

Цитология, 2020, T. 62, № 5, стр. 322-332
Зависимость жизнеспособности Ras-экспрессирующих клеток от повреждения митохондрий, вызванного действием противоопухолевых агентов
Е. Ю. Кочеткова 1, *, Г. И. Блинова 1, А. С. Бойцов 1, В. А. Поспелов 1, Т. В. Поспелова 1
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: lena.linnaea@gmail.com
Аннотация
Опухолевые клетки, экспрессирующие белок Ras, относятся к числу трудно поддающихся терапии, так как способны восстанавливать жизнеспособность в ответ на действие противоопухолевых агентов, активируя цитопротективные процессы. Поиск новых мишеней для более эффективной терапии показал, что такой мишенью могут быть митохондрии, так как Ras-экспрессирующие клетки получают АТФ преимущественно за счет окислительного фосфорилирования, а не гликолиза, как многие другие типы опухолевых клеток. В работе исследована способность рентгеновского облучения вызывать гибель опухолевых клеток, экспрессирующих онкогенный Ras, в сравнении с действием ингибитора целостности митохондрий ABT199 (венетоклакса). Показано, что клетки демонстрируют разные варианты ответа во времени на действие повреждающих агентов. На ранних сроках (2–24 ч после повреждения) активируется аутофагия, которая элиминирует поврежденные митохондрии и повышает жизнеспособность клеток, тогда как появление стареющих клеток через 72 ч относится к позднему ответу, подавляющему пролиферацию. Ингибирование аутофагии и старения повышает гибель клеток при действии повреждающих агентов, что свидетельствует о ключевой роли этих процессов в подавлении гибели после повреждения митохондрий. Таким образом, ингибирование аутофагии и старения может повысить эффективность уничтожения опухолевых клеток при действии противоопухолевых агентов.
Процесс злокачественной трансформации связан с существенными изменениями метаболических процессов клеток и приобретением ими новых признаков. Одной из характеристик процесса канцерогенеза является изменение энергетического метаболизма клеток (Hanahan, Weinberg, 2011). Опухолевые клетки характеризуются особым типом гликолиза, который протекает интенсивно, несмотря на достаточный уровень кислорода (аэробный гликолиз, известный как эффект Варбурга) (Warburg, 1956a, 1956b). Долгое время считалось, что аэробный гликолиз является ключевым источником АТФ для опухолевых клеток. Однако последние данные говорят о том, что митохондрии являются важными участниками поддержания метаболизма и жизнеспособности опухолевых клеток (Porporato et al., 2018).
Опухолевые клетки демонстрируют метаболическую пластичность в поддержании жизнеспособности для получения АТФ после повреждений. В частности, было показано, что при воздействии на клетки меланомы вемурафениба (ингибитора серинтреониновой киназы BRAF) клетки переключаются с гликолиза на окислительное фосфорилирование, что повышает их устойчивость к вемурафенибу, в то время как ингибитор компонентов электроннотранспортной цепи митохондрий повышает клеточную гибель в ответ на действие вемурафениба (Trotta et al., 2017).
Зависимость жизнеспособности от работы митохондрий была показана для опухолевых клеток, характеризующихся наличием онкогенных мутаций в генах, кодирующих малую ГТФазу Ras (Weinberg et et al., 2010; Guo et et al., 2011; Viale et al., 2014). Эти клетки получают АТФ преимущественно за счет окислительного фосфорилирования в митохондриях, а не гликолиза (Weinberg et al., 2010). Онкогенные мутации ras обнаружены примерно в 30% известных опухолей человека (Fernández-Medarde, Santos, 2011). Как было показано, эти опухоли трудно поддаются терапии за счет активации аутофагии, ведущей к восстановлению жизнеспособности и приобретению толерантности к действию противоопухолевых препаратов (Quadir et al., 2008; Altman et al., 2014; Serrano et al., 2016). Для Ras-трансформированных клеток жизненно важным является поддержание пула функционирующих митохондрий, обеспечивающих клетку достаточным уровнем энергии. Важную роль в поддержании пула митохондрий играет аутофагия. Поврежденные митохондрии могут удаляться за счет специфической формы аутофагии – митофагии, которая позволяет таким образом восстановить жизнеспособность после действия повреждающих агентов (Wang, Klionsky, 2011).
В настоящей работе сравнивали способность двух повреждающих воздействий вызывать клеточную гибель Ras-экспрессирующих опухолевых клеток (эмбриональные фибробласты крысы, трансформированные двумя онкогенами, линия ERas) в зависимости от повреждения митохондрий. Для этой цели анализировали повреждения митохондрий и зависимость жизнеспособности клеток от активации аутофагии. В качестве повреждающих агентов были использованы ионизирующее облучение в дозе 3 и 6 Гр и ингибитор целостности митохондрий ABT199 (венетоклакс). Как ингибитор ABT199, так и ионизирующее облучение в обеих дозах вызывают повреждение митохондрий и клеточную гибель.
Показано, что клетки демонстрируют два ответа на повреждение. Активирующаяся через 2 ч после воздействия обоих агентов аутофагия осуществляет удаление поврежденных митохондрий и снижает клеточную гибель. Через 72 ч аутофагия завершается, но часть клеточной популяции претерпевает старение, приводящее к снижению пролиферативной активности.
