроРНК, могут выступать мутации или модификации в 3'-НТР мРНК Zeb1, затрагивающие сайты связывания микроРНК. Например, в клетках аденокарциномы протока поджелудочной железы (PDAC), обработанных гемцитабином, наблюдалось прогрессирующее укорочение транскрипта Zeb1, таким образом, из-за выбора альтернативных сигналов полиаденилирования, это приводило к избирательной делеции сайтов связывания репрессорных микроРНК (Passacantilli et al., 2017).
Недавно был открыт новый класс молекул, способных выступать в качестве потенциальных регуляторов ЭМП, – длинные некодирующие РНК (днРНК, lncRNA). Они представляют собой транскрипты длиной свыше 200 нуклеотидов, обычно транскрибируемые РНК-полимеразой II и не кодирующие белковых молекул. Претерпеваемый ими процессинг с полиаденилированием и сплайсинг приводит к появлению изоформ данных молекул. ДнРНК выступают в качестве регуляторов эпигенетических модификаций, а также на уровне транскрипции и пост-транскрипционных этапах. Накопленные к настоящему времени данные говорят о вовлечении ряда днРНК в канцерогенез, в частности, было показано, что в опухолях молочной железы некоторые днРНК играют важную роль в процессах роста опухоли и метастазирования in vivo (Wang et al., 2018b). В частности, при индукции ЭМП внешними стимулами, такими как TGF-β, или принудительной экспрессией ЭМП-ТФ, задействуются различные днРНК (Yuan et al., 2014; Liao et al., 2017). В основных механизмах регуляции экспрессии генов используется взаимодействие с эпигенетическим комплексом PRC2, а также способность днРНК выступать в качестве конкурирующих эндогенных-РНК (ceRNA) для микроРНК, регулирующих ЭМП (Xu et al., 2016). Исследования последних лет показали, что некоторые хорошо охарактеризованные днРНК оказывают влияние на уровень Zeb1, действуя как конкурирующие эндогенные РНК и снижая активность молекул микроРНК (Li et al., 2016b; Chen et al., 2017; Shen et al., 2017; Wang et al., 2018b; Yang et al., 2018). Например, днРНК XIST и HULC способны регулировать уровень Zeb1 опосредованно, через подавление miR-200b-3p (Li et al., 2016b; Chen et al., 2017). ДнРНК подавляет экспрессию двух микроРНК семейства miR200 (miR-200a/b), что приводит к активации Zeb1 и запуску ЭМП (Chen et al., 2017). Аналогично, в другой работе описана регуляция Zeb1 через связывание miR-574-5p длинной некодирующей РНК linc-ZNF469-3 в клетках TN РМЖ, где последняя может выступать в качестве прогностического маркера метастазирования в легкие (Wang et al., 2018b). В клетках гепатоцеллюлярной карциномы показано, что днРНК HOTAIR связывает miR-23b-3p, предотвращая ее взаимодействие с Zeb1, тем самым, участвуя в запуске ЭМП (Yang et al., 2018). Во всех этих случаях рост и метастатический потенциал опухоли в значительной степени связан с экспрессией данных днРНК, посредством их участия в регуляции Zeb1.
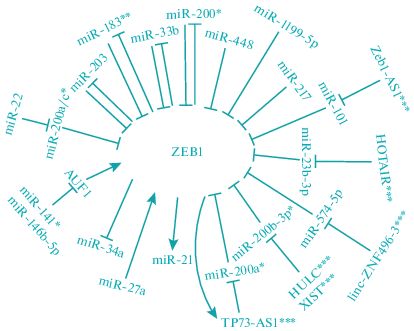
Длинная некодирующая РНК Zeb1 antisense 1 (ZEB1-AS1), которая образуется из промоторной области гена Zeb1, может способствовать запуску ЭМП, путем регуляции уровня Zeb1 (Su et al., 2017; Xiong et al., 2018). В клетках колоректального рака, ZEB1-AS1 связывает miR-101, которая в норме напрямую взаимодействует с мРНК Zeb1, и блокирует ее экспрессию (Xiong et al., 2018). В колоректальных опухолях, по сравнению с нормальной тканью толстой кишки, наблюдался более высокий уровень ZEB1-AS1, который также коррелировал с более поздней стадией развития опухоли. Подавление ZEB1-AS1 в сочетании с оверэкспрессией miR-101 значительно снижало пролиферацию и миграцию клеток колоректального рака (Xiong et al., 2018). Было предложено несколько механизмов регулирования Zeb1 молекулой ZEB1-AS1. Например, на моделях рака простаты показано, что ZEB1-AS1 связывалась с H3K4 метилтрансферазой MLL1 и способствовала метилированию лизина 4 гистона 3 (H3K4me3) в области промотора Zeb1, что приводило к переходу хроматина из неактивной формы в активную (Su et al., 2017). Схема, иллюстрирующая известные к настоящему времени регуляторные взаимодействия транскрипционного фактора Zeb1 с молекулами микроРНК и днРНК, приведена на рис. 1.
Рис. 1.
Спектр регуляторных взаимодействий транскрипционного фактора Zeb1 с молекулами микроРНК и длинными некодирующими РНК. Многие микроРНК способны регулировать уровень экспрессии фактора Zeb1, напрямую или опосредованно, в том числе, выступая в качестве конкурирующих эндогенных РНК, взаимодействуя с различными ТФ или днРНК. Zeb1, в свою очередь, также оказывает влияние на те или иные микроРНК. *Члены семейства miR-200, к которому относятся кластеры miR-200b/miR-200a/miR-429 и miR-200c/miR-141 и др.; **кластер miR-183/miR-96/miR-182; ***длинные некодирующие молекулы РНК. Фигурами ↑ и ⊤ обозначены, соответственно, позитивная и негативная регуляция активности генов и микроРНК.
