

Цитология, 2020, T. 62, № 7, стр. 493-502
Влияние мутации L345P в гене десмина на процесс аутофагии в мышечных клетках линии С2С12
К. С. Сухарева 1, *, Н. А. Смолина 1, А. А. Князева 1, К. К. Калугина 2, А. А. Худяков 1, А. А. Костарева 1
1 Институт молекулярной биологии и генетики Национального медицинского исследовательского центра им. В.А. Алмазова
197341 Санкт-Петербург, Россия
2 Санкт-Петербургский Государственный Университет
199034 Санкт-Петербург, Россия
* E-mail: k.sukhareva@gmail.com
Поступила в редакцию 23.10.2019
После доработки 14.04.2020
Принята к публикации 27.04.2020
Аннотация
Процесс аутофагии играет важную роль в жизнедеятельности клетки, в частности, в процессе поддержания протеостаза. В мышечных клетках процесс аутофагии является высоко динамичным в связи с необходимостью постоянного обновления разрушающихся белков области Z-диска при мышечном сокращении. При этом цитоскелет и, в частности промежуточные филаменты, играют основную роль в поддержании структурной и функциональной целостности мышечной клетки. Нами было предположено, что нарушение структуры промежуточных филаментов мышечной клетки, в частности десмина, ведет к изменению динамики процесса аутофагии. Мы продемонстрировали, что в клетках линии С2С12 мутация гена десмина (DES L345P) приводит к значительному увеличению базового уровня аутофагии и скорости деградации клеточных компонентов за счет процесса аутофагии. Было показано, что в клетках с мутацией DES L345P по сравнению с клетками дикого типа после стимуляции процесса аутофагии сывороточным голоданием в течение 2 и 4 ч количество белка LC3-II снижается в два раза относительно клеток с десмином дикого типа. При этом скорость конверсии формы LC3-I в LC3-II остается неизменной. Это наблюдение может быть связано с увеличение базового уровня процесса аутофагии и скорости деградации аутофагосом для удаления из клетки мутантных агрегантных форм белка десмина вследствие мутации L345P.
Промежуточные филаменты являются одним из компонентов клеточного цитоскелета и необходимы для поддержания структурной и функциональной целостности клетки (Paulin, Li, 2004). Они образуют прочную сеть, волокна которой по диаметру занимают промежуточное положение между нитями актина и микротрубочками, что и обусловливает их название. Основываясь на структуре, выделяют несколько классов промежуточных филаментов, представителями которых являются кератины, ламины, десмин, нейрофиламенты, глиальный фибриллярный белок и другие (Paulin, Li, 2004; Herrmann, Aebi, 2016). В большинстве случаев промежуточные филаменты являются тканеспецифичными, и каждый их тип экспрессируется в определенных клетках и тканях. Исключение составляют белки ламины, которые являются структурным компонентом всех ядер, а также белок синемин, способный кополимеризоваться с большинством других представителей промежуточных филаментов. В настоящее время известно множество заболеваний, связанных с мутациями генов, кодирующих белки промежуточных филаментов (Dalakas, 2000; Worman, Courvalin, 2004). С учетом тканеспецифичной экспрессии данных белков, в случае их структурных нарушений патологический процесс затрагивает преимущественно одну систему органов. Так, например, мутации генов кератинов приводят к развитию кожного заболевания – буллезного эпидермиолиза, а в случае мутаций в гене глиального ацидофильного белка GFAP развивается болезнь Александера – прогрессирующее нейродегенеративное заболевание (Omary, 2009; Jerabkova, Marek et al., 2010).