Таким образом, мы показали способность разных агентов вызывать в клетках ERas повреждения митохондрий и разные варианты ответа: активацию аутофагии, восстанавливающую жизнеспособность и активацию старения, приводящую к снижению пролиферативной активности. Применение как ингибиторов аутофагии, так и ингибиторов старения усиливает гибель облученных клеток. Следовательно, для повышения эффективности терапии необходимо комбинированное воздействие повреждающих митохондрии агентов и ингибиторов аутофагии и старения.
МАТЕРИАЛ И МЕТОДИКА
Клеточные линии. Работа выполнена на эмбриональных фибробластах крысы, трансформированных онкогенами E1A раннего района аденовируса Ad5 и cHa-Ras человека с мутациями в 12 и 65 положениях (линия ERas). Клетки культивировали в среде Дальбекко в модификации Игла (Биолот, Россия), содержащей 10% сыворотки крупного рогатого скота (HyClone, США) и 1 мг/мл гентамицина (Биолот, Россия) при 37°С и 5% CO2.
Рентгеновское облучение клеток. Осуществляли с помощью прибора РАП-150/300-14 (Актюбрентген, Россия). Клетки облучали в дозе 3 и 6 Гр.
Модуляция аутофагии и старения. Использовали следующие ингибиторы: ингибиторы аутофагии на стадии слияния аутофагосом с лизосомами бафиломицин (Sigma-Aldrich, США) и хлороквин (BioVision, США); селективный ингибитор комплекса mTORC1 рапамицин в концентрации 200 нМ (Calbiochem, США), киназный ингибитор mTOR pp242 в концентрации 200 нМ, (Sigma-Aldrich, США). Для нарушения проницаемости митохондрий использован ABT199 (венетоклакс) в концентрации 4 мкM (Selleckchem, США).
Жизнеспособность клеток оценивали по конверсии 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромида в формазан (МТТ-тест). Клетки высевали на 12-луночные платы в плотности 3 × 104 клеток на 1 лунку. В указанные сроки среду заменяли на раствор МТТ (0.5 мг/мл) в фосфатно-солевом буфере и инкубировали клетки в течение 1 ч при 37°С. Конвертированный клетками формазан растворяли в диметилсульфоксиде и наносили по 80 мкл в лунки 96-луночного планшета. Количество формазана определяли спектрометрически на приборе Multiscan Ex (Thermo, США) при длине волны поглощения 572 нм.
Клоногенная выживаемость. Клетки облучали в дозе 3 и 6 Гр и после этого культивировали в течение 24, 72 и 120 ч, затем пересевали на чашки Петри 35 мм в плотности 100 клеток на 1 чашку в 1 мл свежей среды. Далее клетки культивировали в стандартных условиях, ежедневно контролируя рост клонов под микроскопом. Через 7 сут выросшие клоны окрашивали Кристал-Виолетом (позволяет визуализировать выросшие клоны) в течение 30 мин, промывали водой и считали под микроскопом.
Оценка целостности митохондрий. Использовали согласно инструкции производителя 2 митохондриальных красителя: 1) потенциал-зависимый витальный краситель Mitotracker Orange (Thermo Fischer, США), который визуализирует только неповрежденные митохондрии, сохраняющие разницу потенциалов мембран, и 2) Mitotracker Green (Thermo Fischer, США), который окрашивает все митохондрии независимо от повреждения. Клетки высевали на покровные стекла в чашки 3.5 мм при плотности 5 × 104 кл./чашка и подвергали воздействию ингибитора ABT199 или облучения. Через 2, 24, 72 ч после действия агента клетки окрашивали 200 нM Mitotracker Green и 50 нМ Mitotracker Orange в течение 30 мин при 37°С. Затем клетки промывали теплым фосфатно-солевым буфером и получали изображения при помощи микроскопа Leica TSC SP5 (Leica Microsystems, Германия). Ядра окрашивали с помощью Hoechst 33 258 (Thermo Fischer, США). Измерение интенсивности сигнала от обоих красителей производили с помощью программы ImageJ.
Вестерн-блот-анализ. Клетки высевали на 10 мм чашки в плотности 6 × 105 и подвергали обработке. В указанные промежутки времени клетки лизировали в буферном растворе следующего состава: 1% I-GEPAL, 0.5% деоксихолата натрия; 0.1% SDS; 50 мМ трис-HCl, pH 8.0; 150 мМ NaCl; 5 мМ EDTA, pH 8.0; 60 мМ NaF. Количество белка в пробах измеряли по методу Брэдфорд. Для электрофореза в полиакриламидном геле отбирали по 50 мкг белка. После электрофореза пробы переносили на PVDF-мембрану с помощью мокрого переноса, осуществляли блокирование неспецифического связывания с помощью 5%-ного раствора молока (Santa-Cruz Biotechnology, США) в трис-солевом буфере, содержащем 0.1% TWEEN-20, и инкубировали с первыми антителами в течение 12 ч при 4°С. Затем мембраны промывали и инкубировали со вторыми антителами, конъюгированными с пероксидазой хрена, в течение 1 ч при комнатной температуре. Бэнды визуализировали с помощью системы гель-документации SynGene.