РЕГУЛЯЦИЯ Zeb1 НА УРОВНЕ ТРАНСЛЯЦИИ И ПОСТТРАНСЛЯЦИОННЫХ МОДИФИКАЦИЙ
Активность и скорость деградации короткоживущих регуляторных белков в клетке контролируется с помощью ковалентных посттрансляционных модификаций (ПТМ). Для транскрипционного фактора Zeb1 экспериментально обнаружены такие модификации, как фосфорилирование, убиквитилирование и сумоилирование.
Фосфорилирование может как повышать, так и подавлять активность Zeb1, оказывая влияние на стабильность данного белка или его ядерную локализацию. Например, было показано, что фосфорилирование фактора Zeb1 влияет на некоторые его свойства и локализацию (Costantino et al., 2002), а именно, при дефосфорилировании нативного Zeb1 наблюдалось повышение его ДНК-связывающей активности. Обнаружено, что в регуляции фосфорилирования Zeb1 участвуют различные сигнальные пути, а обработка опухолевых клеток специфическими ингибиторами киназ MEK/ERK, PKC или PI3K приводила к усилению связывания Zeb1 с ДНК (Llorens et al., 2016). Фосфорилирование аминокислотной последовательности Zeb1 по различным сайтам по-разному влияло на активность белка Zeb1. При воздействии ионизирующего излучения на клетки РМЖ, в устойчивых к радиации клетках происходила активация серин/треониновой протеинкиназы ATM, которая стабилизировала Zeb1 посредством фосфорилирования по остатку серина 585. Zeb1, в свою очередь, способен к взаимодействию с убиквитин-специфической протеазой 7 (USP7), которая деубиквитилирует и стабилизирует серин/треониновую киназу CHK1, способствуя, тем самым, репарации ДНК и устойчивости к воздействию ионизирующего излучения (Zhang et al., 2014).
Подавляющее большинство внутриклеточных белков утилизируется посредством протеасомной деградации (Konstantinova et al., 2008). Для протеолиза в 26S протеасомах требуется предварительная модификация белка-мишени полиубиквитиновой меткой по остаткам лизина. Не являются исключением и ЭМП-ТФ, к коим принадлежит белок Zeb1: регуляция их стабильности осуществляется посредством убиквитилирования, что вносит вклад в активацию или подавление ЭМП (Díaz et al., 2014). В литературных источниках описана регуляция уровня Zeb1 убиквитилированием (Xu et al., 2015; Chen et al., 2015; Wang et al., 2017а). Например, атипичная E3 убиквитин-лигаза Skp1-Pam-Fbxo45 (SPFFbxo45) опосредует деградацию различных ЭМП-ТФ, в числе которых Zeb1, Zeb2, Snail, Slug и Twist (Xu et al., 2015). На клеточных моделях рака молочной железы продемонстрировано, что регуляция ЭМП путем убиквитилирования Zeb1 осуществляется семейством E3 убиквитин-лигаз Siah. Экспрессия белка Siah снижалась при обработке клеток TGF-β, в то время как его нокдаун активировал экспрессию мезенхимальных генов (Chen et al., 2015). Аналогично, в клетках глиобластомы, убиквитилирование Zeb1 E3 убиквитин-лигазой PARK2 приводило к подавлению его экспрессии, тем самым, препятствуя метастазированию (Wang et al., 2017а). Ожидаемо, обратный эффект наблюдался при действии деубиквитилирующих ферментов: их активность повышала стабильность Zeb1. Убиквитин-специфическая протеаза 51 (USP51), связываясь с Zeb1, стабилизировала данный белок, удаляя полиубиквитиновую метку. В опухолях молочной железы человека отмечена повышенная экспрессия USP51, коррелирующая с пониженным уровнем выживаемости пациентов. При эктопической экспрессии USP51 in vitro наблюдалось повышение уровня Zeb1 и таких мезенхимных маркеров, как N-кадгерин и виментин (Zhou et al., 2017).
Наряду с приведенными выше примерами регуляции экспрессии Zeb1 напрямую, в литературе описан белок FLASH (он же – каспаза-8-ассоциированный белок 2, CASP8AP2), который защищает ЭМП-ТФ Zeb1 от убиквитилирования Е3 лигазами SIAH1 и FBXO45 и последующей неизбежной деградации по убиквитин-протеасомному пути. В отсутствие FLASH происходит снижение уровня белка Zeb1 (но не экспрессии мРНК Zeb1), в результате чего растет экспрессия Е-кадгерина. В клетках PANC-1, не экспрессирующих FLASH, после воздействия TGB-β уровень Zeb1 не повышается, и клетки сохраняют эпителиальный фенотип (Abshire et al., 2016). Это означает, что предотвращение протеасомной деградации Zeb1 необходимо для запуска ЭМП после активации сигнальных путей.