Основным промежуточным филаментом скелетно-мышечных, а также гладкомышечных клеток и кардиомиоцитов является белок десмин. Его филаменты формируют трехмерную сеть, которая обеспечивает взаимодействие сократительного аппарата мышечной клетки с ядром, лизосомами, митохондриями и другими клеточными органеллами (Capetanaki, Bloch et al., 2007). Десмин участвует в процессе механотрансдукции посредством тесного структурного взаимодействия десминовых филаментов с компонентами Z-диска. Эта структура обеспечивает механическую целостность саркомера и содержит в своем составе более 15 различных белков, которые имеют как структурные, так и сигнальные функции, выполняя в дифференцированных мышечных клетках роль центра механочувствительности (Goldfarb, Park et al., 1998; Capetanaki, Bloch et al., 2007; Capetanaki, Papathanasiou et al., 2015). Показано, что нарушение целостности и функций десмина вследствие мутаций приводит к нарушению структуры Z-дисков саркомера и развитию заболеваний сердечной мышцы и скелетно-мышечной мускулатуры (кардиомиопатий), дистальных и миофибриллярных миопатий (Elliott, Andersson et al., 2008; Kley, van der Ven et al., 2013).
В основе развития патологического процесса при мутациях в генах промежуточных филаментов часто лежит способность структурно-аномальных белков формировать нерастворимые белковые агрегаты. Именно поэтому в большинстве случаев при мутациях в генах промежуточных филаментов иммуногистохимическое окрашивание и электронная микроскопия обнаруживают в клетках миофибриллярные белковые массы, состоящие из патологически измененного белка и ассоциированные с ними белки-шапероны (Sanghvi-Shah, Weber, 2017). В большинстве случаев, как, например, при миофибриллярных миопатиях (мутации в гене DES), боковом амиотрофическом склерозе (мутации в гене NEFH), болезни Александера (мутации в гене GFAP) и болезни Шарко−Мари−Тут (мутации в гене NEFL) такие внутриклеточные белковые агрегаты являются диагностическим маркером. В норме их деградация обеспечивается процессом селективной макроаутофагии (Tang, Yue et al., 2008).
Особенно значимым этот процесс для мышечных клеток, в которых структурные компоненты Z-диска постоянно подвергаются механическому воздействию, разрушаются и должны быть своевременно утилизированы посредством селективной шаперон-ассистированной аутофагии (Nair, Klionsky, 2011). Поддержание протеостаза в данном компартменте клетки является обязательным условием физиологического функционирования миоцитов, в связи с чем процесс аутофагии является неотъемлемой частью системы контроля качества белков в кардиомиоцитах в норме и при развитии патологического процесса (Grumati, Coletto et al., 2011; Jia, Xue et al., 2018). Нарушение этого процесса вследствие мутаций генов, кодирующих белки Z-диска и факторы селективной макроаутофагии, такие как BAG3, DNAJB6 и HSPB8 приводит к формированию миофибриллярных внутриклеточных агрегатов и, как следствие, развитию миопатии и кардиомиопатии.
Примером может являться мутация L345P гена десмина (DES), затрагивающая структуру головного домена белка (Sjoberg, Saavedra-Matiz et al., 1999). При наличии этой мутации нарушается процесс полимеризации десмина и происходит образование агрегатов, препятствующих взаимодействию десмина с клеточными органеллами. Белковые агрегаты, имеющие в составе белки десмин, филамин, кристаллин и ряд белков-шаперонов области Z-диска подвергаются деградации посредством селективной аутофагии. В то же время, сам процесс аутофагии может быть нарушен вследствие дисфункции десминовых филаментов, поскольку слияние аутофагосомы с лизосомами происходит с опосредованным участием десмина. Детальные механизмы влияния дисфункции десминовых филаментов на процесс аутофагии в мышечных клетках не изучены. В связи с этим цель нашего исследования заключалась в изучении влияния мутации L345P в гене DES на процесс аутофагии в клетках мышиных миобластов линии С2С12.
МАТЕРИАЛ И МЕТОДИКА
Клетки. Работу проводили на мышиных иммортализованных мышечных сателитных клетках линии С2С12 (ATCC, США, каталожный номер CRL 1772). Для культивирования клеток использовали среду на основе DMEM (Invitrogen, США), содержащую 10% телячьей эмбриональной сыворотки (FCS; Invitrogen, США), 1% смеси антибиотиков пенициллина и стрептомицина (Invitrogen, США) и 1% L-глутамата (Invitrogen, США) при температуре 37°С, 5%-ном содержании CO2 и влажности 99%. Клетки пересевали каждые 72 ч по 100 тыс. на чашку диаметром 100 мм.