Выявление ассоциированной со старением β-галаксидазной активности. Использовали описанную методику (Dimri et al., 1995). Клетки высевали на покровные стекла в плотности 5 × 104 кл./чашка (чашка 3.5 мм) и подвергали воздействию. Через 120 ч клетки фиксировали в 3.7%-ном формальдегиде. На стекла наносили раствор 1мг/мл субстрата β-галактозидазы в буфере (лимонная кислота/фосфат натрия (0.2 M, рН 6.0), 5 мМ NaCl, 2 мМ MgCl2) и инкубировали в течение 18 ч при температуре 37°С. Об активности ассоциированной со старением β-галактозидазы судили по выявлению в цитоплазме клеток синих гранул. Изображения получали на микроскопе Axiovert 200M (Carl Zeiss, Германия) + Leica DFC 420C (Leica Microsystems, Германия) при увеличении объектива 40×.
Аутофагия. Об инициации процесса аутофагии при действии облучения и ингибитора ABT199 судили по конверсии маркера аутофагии LC3 из цитоплазматической формы LC3I в форму LC3II, ассоциированную с мембранами созревающих аутофагосом. LC3I и LC3II выявляли с помощью Вестерн-блот-анализа в 12.5%-ном полиакриламидном геле.
Антитела. В работе использованы следующие антитела: первичные (Cell Signalling, США) к фосфорилированному гистону фосфо-H2AX и пан-LC3 (соответственно номера по каталогу 9718 и 4108S). Вторичные антитела: козьи анти-кроличьи, конъюгированные с пероксидазой хрена (Sigma, США) или с Alexa Fluor 488 (Thermo Fischer, США).
Статистическая обработка. Использовали программу Microsoft Excel 2016 и Statistica 7.0 (Statsoft). Для оценки достоверности различий между контрольными и подвергавшимися обработке клетками использовался One-Way Anova. Данные представлены средним значением из трех независимых экспериментов и его ошибкой. Различия считали достоверными при значениях Р ≤ 0.05.
РЕЗУЛЬТАТЫ
Работа выполнена на эмбриональных фибробластах крысы, трансформированных онкогенами E1A + cHa-Ras (клетки ERas). Клетки подвергали облучению в дозе 3 и 6 Гр. Обе дозы вызывают повреждения ДНК, как следует из данных по накоплению фокусов фосфорилированного гистона H2AX в ядрах клеток (рис 1). Согласно данным с использованием Mitotracker Orange, облучение в обеих дозах приводит к повреждению митохондрий (рис. 2а). Через 2 ч после облучения интенсивность сигнала этого красителя снижается в 2.5 раза при использовании обеих доз (Р ≤ 0.05). В это же время повреждение митохондрий индуцирует аутофагию.
Рис. 1.
Формирование фокусов фосфорилированного гистона H2AX (а) в ядрах клеток ERas, облученных в дозе 3 и 6 Гр через 1, 3 и 24 ч после облучения. Иммунофлуоресценция с антителами к фосфо-H2AX (зеленый цвет), ядра окрашены DAPI (синий цвет); увел. об.: 40×. б – График, показывающий изменение числа фокусов в контрольных клетках (0 ч) и в облученных через 1, 3 и 24 ч; данные представлены средним значением и его стандартной ошибкой. Здесь и на рис. 2, 3 достоверность различий по сравнению с контролем при P < 0.05 показана звездочкой.
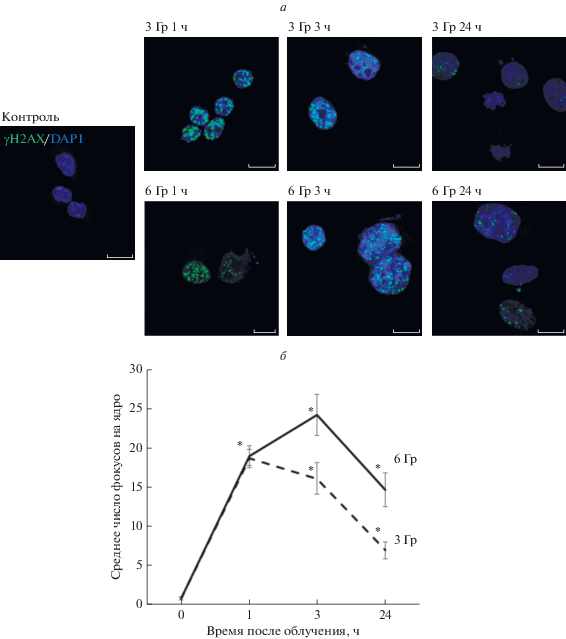
Рис. 2.
Повреждение митохондрий и активация аутофагии в клетках ERas, подвергнутых воздействию ионизирующего облучения в дозе 3 и 6Гр через 2, 24 и 72 ч после облучения. а – Неповрежденные митохондрии, выявленные с помощью прижизненного потенциал-зависимого красителя Mitotracker Orange окрашены в красный цвет; потенциал-независимый Mitotracker Green окрашивает все митохондрии в зеленый цвет; ядра окрашены Hoechst33258 (синий цвет); видно, что через 2–24 ч количество неповрежденных митохондрий снижается. Увел. об.: 40×. б – График временной зависимости свечения Mitotracker O-range в необлученных контрольных (0 ч) и облученных клетках. в – Конверсия цитоплазматической формы LC3I в мембран-ассоциированную LC3II. Вестерн-блотинг с антителами к LC3 и альфа-тубулину.