Помимо деградации по убиквитин-зависимому пути, реализуемой через 26S протеасомы, многие внутриклеточные белки могут претерпевать и убиквитин-независимый протеолиз (Erales, Coffino, 2014), осуществляемый без участия 19S регуляторных комплексов протеасом, тем более, что по последним данным, лишь четверть коровых протеасомных комплексов ассоциирована с РА700, в то время как остальные 20S протеасомы находятся в клетке либо в свободном виде, либо в ассоциации с PA28 или РА200 регуляторами (Fabre et al., 2013). Именно такие протеасомные комплексы и осуществляют убиквитин-независимый протеолиз. Мишенями для данного альтернативного способа утилизации белков могут выступать участки внутренней неупорядоченности в аминокислотной последовательности (Baugh et al., 2009; Erales, Coffino, 2014; Mittenberg et al., 2018). В недавних исследованиях обнаружено, что ЭМП-ТФ Zeb1 содержит такие участки в своей последовательности (Mooney et al., 2016), что, наряду с обнаружением в интерактоме ЭМП-ТФ Zeb1 в клетках карциномы молочной железы субъединиц 11S (РА28) регуляторного комплекса протеасом, дает основания рассматривать Zeb1 в качестве мишени и для убиквитин-независимого протеолиза.
ЗАКЛЮЧЕНИЕ. Zeb1, микро-РНК, АНГИОГЕНЕЗ И СТВОЛОВЫЕ КЛЕТКИ ОПУХОЛИ
По мере роста, клетки опухоли исчерпывают запасы кислорода и питательных веществ микроокружения, что вызывает стресс, каноническим ответом на который является усиление ангиогенеза, обеспечивающего перестройки сосудистой сети (Weis, Cheresh, 2011). В то время как зарождающиеся опухолевые сосуды часто дезорганизованы и малофункциональны, они, тем не менее, позволяют пополнять запасы кислорода и питательных веществ, необходимых для обеспечения роста опухоли, а также являются путем, по которому опухолевые клетки могут метастазировать и расселяться (Folkman, 2002). Для терапии опухолей были разработаны ингибиторы ангиогенеза, но они лишь незначительно влияли на уровень выживаемости пациентов со многими типами злокачественных новообразований, включая рак молочной железы (Quesada et al., 2010). Это связано, по крайней мере, отчасти, с дефицитом питательных веществ и кислорода, возникшим вследствие такой терапии. Например, повышенная гипоксия, вызванная антиангиогенными препаратами, может вызывать запуск процессов ЭМП (Yang et al., 2008; Conley et al., 2012; Zhang et al., 2013). Последующие исследования показали, что данный вид терапии может приводить к усилению альтернативных путей васкуляризации, включающих приобретение раковыми клетками эндотелиального фенотипа посредством васкулярной мимикрии (процесса формирования сосудистоподобных структур без эндотелиальной выстилки) и (или) эндотелиальной трансдифференцировки (Soda et al., 2011; Xu et al., 2012). Zeb1 участвует в регуляции процессов васкулярной мимикрии (ВМ) через взаимодействия с молекулами микроРНК. Повышенная экспрессия кластеров miR-200c/miR-141 и miR-183/miR-96/miR-182 может приводить к снижению синтеза Zeb1, фибронектина и SEC23A, а также повышению экспрессии Е-кадгерина. Zeb1 участвует в подавлении экспрессии данных кластеров микроРНК и обеспечивает компенсацию дефицита кислорода и питательных веществ через ВМ (Langer et al., 2018). Способность Zeb1 к формированию ВМ и запуску ЭМП была также продемонстрирована на клетках колоректального рака (de Barrios et al., 2017), рака мочевого пузыря (Li et al., 2018a) и рака простаты (Wang et al., 2018a).
Во многих исследованиях отмечено образование в процессе ЭМП популяции стволовых клеток опухоли, для которых характерны два основных свойства: способность к самовоспроизведению и потенциал к восстановлению фенотипической гетерогенности родительской опухоли (Badve, Nakshatri, 2012). Данный тип клеток, который, как предполагается, поддерживает рост опухоли и обеспечивает процессы распространения опухолевых клеток и образования метастазов в новых сайтах, представляет собой минорную фракцию от всей популяции раковых клеток, однако связь между ЭМП и образованием стволовых клеток опухоли подразумевает, что последние могут быть образованы de novo из обычной опухолевой клетки, что демонстрирует невероятный уровень пластичности раковых клеток в отдельных опухолях. Zeb1 играет важнейшую роль в процессах динамичного взаимопревращения стволовых и нестволовых клеток опухоли. Как иллюстрация, на модели базального типа РМЖ было показано, что, в ответ на стимулы микроокружения, такие как TGF-β, Zeb1 способствует превращению нестволовых клеток опухоли в стволовые (Chaffer et al., 2013). На молекулярном уровне это превращение происходит через опосредованное Zeb1 подавление экспрессии микроРНК (miR-200, а также miR-183 и miR-203), мишенью которых являются белок BMI1 и, возможно, другие факторы, ассоциированные со “стволовостью”, такие как SOX2 и KLF4 (Wellner et al., 2009). В отсутствие р53 дикого типа происходит значительное увеличение популяции CD24–CD44+ стволовых клеток опухоли и повышается экспрессия BMI1. В свою очередь, экспрессия р53 блокирует рост популяции стволовых клеток опухоли, индуцированный TGF-β, т.е. р53 подавляет образование стволовых клеток опухоли, ассоциированных с ЭМП. Если в клетках, в которых по тем или иным причинам не происходит синтез р53, индуцировать экспрессию miR-200с, то уровень факторов Zeb1, KLF4 и BMI1 значительно снижается, в то время как экспрессия Е-кадгерина, синтез которого был подавлен вследствие нокдауна р53, усиливается. Реэкспрессия miR-200с также значительно уменьшает процент CD24–CD44+ стволовых клеток опухоли. Это указывает на то, что р53 модулирует ЭМП и образование стволовых клеток опухоли путем регуляции miR-200с. Есть данные, что р53 напрямую активирует экспрессию miR-200с, связываясь с ее промотором (Chang et al., 2011).