Дизайн эксперимента. Клетки мышечной линии С2С12 трансдуцировали геном десмина дикого типа и геном десмина, несущим мутацию L345P. После стимуляции процесса аутофагии в течение 2–8 ч с помощью Вестерн-блотинга и иммуноцитохимии оценивали содержание в клетках изоформ белка LC3 – белкового компонента аутофагосом, который имеет две изоформы: LC3-I – растворимая или свободная фракция белка, содержащаяся в цитоплазме и изоформа LC3-II – связанная или интергрированная в мембрану аутофагосомы фракция белка.
Культивирование и вирусная трансдукция клеток С2С12. Для наработки лентивирусных конструкций клетки линии НЕК293-Т котрансфецировали тремя плазмидами на основе плазмиды pLVNHM: смысловой плазмидой с геном интереса дикого типа и заменой L345P (DES L345P) плазмидой, кодирующей белки вирусной оболочки (pMD2.G), и плазмидой, кодирующей пакующие белки (psPAX2). Для увеличения эффективности трансдукции реагент PEI (Sigma, USA) добавляли в среду DMEM из расчета 2 : 1 на общее количество пДНК. Трансдукцию клеток С2С12 проводили вирусными частицами из расчета 20 мкл вируса на 60 тыс. клеток в присутствии полибрена (Sigma, Германия) в концентрации 8 мкг/мл на весь объем среды для культивирования.
Стимуляция процесса аутофагии. Клетки линии С2С12 после трансдукции генами дикого типа и DES L345P высевали в количестве 50 или 30 тыс. на чашку диаметром 60 мм для последующей детекции методами Вестерн-блотинга и иммуноцитохимии соответственно. Через 72 ч после проведения трансдукции для индукции аутофагии проводили замену клеточной среды на бессыворочную среду EBSS (Earle’s Balanced Salt Solution) для глодания клеток. Стимуляцию аутофагии проводили в течение 2–8 ч.
Вестерн-блот-анализ. Для количественной детекции изоформ белка LC3 использовали протокол, отработанный ранее для клеток линии С2С12 (Сухарева, Смолина и др., 2016; Klionsky, Abdelmohsen et al., 2016) После стимуляции аутофагии в течении 2–8 ч клеточную суспензию помещали в пробирки и центрифугировали при 1000 g в течении 5 мин. Пермеабилизацию клеточного осадка проводили с использованием детергента дигитонина (Sigma, USA) в PBS в концентарции 0.025% в течении 5 мин на льду. После центрифугирования при 2000 g в течении 5 мин надосадочную жидкость собирали для дальнейшего анализа растворимой изоформы белка LC3 (LC3-I). Экстракцию нерастворимой фракции белка LC3 (LC3-II) проводили путем лизиса клеточного осадка в буфере RIPA, содержащем коктейль ингибиторов протеаз (Roche, США) и модификации нескольких компонентов для увеличения эффекивности лизиса (1% NP-40, 0.5% диоксихолата натрия; 1% SDS; 1% Triton-X; 5 мМ EDTA) в течение 10 мин на льду. Белковый лизат центрифугировали при 16000 g в течении 10 мин, после чего полученную надосадочную жидкость собирали в новую пробирку для дальнейшего анализа нерастворимой изоформы LC3-II, связанной с мембраной аутофагосом.