Процесс аутофагии после воздействия рентгеновского облучения анализировали по маркеру LC3, который при активации аутофагии претерпевает конверсию из цитоплазматической формы LC3I в мембран-ассоциированную форму LC3II. Конверсию LC3I в LC3II анализировали методом Вестерн-блотинга, используя антителами против LC3I и LC3II. Вестерн-блот-анализ показал, что через 2 ч в клетках активируется аутофагия – выявляется формa LC3II, ассоциированная с мембранами созревающих аутофагосом, и эта аутофагия завершается через 72 ч (рис. 2в). Аутофагия удаляет поврежденные митохондрии, судя по данным с красителем Mitotracker Orange, показавшим почти полное восстановление интенсивности сигнала через 72 ч культивирования клеток после облучения (рис. 2а).
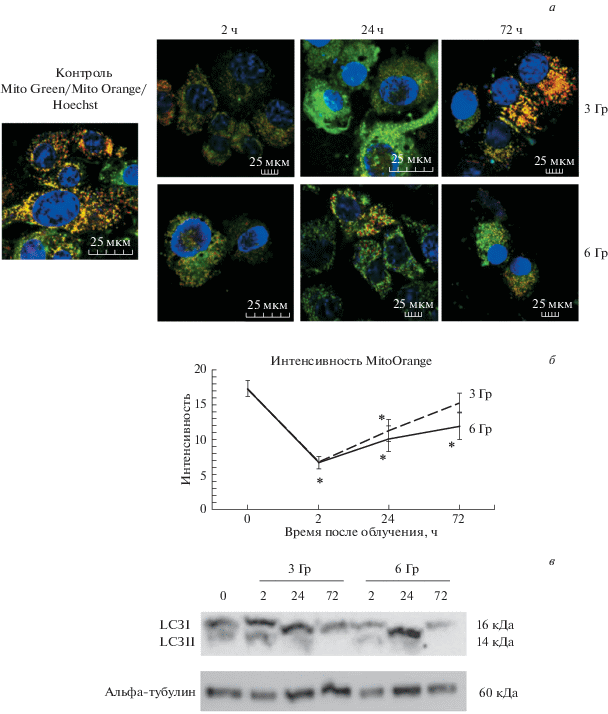
Анализ жизнеспособности клеток показал, что облучение снижает жизнеспособность клеток через 24 ч после облучения, однако через 72 ч процесс клеточной гибели прекращается. При этом облучение в обеих дозах снижает пролиферативную активность клеток через 120 ч культивирования, что следует из кривых роста и способности клеток пролиферировать в клональном посеве (рис. 3).
Рис. 3.
Изменение жизнеспособности (а) и пролиферативной активности (б–г) клеток после воздействия ионизирующего облучения в дозе 3 и 6 Гр. а – Жизнеспособность по МТТ-тесту. Данные представлены средним значением и его стандартной ошибкой (вертикальные отрезки). б – Кривые пролиферативной активности клеток в контроле (без облучения, 0 ч) и через 24–120 ч после облучения. в – Клетки в клональном посеве: через 24, 72 и 120 ч после облучения клетки высевали в плотности 100 кл./мл в чашку 35 мм, через 7 сут клоны окрашивали Кристалл-Виолетом и считали. г – Жизнеспособность клеток через 24 и 72 ч после облучения и после замены среды культивирования на свежую (через 24ч после облучения); окраска Кристал-Виолетом; видно, что через 72 ч после облучения популяция клеток восстанавливается по сравнению со сроком 24 ч.
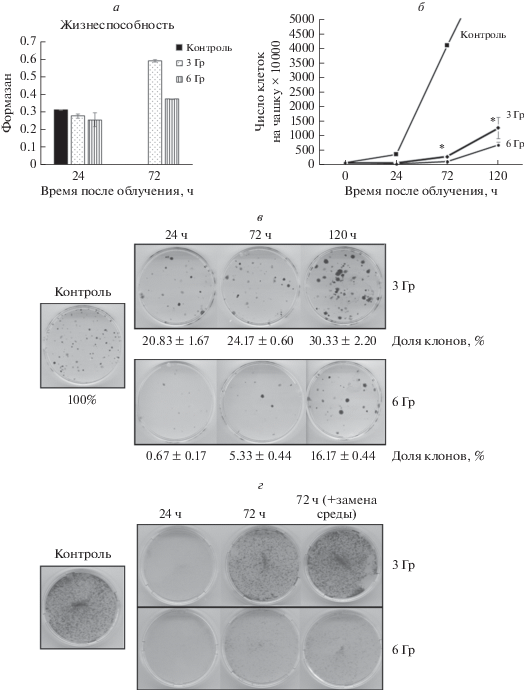
Ингибирование аутофагии с помощью бафиломицина А1 (подавляющего ацидификацю лизосом) и хлороквина (подавляющего слияние аутофагосом с лизосомами) снижает жизнеспособность облученных клеток (рис. 4а, б), что подтверждает ключевую роль аутофагии в уменьшении гибели клеток. Бафиломицин и хлороквин ингибируют аутофагию на финальной стадии – слиянии аутофагосом с лизосомами. Киназный ингибитор рр242, мишенью которого является комплекс mTORC1, активирует аутофагию на стадии инициации (Ganley et al., 2009; Hosokawa et al., 2009). Однако действие ингибиторов комплекса mTORC1 рр242 и рапамицина на облученные клетки приводит не к восстановлению, а к снижению их жизнеспособности (рис. 5б).