Недавние исследования продемонстрировали, что экспрессия miR-200с приводит к подавлению способности нормальных стволовых клеток молочной железы к формированию протоков, а также способности к формированию опухоли за счет СКО. Уровень экспрессии кластеров miR-200c/miR-141, miR-200b/miR-200a/miR-429 и miR-183/miR-96/miR-182 снижен в нормальных стволовых клетках молочной железы, в стволовых клетках РМЖ и в клетках эмбриональной карциномы, поскольку данные микроРНК предотвращают генерацию стволовых клеток. И, как уже отмечалось ранее, miR-200с регулирует экспрессию BMI1 (Shimono et al., 2009). BMI1 в стволовых клетках подавляет сигнальные пути программированной клеточной гибели, старения и дифференцировки (Park et al., 2003); так, у мышей с нокаутом BMI1 нарушены самовоспроизведение и пролиферация гематопоэтических стволовых клеток, нормальных стволовых клеток молочной железы, нейронных стволовых клеток (Molofsky et al., 2003; Park et al., 2003; Pietersen et al., 2008).
Важно подчеркнуть, что хотя экспрессия Zeb1 в ответ на сигналы микроокружения является инструментом обеспечения пластичности раковых клеток, ЭМП также наблюдается и в нормальных стволовых клетках молочной железы, где Zeb1 обеспечивает запуск антиоксидантных программ через метионинсульфоксидредуктазу MSRB3, что защищает клетки от окислительного стресса, в норме возникающего при неправильной митогенной активации (Morel et al., 2017). Как следствие, экспрессия Zeb1 предотвращает активацию р53-зависимого ответа на повреждения ДНК и последующую индукцию апоптоза и преждевременного старения, двух важнейших барьеров на пути опухолеобразования. Поскольку двунитевые разрывы ДНК, генерируемые после онкогенной активации, являются основной причиной нестабильности генома, эндогенная экспрессия Zeb1 обеспечивает поддержание геномной стабильности на всем протяжении развития опухоли (Halazonetis et al., 2008).
Таким образом, данные о транскрипционном факторе Zeb1 и различных путях регуляции его активности, используемых опухолевыми клетками в процессах поддержания эпителиально-мезенхимной пластичности и метастазирования, а также исследование биологических функций микроРНК, позволят разработать новые стратегии в диагностике, прогнозировании и лечении злокачественных новообразований человека, в том числе и рака молочной железы. В последние несколько лет уже проводятся клинические испытания возможности использования профилирования микроРНК для прогнозирования развития опухоли и клинического ответа у конкретных пациентов, а первые препараты на основе микроРНК уже применяются в клинической практике для лечения рака.
Список литературы
Гервас П.А., Молоков А.Ю., Панферова Е.В., Писарева Л.Ф., Чердынцева Н.В. 2019. Этнические аспекты наследственного рака молочной железы. Сибирский онкологический журнал. Т. 18. № 2. С. 102. (Gervas P.A., Molokov A.Yu., Panpherova E.V., Pisareva L.Ph., Cherdyntseva N.V. 2019. Ethnic aspects of hereditary breast cancer. Siberian J. Oncology. V. 18. № 2. P. 102.)
Поздняков Д.Ю., Шувалов О.Ю., Барлев Н.А., Миттенберг А.Г. 2019. Транскрипционный фактор Zeb1 и его роль в процессах метастазирования и онкогенеза. Цитология. Т. 61. № 11. С. 915. (Pozdnyakov D.Y., Shuvalov O.Y., Barlev N.A., Mittenberg A.G. 2019. Role of Zeb1 ЕМТ-TF in metastasis and carcinogenesis. Tsitologiya. V. 61. № 11. P. 915.)
Abshire C.F., Carroll J.L., Dragoi A.M. 2016. FLASH protects ZEB1 from degradation and supports cancer cells epithelial-to-mesenchymal transition. Oncogenesis. V. 5. P. e254.
Ahn Y.H., Gibbons D.L., Chakravarti D., Creighton C.J., Rizvi Z.H., Adams H.P., Pertsemlidis A., Gregory P.A., Wright J.A., Goodall G.J., Flores E.R., Kurie J.M. 2012. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Invest. V. 122. P. 3170.
Al-Khalaf H.H., Aboussekhra A. 2014. MicroRNA-141 and microRNA-146b-5p inhibit the prometastatic mesenchymal characteristics through the RNA-binding protein AUF1 targeting the transcription factor ZEB1 and the protein kinase AKT. J. Biol. Chem. V. 289. P. 31433.
Badve S., Nakshatri H. 2012. Breast-cancer stem cells-beyond semantics. Lancet Oncol. V. 13. P. e43.
Baugh J.M., Viktorova E.G., Pilipenko E.V. 2009. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. V. 386. P. 814.
Beerling E., Seinstra D., de Wit E., Kester L., van der Velden D., Maynard C., Schäfer R., van Diest P., Voest E., van Oudenaarden A., Vrisekoop N., van Rheenen J. 2016. Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis-enhancing stem cell capacity. Cell Rep. V. 14. P. 2281.
Brabletz S., Brabletz T. 2010.The ZEB/miR-200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. V. 11. P. 670.