Для электрофореза в концентрирующем геле использовали постоянный ток 20 мА, в разделяющем геле – 40 мА. Перенос разделенных по молекулярной массе белков на нитроцеллюлозную мембрану (Applichem, США) с диаметром пор 0.45 мкм проводили в течение 1 ч при постоянном напряжение 100 В с использованием буфера для переноса (49.9 мМ TrisHCl, 38.6 мМ глицин, 0.0385% SDS, 20% метанола). Для оценки качества процесса переноса белков мембрану обратимо окрашивали красителем Ponceau S (Sigma-Aldrich, США) с последующей отмывкой в PBS, содержащем 0.05% Tween 20 (PBS-Tween). Блокировку неспецифического связывания антител осуществляли путем икубирования в 5%-ном растворе обезжиренного молока, разведенного в PBS-Tween 0.05% в течение 2 ч при комнатной температуре. Инкубацию мембраны в растворе первичных поликлональных антител анти-LC3 (MBL International Corporation, США) в разведении 1 : 7000 в 5%-ном растворе молока на PBS-Tween проводили в течение ночи при температуре 4°С. В качестве вторичных использовалиись антител против иммуноглобулинов кролика, конъюгированные c пероксидазой хрена (BioRad, США) в концентрации 1 : 20 000, разведенные в растворе PBS-Tween. Инкубацию проводили в течении 1 ч при комнатной температуре. Детекцию сигнала пероксидазной активности осуществляли с использованием реактива для усиленной хемилюминисценции Super signal west femto maximum sensitivity substrate (Thermo Fisher Scientific, США) согласно инструкции производителя в камере для детекции люминесцентного сигнала Vilber Lourmat (BioRad, США).
Иммуноцитохимия. Количественный анализ образовавшихся в клетках аутофагосом проводили методом иммуноцитохимического окрашивания с применением антител к LC3. Клеточную культуру, посеянную на культуральные стекла, предварительно подвергали стимуляции процесса аутофагии в течение 2–4 ч. Без предварительной фиксации клетки инкубировали 5 мин в 0.005%-ном растворе дигитонина (Sigma, Германия) в PBS на льду для экстракции растворимой изоформы LC3-I. После трехкратной отмывки в PBS в течение 5 мин клетки фиксировали 4%-ным раствором парафармальдегида PFA (Sigma, США) в течение 5 мин на льду. Клетки блокировали 15%-ным раствором сыворотки FCS (Gibco, США) при комнатной температуре 30 мин. Инкубацию в растворе первичных поликлональных антител анти-LC3 (MBL International Corporation, США) при разведении 1 : 500 проводили при комнатной температуре в течение 1 ч. В качестве вторичных антител использовали антитела Alexa-Fluor 560 против иммуноглобулинов кролика (Thermo Fisher Scientific, США) в разведении 1 : 200 в течение 45 мин при комнатной температуре в темноте. Для визуализации ядер испольовали краситель DAPI (Invitrogen, США) в течение 30 с.
Обработка данных. Обработка данных, полученных методом Вестерн блот проводилась с использованием программы Fusion в режиме количественной оценки белка в пробе. Оценка количества белка в банде проводилась c помощью оценки оптической плотности пикселей на выбранную область образца. Нормализация концентрации образцов проводилась по анализу снимков мембраны с использованием программы Fusion после окрашивание красителем Ponceau S (Sigma-Aldrich, США).
Количественная оценка сигнала LC3-II в клетках после проведения иммуноцитохимического окращивания проводилась методом подсчета флуоресцентных точек на фотографиях исследуемых образцов, сделанных с помощью флуоресцентного микроскопа Observer.D1, в программе MosaiX (Carl Zeiss Microsystems, Германия).
Для статистической обработки данных и построения графиков использовали программу GraphPad. Полученные результаты представлены в виде среднего значения по всем проведённым экспериментам. Достоверность различий между группами оценивали при помощи непараметрического критерия Манна−Уитни. Достоверными считали различия при уровне значимости P < 0.05.
РЕЗУЛЬТАТЫ
Динамика процесса аутофагии в клетках линии С2С12. На начальном этапе нами была проведена оценка динамики процесса аутофагии в нетрансдуцированных клетках линии С2С12 при стимуляции процесса бессывороточным голоданием в течение 2–8 ч. Был выявлен переход изоформы LC3-I в изоформу LC3-II и уменьшение через 8 ч голодания изофрмы LC3-I более чем в 2 раза относительно контрольных клеток (рис. 1). В то же время, динамика формирования и утилизации нерастворимой изоформы LC3-II показала накопление ее в течение 2 ч голодания с последующей утилизацией и уменьшением количества LC3-II до минимальных значений к 8 ч голодания (рис. 1). При стимуляции аутофагии в течении 2–8 ч в клетках линии С2С12, трансдуцированных геном DES дикого типа и при трансдукции конструкцией, несущей мутацию L345P этого гена была отмечена сходная динамика изоформ LC3-I и LC3-II (рис. 2а–в). Таким образом, для дальнейшей оценки процесса аутофагии в клетках линии С2С12 и взаимного перехода изоформ белка LC3 использовали временные точки 2 и 4 ч.