Рис. 4.
Изменение жизнеспособности облученных клеток ERas при ингибировании аутофагии бафиломицином (Баф, а) или хлороквином (Хл, б). Данные МТТ-теста. Различия между вариантами облучения в отсутствие и в присутствии Баф или Хл достоверны (*) при P < 0.05.
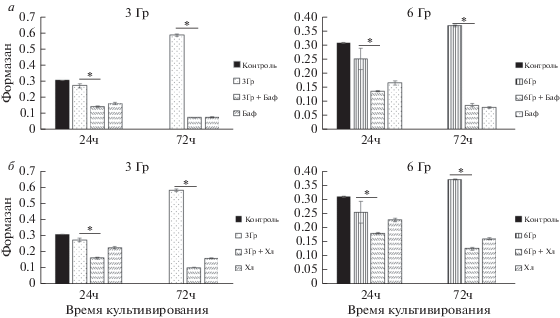
Рис. 5.
Ассоциированная со старением β-галактозидазная активность в клетках Eras через 72 ч после облучения в дозе 3 и 6 Гр (а) и жизнеспособность облученных клеток при ингибировании mTORC1 ингибитором рр242 и рапамицином (Рап, б) через 24 и 72 ч после облучения. а – Увел. об.: 40×. б – По данным МТТ-теста; различия между вариантами облучения в отсутствие и в присутствии ингибитора достоверны (*) при P < 0.05.
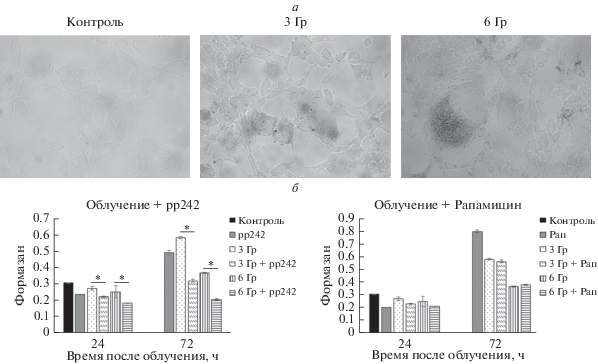
Наиболее вероятно, что эти результаты связаны с аддитивным действием облучения и ингибитора рр242 на повреждение митохондрий (Gordeev et al., 2015), которое не может компенсировать аутофагия. Кроме того, комплекс mTORC1 является не только регулятором аутофагии, но и ключевым регулятором старения (Sarbassov et al., 2005; Blagosklonny, 2011a, 2011b, 2012), которое тоже вносит вклад в жизнеспособность популяции.
Анализ маркеров старения показал появление в популяции клеток с повышенной активностью β-галактозидазы, ассоциированной со старением, и снижение пролиферативной активности по сравнению с контрольными необлученными клетками (кривые роста на рис. 5а). При подавлении активности комплекса mTORC1 с помощью ингибиторов рр242 и рапамицина через 72 ч наблюдается снижение жизнеспособности облученных клеток, что подтверждает роль старения в снижении гибели клеток после облучения.
Повреждающее действие облучения сравнивали с действием ингибитора, действующего непосредственно на митохондрии. Был использован ингибитор целостности митохондрий BH3-миметик ABT199 в концентрации 4 мкM. Действие ABT199 приводит к повреждению митохондрий, что следует из данных по падению свечения прижизненного красителя Mitotracker Orange (рис. 6а). Через 24 ч культивирования ABT199 в дозе 4 мкМ приводит к снижению жизнеспособности на 33% (рис. 6б). Однако уже через 2 ч действия ингибитора активируется аутофагия, которая удаляет поврежденные митохондрии и полностью восстанавливает жизнеспособность клеток через 72 ч (рис 6д).
Рис. 6.
Повреждение митохондрий, активация цитопротективной аутофагии и подавление пролиферации в клетках Eras при действии ингибитора целостности митохондрий ABT199 в течение разного времени. а – Выявление неповрежденных митохондрий с помощью прижизненного красителя Mitotracker Orange (красный цвет); потенциал-независимый Mitotracker Green окрашивает все митохондрии в зеленый цвет: ядра окрашены Hoechst33258 (синий цвет); увел. об.: 40×. б – Жизнеспособность по МТТ тесту; данные представлены как среднее ± стандартная ошибка среднего; различия между экспериментом и контролем достоверны (*) при P < 0.05. в – Кривая пролиферативной активности (представлены средние ± стандартная ошибка; отличия от контроля (К) в точках 72 (*) и 120 (**) ч достоверны при P < 0.05). г – Распределение клеток по фазам клеточного цикла в контроле и через 24 и 72 ч действия ингибитора ABT199; указана доля клеток в S-фазе. д – Конверсия цитоплазматической формы LC3I в LC3II, ассоциированную с мембраной; Вестерн-блотинг с антителами к LC3 и альфа-тубулину.