Bracken C.P., Gregory P.A., Kolesnikoff N., Bert A.G., Wang J., Shannon M.F., Goodall G.J. 2008. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. V. 68. P. 7846.
Bryzgunova O.E., Konoshenko M.Y., Laktionov P.P. 2018. MicroRNA-guided gene expression in prostate cancer: Literature and database overview. J. Gene. Med. V. 20 : e3016.
Burk U., Schubert J., Wellner U., Schmalhofer O., Vincan E., Spaderna S., Brabletz T. 2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. V. 9. P. 582.
Chaffer C.L., Marjanovic N.D., Lee T., Bell G., Kleer C.G., Reinhardt F., D’Alessio A.C., Young R.A., Weinberg R.A. 2013. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. V. 154. P. 61.
Chandra Mangalhara K., Manvati S., Saini S.K., Ponnusamy K., Agarwal G., Abraham S.K., Bamezai R.N.K. 2017. ERK2-ZEB1-miR-101-1 axis contributes to epithelial-mesenchymal transition and cell migration in cancer. Cancer Lett. V. 391. P. 59.
Chang C.J., Chao C.H., Xia W., Yang J.Y., Xiong Y., Li C.W., Yu W.H., Rehman S.K., Hsu J.L., Lee H.H., Liu M., Chen C.T., Yu D., Hung M.C. 2011. p53 regulates epithelial–mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. V. 13. P. 317.
Chen A., Wong C.S., Liu M.C., House C.M., Sceneay J., Bowtell D.D., Thompson E.W., Möller A. 2015. The ubiquitin ligase Siah is a novel regulator of Zeb1 in breast cancer. Oncotarget. V. 6. P. 862.
Chen D.L., Chen L.Z., Lu Y.X., Zhang D.S., Zeng Z.L., Pan Z.Z., Huang P., Wang F.H., Li Y.H., Ju H.Q., Xu R.H. 2017. Long noncoding RNA XIST expedites metastasis and modulates epithelial-mesenchymal transition in colorectal cancer. Cell Death Dis. V. 8 : e3011.
Conley S.J., Gheordunescu E., Kakarala P., Newman B., Korkaya H., Heath A.N., Clouthier S.G., Wicha M.S. 2012. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA. V. 109. P. 2784.
Costantino M.E., Stearman R.P., Smith G.E., Darling D.S. 2002. Cell-specific phosphorylation of Zfhep transcription factor. Biochem. Biophys. Res. Commun. V. 296. P. 368.
de Barrios O., Győrffy B., Fernández-Aceñero M.J., Sánchez-Tilló E., Sánchez-Moral L., Siles L., Esteve-Arenys A., Roué G., Casal J.I., Darling D.S., Castells A., Postigo A. 2017. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut. V. 66. P. 666.
Díaz V.M., Viñas-Castells R., García de Herreros A. 2014. Regulation of the protein stability of EMT transcription factors. Cell Adh. Migr. V. 8. P. 418.
Diepenbruck M., Tiede S., Saxena M., Ivanek R., Kalathur R.K.R., Lüönd F., Meyer-Schaller N., Christofori G. 2017. miR-1199-5p and Zeb1 function in a doublenegative feedback loop potentially coordinating EMT and tumour metastasis. Nat. Commun. V. 8. P. 1168.
Dong P., Karaayvaz M., Jia N., Kaneuchi M., Hamada J., Watari H., Sudo S., Ju J., Sakuragi N. 2013. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. V. 32. P. 3286.
Erales J., Coffino P. 2014. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta. V. 1843. P. 216.
Fabre B., Lambour T., Delobel J., Amalric F., Monsarrat B., Burlet-Schiltz O., Bousquet-Dubouch M.P. 2013. Subcellular distribution and dynamics of active proteasome complexes unraveled by a workflow combining in vivo complex cross-linking and quantitative proteomics. Mol. Cell. Proteomics. V. 12. P. 687.
Folkman J. 2002. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. V. 29. Suppl 16. P. 15.
Fontemaggi G., Gurtner A., Damalas A., Costanzo A., Higashi Y., Sacchi A., Strano S., Piaggio G., Blandino G. 2005. deltaEF1 repressor controls selectively p53 family members during differentiation. Oncogene. V. 24. P. 7273.
Halazonetis T.D., Gorgoulis V.G., Bartek J. 2008. An oncogene-induced DNA damage model for cancer development. Science. V. 319. P. 1352.
Harb O.A., Elfeky M.A., El Shafaay B.S., Taha H.F., Osman G., Harera I.S., Gertallah L.M., Abdelmonem D.M., Embaby A. 2018. SPOP, ZEB-1 and E-cadherin expression in clear cell renal cell carcinoma (cc-RCC): clinicopathological and prognostic significance. Pathophysiology. V. 25. P. 335.
Horiguchi K., Sakamoto K., Koinuma D., Semba K., Inoue A., Inoue S., Saitoh M. 2012. TGF-β drives epithelial-mesenchymal transition through ΔEF1-mediated downregulation of ESRP. Oncogene. V. 31. P. 3190.
Hou P., Li L., Chen F., Chen Y., Liu H., Li J., Bai J., Zheng J. 2018. PTBP3-mediated regulation of ZEB1 mRNA stability promotes epithelial-mesenchymal transition in breast cancer. Cancer Res. V. 78. P. 387.
Hugo H., Ackland M.L., Blick T., Lawrence M.G., Clements J.A., Williams E.D., Thompson E.W. 2007. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J. Cell. Physiol. V. 213. P. 374.