Рис. 1.
Динамика процесса ауатофагии в нетрансдуцированных клетках линии С2С12, показанная по динамике растворимой (LC3-I) и нерастворимой (LC3-II) фракций белка LC3 в течение 2, 4, 6 и 8 ч голодания по данным Вестерн-блот-анализа (а, мембрана окрашена анти Ponceau S) и ее графическое представление по данным денситометрии (оптической плотности D) (б). К – контроль (в отсутствие голодания).
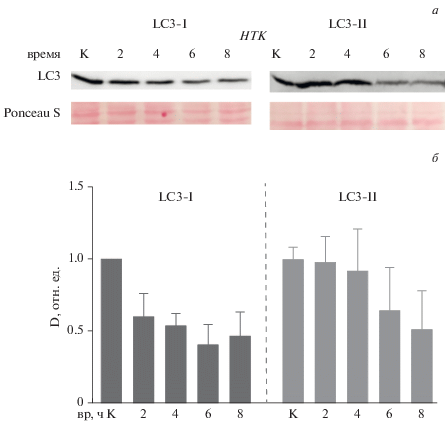
Рис. 2.
Сравнение процесса аутофагии клеток С2С12, трансдуцированных конструкцией с геном десмина DES дикого типа (ДТ) и клеток, трансдуцированных конструкцией с мутантным DES L345P. а, в – Динамика изоформ LC3-I и LC3-II в течение 2–8 ч голодания. б, г – Графическое представление динамики, показанной на рисунке а, б соответственно, по данным десмитометрии (D); К – контроль (отсутствие голодания); показаны средние значения и среднеквадратичные отклонения (вертикальные отрезки); достоверность различий Р оценивали при помощи непараметрического критерия Манна−Уитни.
Вестерн-блот-анализ аутофагии в клетках С2С12. После индукции аутофагии в течении 2 ч нами было выявлено, что растворимая форма белка LC3 (LC3-I) расходуется с одинаковой скоростью в образцах дикого типа и в образцах с мутацией L345P гена десмина, что обусловлено образованием новых аутофагосом и переходом растворимой изоформы LC3-I в связанную с мембраной аутофагосомы изоформy LC3-II (рис. 3 Iа, в). В то же время, по количеству изоформы LC3-II были выявлены значимые различия. В отличие от клеток дикого типа, в которых наблюдали незначительное повышение уровня белка LC3-II через 2 ч голодания, в клетках с мутацией L345P гена DES происходило значимое снижение количества изоформы LC3-II (рис. 3 Iб–г).
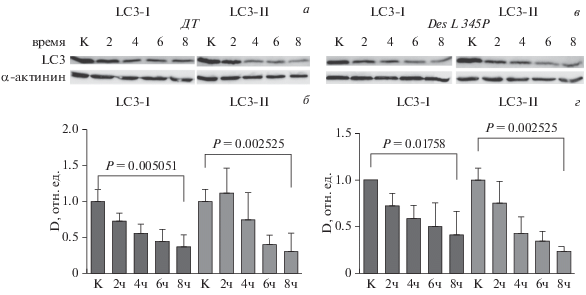
Рис. 3.
Количество растворимой (LC3-I; а, б) и нерастворимой (LC3-II; в, г) фракций белка LC3 в клетках С2С12, трансдуцированных конструкцией, несущей ген DES дикого типа (ДТ) или его мутант DES L345P (L345P) через 2 (I) и 4 (II) ч после индукции аутофагии бессывороточным голоданием. а, в – По данным Вестерн-блот-анализа. б, г – Графическое представление по данным денситометрии (D); показаны средние значения и среднеквадратичные отклонения (вертикальные отрезки), достоверность различий Р оценивали при помощи непараметрического критерия Манна–Уитни.