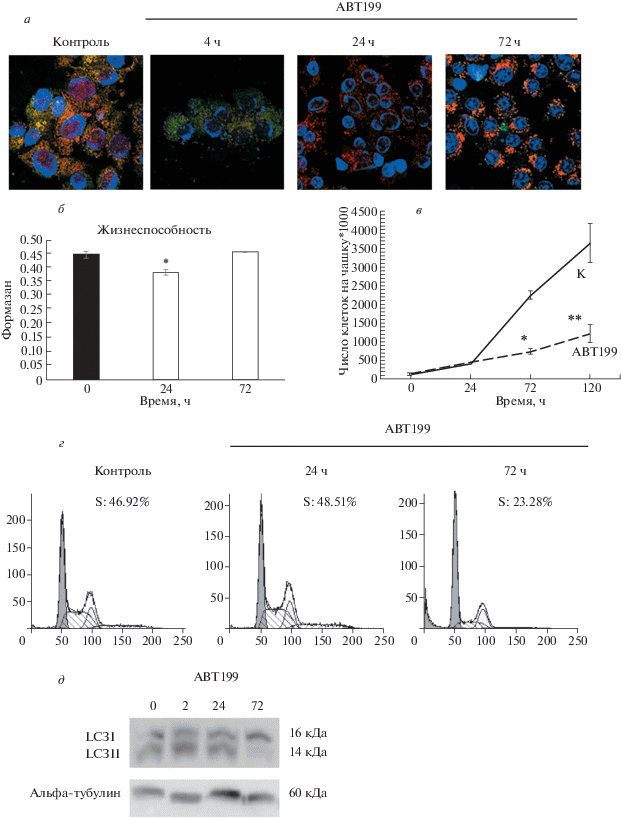
Ингибитор ABT199 оказывает влияние на пролиферацию клеток. Согласно данным кривых роста, через 72 и 120 ч пролиферативная активность клеток снижена вдвое по сравнению с контрольными клетками на этом же сроке (Р ≤ 0.05) (рис. 6в). Эти данные подтверждаются данными проточной цитометрии, согласно которым через 72 ч культивирования в присутствии ABT199 доля клеток в S-фазе снижена до 23% по сравнению с с контрольными клетками (47%) (рис. 6г). Таким образом, действие ингибитора ABT199 и ионизирующего облучения вызывает повреждение митохондрий и гибель части популяции, снижение пролиферативной активности и активацию аутофагии. Аутофагия играет цитопротективную роль, удаляя поврежденные митохондрии и восстанавливая жизнеспособность клеток.
ОБСУЖДЕНИЕ
Активация в опухолевых клетках цитопротективных процессов является фактором, осложняющим терапию злокачественных опухолей. К числу основных процессов относится аутофагия (Amaravadi et al., 2007; Fan et al., 2010). Нами было показано, что действие облучения в дозе 3 и 6 Гр, как и митохондриального ингибитора ABT199, вызывает повреждение митохондрий в клетках ERas. Ключевым механизмом, позволяющим клеткам удалить поврежденные митохондрии, является аутофагия. Ранее было показано, что к повреждению митохондрий приводит также действие киназных ингибиторов Ras/Raf/MEK/ERK-пути (Monick et al., 2008). Подавление аутофагии с помощью ингибиторов бафиломицина и хлороквина, как мы показали (рис. 4), усиливает гибель облученных клеток. Таким образом, применение ингибиторов аутофагии позволяет повысить способность повреждающих агентов элиминировать опухолевые клетки.
Действие рентгеновского облучения, помимо повреждений ядерной ДНК, приводит также к нарушению целостности мембран митохондрий, судя по снижению интенсивности сигнала красителя Mitotracker Orange. Ядерный геном кодирует ряд белков, регулирующих функционирование митохондрий, в частности, белок NRF1, который контролирует экспрессию компонентов электроннотранспортной цепи (Niu et al., 2019). В число его мишеней входит также антиапоптотический белок Bcl2, локализующийся на мембранах митохондрий (Niu et al, 2019).
Потребность опухолевых клеток в стабильном функционировании митохондрий для поддержания неограниченной пролиферации (Porporato et al., 2018) может быть связана с тем, что кроме большего, чем при гликолизе, количества АТФ, в ходе цикла трикарбоновых кислот производится также ацетил-коэнзим А, играющий важную роль в многочисленных процессах раковой клетки, таких как синтез холестерина для плазматических мембран или реакции ацетилирования, вовлеченные в регуляцию аутофагии (Mariño et al., 2014).