Imani S., Hosseinifard H., Cheng J., Wei C., Fu J. 2016. Prognostic value of EMT-inducing transcription factors (EMT-TFs) in metastatic breast cancer: A systematic review and meta-analysis. Sci. Rep. V. 6 : 28587.
Jang S.G., Yoo C.W., Park S.Y., Kang S., Kim H.K. 2014. Low expression of miR-449 in gynecologic clear cell carcinoma. Int. J. Gynecol. Cancer. V. 24. P. 1558.
Karihtala P., Auvinen P., Kauppila S., Haapasaari K.M., Jukkola-Vuorinen A., Soini Y. 2013. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res. Treat. V. 138. P. 81.
Kim T., Veronese A., Pichiorri F., Lee T.J., Jeon Y.J., Volinia S., Pineau P., Marchio A., Palatini J., Suh S.S., Alder H., Liu C.G., Dejean A, Croce C.M. 2011. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. V. 208. P. 875.
Konstantinova I.M., Tsimokha A.S., Mittenberg A.G. 2008. Role of proteasomes in cellular regulation. Int. Rev. Cell Mol. Biol. V. 267. P. 59.
Krebs A.M., Mitschke J., Lasierra Losada M., Schmalhofer O., Boerries M., Busch H., Boettcher M., Mougiakakos D., Reichardt W., Bronsert P., Brunton V.G., Pilarsky C., Winkler T.H., Brabletz S., Stemmler M.P., Brabletz T. 2017. The EMT-activator ZEB1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. V. 19. P. 518.
Langer E.M., Kendsersky N.D., Daniel C.J., Kuziel G.M., Pelz C., Murphy K.M., Capecchi M.R., Sears R.C. 2018. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene. V. 37. P. 1005.
Li B., Mao X., Wang H., Su G., Mo C., Cao K., Qiu S. 2018a. Vasculogenic mimicry in bladder cancer and its association with the aberrant expression of ZEB1. Oncol. Lett. V. 15. P. 5193.
Li J., Choi P.S., Chaffer C.L., Labella K., Hwang J.H., Giacomelli A.O., Kim J.W., Ilic N., Doench J.G., Ly S.H., Dai C., Hagel K., Hong A.L., Gjoerup O., Goel S., et al. 2018b. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife. V. 7 : e37184.
Li S., Zhang H.Y., Li C., Du Z.X., An M.X., Zong Z.H., Liu B.Q., Wang H.Q. 2016a. Induction of epithelial-mesenchymal transition (EMT) by Beclin 1 knockdown via posttranscriptional upregulation of ZEB1 in thyroid cancer cells. Oncotarget. V. 7. P. 70364.
Li S.P., Xu H.X., Yu Y., He J.D., Wang Z., Xu Y.J., Wang C.Y., Zhang H.M., Zhang R.X., Zhang J.J., Yao Z., Shen Z.Y. 2016b. LncRNA HULC enhances epithelialmesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget. V. 7. P. 42431.
Liao J.Y., Wu J., Wang Y.J., He J.H., Deng W.X., Hu K., Zhang Y.C., Zhang Y., Yan H., Wang D.L., Liu Q., Zeng M.S., Phillip Koeffler H., Song E., Yin D. 2017. Deep sequencing reveals a global reprogramming of lncRNA transcriptome during EMT. Biochem. Biophys. Acta. V. 1864. P. 1703.
Llorens M.C., Lorenzatti G., Cavallo N.L., Vaglienti M.V., Perrone A.P., Carenbauer A.L., Darling D.S., Cabanillas A.M. 2016. Phosphorylation regulates functions of ZEB1 transcription factor. J. Cell. Physiol. V. 231. P. 2205.
Lu M., Jolly M.K., Levine H., Onuchic J.N., Ben-Jacob E. 2013. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA. V. 110. P. 18144.
Ma P., Ni K., Ke J., Zhang W., Feng Y., Mao Q. 2018. miR-448 inhibits the epithelial-mesenchymal transition in breast cancer cells by directly targeting the E-cadherin repressor ZEB1/2. Exp. Biol. Med. (Maywood). V. 243. P. 473.
Ma Y., Zheng X., Zhou J., Zhang Y., Chen K. 2015. ZEB1 promotes the progression and metastasis of cervical squamous cell carcinoma via the promotion of epithelialmesenchymal transition. Int. J. Clin. Exp. Pathol. V. 8. P. 11258.
Meidhof S., Brabletz S., Lehmann W., Preca B.T., Mock K., Ruh M., Schüler J., Berthold M., Weber A., Burk U., Lübbert M., Puhr M., Culig Z., Wellner U., Keck T. et al. 2015. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. V. 7. P. 831.
Mishra A., La Perle K., Kwiatkowski S., Sullivan L.A., Sams G.H., Johns J., Curphey D.P., Wen J., McConnell K., Qi J., Wong H., Russo G., Zhang J., Marcucci G., Bradner J.E., et al. 2016. Mechanism, consequences, and therapeutic targeting of abnormal IL15 signaling in cutaneous T-cell lymphoma. Cancer Discov. V. 6. P. 986.
Mittenberg A.G., Kuzyk V.O., Shabelnikov S.V., Gorbach D.P., Fedorova O.A., Shatrova A.N., Barlev N.A. 2018. Combined treatment of human multiple myeloma cells with bortezomib and doxorubicin alters the interactome of 20S proteasomes. Cell Cycle. V. 17. P. 1745.
Mock K., Preca B.T., Brummer T., Brabletz S., Stemmler M.P., Brabletz T. 2015. The EMT-activator ZEB1 induces bone metastasis associated genes including BMP-inhibitors. Oncotarget. V. 6. P. 14399.
Molofsky A.V., Pardal R., Iwashita T., Park I.K., Clarke M.F., Morrison S.J. 2003. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. V. 425. P. 962.
Mooney S.M., Jolly M.K., Levine H., Kulkarni P. 2016. Phenotypic plasticity in prostate cancer: role of intrinsically disordered proteins. Asian J. Andrology. V. 18. P. 704.
Morel A.P., Ginestier C., Pommier R.M., Cabaud O., Ruiz E., Wicinski J., Devouassoux-Shisheboran M., Combaret V., Finetti P., Chassot C., Pinatel C., Fauvet F., Saintigny P., Thomas E., Moyret-Lalle C. et al. (2017). A stemness-related ZEB1–MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat. Med. V. 23. P. 568.
Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P. 2016. EMT: 2016. Cell. V. 166. P. 21.
Nishijima N., Seike M., Soeno C., Chiba M., Miyanaga A., Noro R., Sugano T., Matsumoto M., Kubota K., Gemma A. 2016. miR-200 / ZEB axis regulates sensitivity to nintedanib in non-small cell lung cancer cells. Int. J. Oncol. V. 48. P. 937.
Park I.K., Qian D., Kiel M., Becker M.W., Pihalja M., Weissman I.L., Morrison S.J., Clarke M.F. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. V. 423. P. 302.
Passacantilli I., Panzeri V., Bielli P., Farini D., Pilozzi E., Fave G.D., Capurso G., Sette C. 2017. Alternative polyadenylation of ZEB1 promotes its translation during genotoxic stress in pancreatic cancer cells. Cell Death Dis. V. 8 : e3168.
Pietersen A.M., Evers B., Prasad A.A., Tanger E., Cornelissen-Steijger P., Jonkers J., van Lohuizen M. 2008. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr. Biol. V. 18. P. 1094.
Qu J., Li M., An J., Zhao B., Zhong W., Gu Q., Cao L., Yang H., Hu C. 2015. MicroRNA-33b inhibits lung adenocarcinoma cell growth, invasion, and epithelial-mesenchymal transition by suppressing Wnt/β-catenin/ZEB1 signaling. Int. J. Oncol. V. 47. P. 2141.
Quesada A.R., Medina M.A., Munoz-Chapuli R., Ponce A.L. 2010. Do not say ever never more: the ins and outs of antiangiogenic therapies. Curr. Pharm. Des. V. 16. P. 3932.
Riaz M., van Jaarsveld M.T., Hollestelle A., Prager-van der Smissen W.J., Heine A.A., Boersma A.W., Liu J., Helmijr J., Ozturk B., Smid M., Wiemer E.A., Foekens J.A., Martens J.W. 2013. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. V. 15. P. R33.
Sahay D., Leblanc R., Grunewald T.G., Ambatipudi S., Ribeiro J., Clézardin P., Peyruchaud O. 2015. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget. V. 6. P. 20604.
Shapiro I.M., Cheng A.W., Flytzanis N.C., Balsamo M., Condeelis J.S., Oktay M.H., Burge C.B., Gertler F.B. 2011. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. V. 7 : e1002218.
Shen Y., Liu S., Yuan H., Ying X., Fu H., Zheng X. 2017. A long non-coding RNA lncRNAPE promotes invasion and epithelial-mesenchymal transition in hepatocellular carcinoma through the miR-200a/b-ZEB1 pathway. Tumour Biol. V. 39 : 1010428317705756.
Shimono Y., Zabala M., Cho R.W., Lobo N., Dalerba P., Qian D., Diehn M., Liu H., Panula S.P., Chiao E., Dirbas F.M., Somlo G., Pera R.A., Lao K., Clarke M.F. 2009. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. V. 138. P. 592.
Siemens H., Jackstadt R., Hünten S., Kaller M., Menssen A., Götz U., Hermeking H. 2011. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. V. 10. P. 4256.
Soda Y., Marumoto T., Friedmann-Morvinski D., Soda M., Liu F., Michiue H., Pastorino S., Yang M., Hoffman R.M., Kesari S., Verma I.M. 2011. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA. V. 108. P. 4274.
Song S.J., Poliseno L., Song M.S., Ala U., Webster K., Ng C., Beringer G., Brikbak N.J., Yuan X., Cantley L.C., Richardson A.L., Pandolfi P.P. 2013. MicroRNA antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. V. 154. P. 311.
Su W., Xu M., Chen X., Chen N., Gong J., Nie L., Li L., Li X., Zhang M., Zhou Q. 2017. Long noncoding RNA ZEB1-AS1 epigenetically regulates the expressions of ZEB1 and downstream molecules in prostate cancer. Mol. Cancer. V. 16 : 142.
Suzuki K., Kawataki T., Endo K., Miyazawa K., Kinouchi H., Saitoh M. 2018. Expression of ZEBs in gliomas is associated with invasive properties and histopathological grade. Oncol. Lett. V. 16. P. 1758.
Tevaarwerk A.J., Gray R.J., Schneider B.P., Smith M.L., Wagner L.I., Fetting J.H., Davidson N., Goldstein L.J., Miller K.D., Sparano J.A. 2013. Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: little evidence of improvement over the past 30 years. Cancer. V. 119. P. 1140.
Tian X.J., Zhang H., Xing J. 2013. Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophys. J. V. 105. P. 1079.