Как показывает настоящая работа, клетки отвечают разными событиями на ионизирующее облучение. На ранних сроках после облучения (2–24 ч) клетки отвечают активацией аутофагии, связанной с удалением поврежденных митохондрий. Кроме того, через 72 ч после облучения в популяции выявляются клетки, претерпевающие старение, согласно накоплению в них активности β-галактозидазы. Интересно отметить, что через 72 ч в клетках, облученных в дозе 6 Гр, выявлено больше клеток, позитивных по маркеру старения, чем в клетках, облученных в дозе 3 Гр, что говорит о дозовой зависимости процесса старения при действии ионизирующего облучения.
Известно, что индукция клеточного старения является одним из способов ответа клетки на стресс, в частности, на повреждения ДНК (Toussaint et al., 2000; von Zglinicki et al., 2015), а также на противоопухолевую терапию (Ewald et al., 2010). Несмотря на подавление пролиферации, стареющие клетки сохраняют жизнеспособность и обладают устойчивостью к апоптотической гибели (Ryu et al., 2007). Одной из характеристик старых клеток является ассоциированный со старением секреторный фенотип (senescence-associated secretory phenotype, SASP). Старые клетки секретируют интерлейкины (в частности, IL1a, IL6, IL8), хемокины и металлопротеиназы (Hernandez-Segura et al., 2018). Секретируемые старыми клетками цитокины могут способствовать поддержанию пролиферативной активности клеток микроокружения (Campisi, 2011). Следовательно, не только аутофагия, но и старение является механизмами развития толерантности опухолевых клеток к повреждающим агентам.
Оценка клеточной гибели после действия облучения и после действия ингибитора ABT199 говорит о том, что митохондрии играют ключевую роль в поддержании жизнеспособности Ras-экспрессирующих опухолевых клеток. Однако действие обоих повреждающих агентов приводит к быстрой активации цитопротективной аутофагии. Из всех разработанных к настоящему времени ингибиторов, воздействующих на митохондрии через Bcl2, ABT199 (венетоклакс) показал наивысшую эффективность как в монотерапии, так и в комбинации с другими препаратами при лечении хронической лимфоцитарной лейкемии (Lampson, Davids, 2017). Тем не менее, ряд исследований показал, что опухолевые клетки могут демонстрировать устойчивость к этому агенту. В частности, было показано, что ABT199 имеет ограниченную эффективность против некоторых линий клеток не Ходжкинской лимфомы (Choudhary et al., 2015).
Исследование механизмов устойчивости к ABT199 выявило таких участников, как Bcl-XL и PI3K/AKT/mTOR-сигнальный путь (Choudhary et al., 2015). Линия ERas имеет высокую активность PI3K/mTORC1-сигнального пути (Гордеев и др., 2015), а действие ABT199 активирует в этих клетках аутофагию (рис. 6в). В большинстве исследований показана способность ABT199 (одного или в комбинации с другим противоопухолевым агентом) вызывать апоптотическую гибель опухолевых клеток, в то время как для линии ERas ABT199 оказал только антипролиферативное действие.
Надо отметить, что активировавшаяся при действии ингибитора ABT199 аутофагия восстанавливает до контрольного уровня жизнеспособность, но не пролиферативную активность клеток. Таким образом, для увеличения гибели клеток при действии этих повреждающих агентов необходимо найти способ блокировать цитопротективные процессы и, в первую очередь, аутофагию. Понимание того, как можно блокировать этот процесс, позволит получить новые данные, полезные для разработки новых способов уничтожения опухолевых клеток.
Список литературы
Гордеев С.А., Быкова Т.В., Зубова С.Г., Аксенов Н.Д., Поспелова Т.В. 2015. Антиапоптотический ген bcl-2 препятствует реактивации программы старения, индуцированной ингибитором гистоновых деацетилаз бутиратом натрия в фибробластах крысы, трансформированных онкогенами E1a и c-Ha-Ras. Цитология. Т. 57. № 2. С. 135. (Gordeev S.A., Bykova T.V., Zubova S.G., Aksenov N.D., Pospelova T.V. 2015. Antiapoptotic gene bcl-2 prevents cellular senescence program reactivation induced by histone deacetylase inhibitor sodium butyrate in E1A and cHa-ras transformed rat fibroblasts. Tsitologiya, V. 57. № 2. P. 135.)
Altman J.K., Szilard A., Goussetis D.J., Sassano A., Colamonici M., Gounaris E., Frankfurt O., Giles F.J., Eklund E.A., Beauchamp, E.M., Platanias L.C. 2014. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. V. 20. P. 2400.
Amaravadi R.K., Yu D., Lum J.J., Bui T., Christophorou M.A., Evan G.I., Thomas-Tikhonenko A., Thompson C.B. 2007. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clinical Invest. V. 117. P. 326.
Blagosklonny M.V. 2011a. Cell cycle arrest is not senescence. Aging (Albany NY). V. 3. P. 94.
Blagosklonny M.V. 2011b. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). V. 3. P. 1130.
Blagosklonny M.V. 2012. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). V. 4. P. 159.
Campisi J. 2011. Cellular senescence: putting the paradoxes in perspective. Curr. Opin. Genet. Dev. V. 21. P. 107.
Choudhary G.S., Al-Harbi S., Mazumder S., Hill B.T., Smith M.R., Bodo J., His E.D., Almasan A. 2015. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death & Disease 6(1), e1593. https://doi.org/10.1038/cddis.2014.525
Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Liskens M., Rubelj I., Pereira-Smith O. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. V. 92. P. 9363.