Tian Y., Pan Q., Shang Y., Zhu R., Ye J., Liu Y., Zhong X., Li S., He Y., Chen L., Zhao J., Chen W., Peng Z., Wang R. 2014. MicroRNA-200 (miR-200) cluster regulation by achaete scute-like 2 (Ascl2): impact on the epithelial-mesenchymal transition in colon cancer cells. J. Biol. Chem. V. 289. P. 36 101.
Wang H., Huang B., Li B.M., Cao K.Y., Mo C.Q., Jiang S.J., Pan J.C., Wang Z.R., Lin H.Y., Wang D.H., Qiu S.P. 2018a. ZEB1-mediated vasculogenic mimicry formation associates with epithelial-mesenchymal transition and cancer stem cell phenotypes in prostate cancer. J. Cell. Mol. Med. V. 22. P. 3768.
Wang H., Jiang Z., Na M., Ge H., Tang C., Shen H., Lin Z. 2017a. PARK2 negatively regulates the metastasis and epithelial-mesenchymal transition of glioblastoma cells via ZEB1. Oncol. Lett. V. 14. P. 2933.
Wang Y., Xu X., Yu S., Jeong K.J., Zhou Z., Han L., Tsang Y.H., Li J., Chen H., Mangala L.S., Yuan Y., Eterovic A.K., Lu Y., Sood A.K., Scott K.L., Mills G.B., Liang H. 2017b. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. V. 27. P. 1112.
Wang P.S., Chou C.H., Lin C.H., Yao Y.C., Cheng H.C., Li H.Y., Chuang Y.C., Yang C.N., Ger L.P., Chen Y.C., Lin F.C., Shen T.L., Hsiao M., Lu P.J. 2018b. A novel long non-coding RNA linc-ZNF469-3 promotes lung metastasis through miR-574-5p-ZEB1 axis in triple negative breast cancer. Oncogene. V. 37. P. 4662.
Weis S.M., Cheresh D.A. 2011. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. V. 17. P. 1359.
Wellner U., Schubert J., Burk U.C., Schmalhofer O., Zhu F., Sonntag A., Waldvogel B., Vannier C., Darling D., zur Hausen A., Brunton V.G., Morton J., Sansom O., Schüler J., Stemmler M.P. et al. 2009. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness inhibiting microRNAs. Nat. Cell Biol. V. 11. P. 1487.
Wu W.S., You R.I., Cheng C.C., Lee M.C., Lin T.Y., Hu C.T. 2017. Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep. V. 7 : 17 753.
Xiong W.C., Han N., Wu N., Zhao K.L., Han C., Wang H.X., Ping G.F., Zheng P.F., Feng H., Qin L., He P. 2018. Interplay between long noncoding RNA ZEB1-AS1 and miR-101/ZEB1 axis regulates proliferation and migration of colorectal cancer cells. Am. J. Transl. Res. V. 10. P. 605.
Xu M., Zhu C., Zhao X., Chen C., Zhang H., Yuan H., Deng R., Dou J., Wang Y., Huang J., Chen Q., Jiang B., Yu J. 2015. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget. V. 6. P. 979.
Xu Q., Deng F., Qin Y., Zhao Z., Wu Z., Xing Z., Ji A., Wang Q.J. 2016. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. V. 7 : e2254.
Xu Y., Li Q., Li X.Y., Yang Q.Y., Xu W.W., Liu G.L. 2012. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. V. 31 : 16.
Yang M.H., Wu M.Z., Chiou S.H., Chen P.M., Chang S.Y., Liu C.J., Teng S.C., Wu K.J. 2008. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. V. 10. P. 295.
Yang T., He X., Chen A., Tan K., Du X. 2018. LncRNA HOTAIR contributes to the malignancy of hepatocellular carcinoma by enhancing epithelial-mesenchymal transition via sponging miR-23b-3p from ZEB1. Gene. V. 670. P. 114.
Yang Y., Park J.W., Bebee T.W., Warzecha C.C., Guo Y., Shang X., Xing Y., Carstens R.P. 2016. Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol. Cell. Biol. V. 36. P. 1704.
Yuan J.H., Yang F., Wang F., Ma J.Z., Guo Y.J., Tao Q.F., Liu F., Pan W., Wang T.T., Zhou C.C., Wang S.B., Wang Y.Z., Yang Y., Yang N., Zhou W.P., Yang G.S., Sun S.H. 2014. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. V. 25. P. 666.
Zhang L., Huang G., Li X., Zhang Y., Jiang Y., Shen J., Liu J., Wang Q., Zhu J., Feng X., Dong J., Qian C. 2013. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1alpha in hepatocellular carcinoma. BMC Cancer. V. 13 : 108.
Zhang J., Ma L. 2012. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. V. 31. P. 653.
Zhang P., Wei Y., Wang L., Debeb B.G., Yuan Y., Zhang J., Yuan J., Wang M., Chen D., Sun Y., Woodward W.A., Liu Y., Dean D.C., Liang H., Hu Y. et al. 2014. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. V. 16. P. 864.
Zheng X., Carstens J.L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C.C., LeBleu V.S., Kalluri R. 2015. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. V. 527. P. 525.
Zhou Z., Zhang P., Hu X., Kim J., Yao F., Xiao Z., Zeng L., Chang L., Sun Y., Ma L. 2017. USP51 promotes deubiquitination and stabilization of ZEB1. Am. J. Cancer Res. V. 7. P. 2020.
Дополнительные материалы отсутствуют.
Инструменты
Цитология