Ewald J.A., Desotelle J.A., Wilding G., Jarrard D.F. 2010. Therapy-induced senescence in cancer. J. Natl. Cancer Institute. V. 102. P. 1536.
Fan Q.W., Cheng C., Hackett C., Feldman M., Houseman B.T., Nicolaides T., Haas-Kogan D., James C.D., Oakes S.A., Debnath J., Shokat K.M., Weiss W.A. 2010. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. V. 3. P. ra81. https://doi.org/10.1126/scisignal.2001017
Fernández-Medarde A., Santos E. 2011. Ras in cancer and developmental diseases. Genes Cancer. V. 2. P. 344.
Ganley I.G., Lam du H., Wang J., Ding X., Chen S., Jiang X. 2009. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. V. 284. P. 12 297.
Gordeev S.A., Bykova T.V., Zubova S.G., Bystrova O.A., Martynova M.G., Pospelov V.A., Pospelova T.V. 2015. mTOR kinase inhibitor pp242 causes mitophagy terminated by apoptotic cell death in E1A-Ras transformed cells. Oncotarget. V. 6. P. 44905.
Guo J.Y., Chen H., Mathew R., Fan J., Strohecker A.M., Karsli-Uzunbas G., Kamphorst J.J., Chen G., Lemons J.M.S., Karantza V., Coller H.A., Dipaola R.S., Gelinas C. et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes&Devel. V. 25. P. 460.
Hanahan D., Weinberg R.A. 2011. Hallmarks of cancer: The next generation. Cell. V. 144. P. 646.
Hernandez-Segura A., Nehme J., Demaria M. 2018. Hallmarks of cellular senescence. Trends Cell Biol. V. 28. P. 436.
Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S., Natsume T., Takehana K., Yamada N., Guan J.L., Oshiro N., Mizushima N. 2009. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. V. 20. P. 1981.
Lampson B.L., Davids M.S. 2017. The development and current use of BCL-2 inhibitors for the treatment of chronic lymphocytic leukemia. Curr. Hematol. Malig. Rep. V. 12. P. 11.
Mariño G., Pietrocola F., Eisenberg T., Kong Y., Malik S. A., Andryushkova A., Schroeder S., Pendl T., Harger A., Niso-Santano M., Zamzami N., Scoazec M., Durand S., Enot D.P., Fernández Á.F. et al. 2014. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell. V. 53. P. 710.
Monick M.M., Powers L.S., Barrett C.W., Hinde S., Ashare A., Groskreutz D.J., Nyunoya T., Coleman M., Spitz D.R., Hunninghake G.W. 2008. Constitutive ERK MAPK activity regulates macrophage ATP production and mitochondrial integrity. J. Immunol. V. 180. P. 7485.
Niu N., Li Z., Zhu M., Sun H., Yang J., Xu S., Zhao W., Song R. 2019. Effects of nuclear respiratory factor-1 on apoptosis and mitochondrial dysfunction induced by cobalt chloride in H9C2 cells. Mol. Med. Rep. V. 19. P. 2153.
Porporato P.E., Filigheddu N., Bravo-San Pedro J.M., Kroemer G., Galluzzi L. 2018. Mit. Metabol. Cancer. Cell Res. V. 28. P. 265.
Quadir M.A., Kwok B., Dragowska W.H., To K.H., Le D., Bally M.B., Gorski S.M. 2008. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res. Treat. V. 112. P. 389.
Ryu S.J., Oh Y.S., Park S.C. 2007. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. V. 14. P. 1020.
Sarbassov D.D., Ali S.M., Sabatini D.M. 2005. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. V. 17. P. 596.
Serrano C., Romagosa C., Hernández-Losa J., Simonetti S., Valverde C., Moliné T., Somoza R., Pérez M., Vélez R., Vergés R., Domínguez R., Carles J., Ramón Y Cajal S. 2016. RAS/MAPK pathway hyperactivation determines poor prognosis in undifferentiated pleomorphic sarcomas. Cancer. V. 122. P. 99.
Toussaint O., Medrano E.E., von Zglinicki T. 2000. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exper. Gerontol. V. 35. P. 927.
Trotta A.P., Gelles J.D., Serasinghe M.N., Loi P., Arbiser J.L., Chipuk J.E. 2017. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. V. 292. P. 11727.
Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V., Kost-Alimova M., Muller F., Colla S., Nezi L., Genovese G. et al. 2014. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. V. 514. P. 628.
Von Zglinicki T., Saretzki G., Ladhoff J., d’Adda di Fagagna F., Jackson S. 2005. Human cell senescence as a DNA damage response. Mech. Ageing Dev. V. 126. P. 111.
Wang K., Klionsky D.J. 2011. Mitochondria removal by autophagy. Autophagy. V. 7. P. 297.
Warburg O. 1956a. On the origin of cancer cells. Science. V. 123. P. 309.
Warburg O. 1956b. On respiratory impairment in cancer cells. Science. V. 124. P. 269.
Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G.M., Budinger G.R.S., Chandel N.S. 2010. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA. V. 107. P. 8788.
Дополнительные материалы отсутствуют.