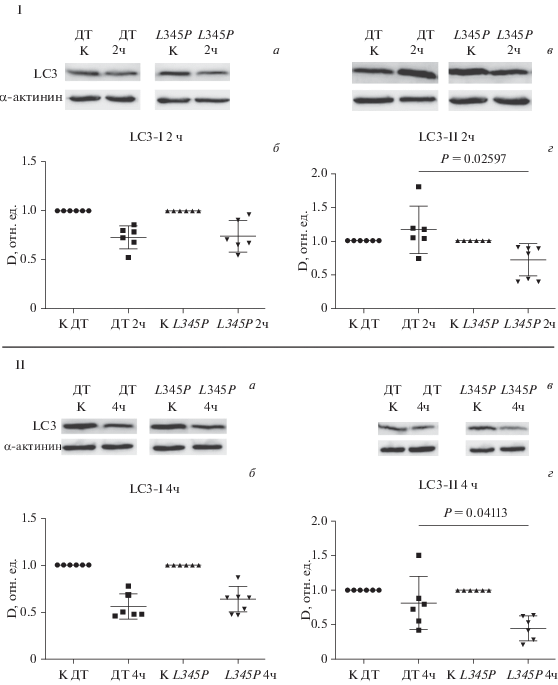
При 4-часовой стимуляции аутофагии количество белка LC3-I уменьшилось относительно точки 2 ч как в клетках дикого типа, так и в L345P-мутантах, значимо не различаясь между собой (рис. 3 IIа–в). Накопление нерастворимой изоформы LC3-II за 4 ч сывороточного голодания прекратилось, и наступил этап деградации содержимого аутофагасом лизосомальными ферментми. В образцах с мутацией L345P гена DES наблюдали значимо меньшее количество белка LC3-II относительно его содержания в клетках дикого типа после 4-часового голодания (рис. 3 IIб–г).
Динамики аутофагии в клетках С2С12 по данным иммуноцитохимии. Предварительные результаты, полученные методом Вестерн-блот-анализа, были далее подтверждены методом иммуноцитохимии. Нами была выявлена схожая с данными Вестерн-блот-анализа зависимость количества белка LC3-II в клетках линии С2С12 от продолжительности стимуляции процесса аутофагии. В клетках, несущих мутацию DES L345P, через 4 ч сывороточного голодания наблюдали уменьшение уровня LС3-II в 2 раза относительно клеток с DES дикого типа, что может быть связано либо со снижением скорости образования аутофагосом, либо с ускорением процесса их деградации лизосомальными ферментами (рис. 4).
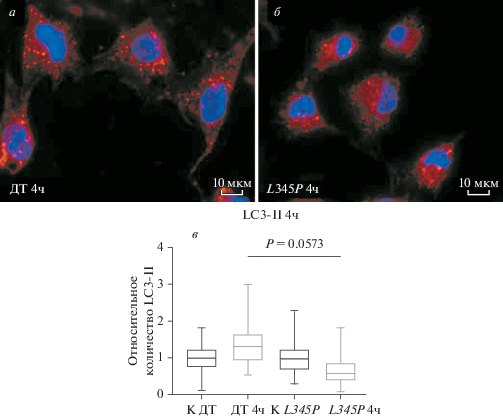
Рис. 4.
Аутофагосомы в клетках С2С12, трансдуцированных DES дикого типа (ДТ, а) и его мутантом DES L345P (L345P, б), после 4-часового сывороточного голодания. а, б – По данным иммуноцитохимии с окраской только нерастворимой фракции белков, содержащей LC3-II, с использованием анти-LС3-антител; ядра окрашены DAPI (синий цвет), аутофагомомы – анти-LC3-антителами (красный цвет); об.: 100×. в – График, отражающий количество LC3-II, которое определяли подсчетом числа аутофагосом в клетках с DES дикого типа (ДТ), в клетках с DES L345P (L345P) в отсутствие голодания (К, контроль) и после 4-часового сывороточного голодания; показаны средние значения и среднеквадратичные отклонения (вертикальные отрезки), достоверность различий Р оценивали при помощи непараметрического критерия Манна–Уитни.
Базальный уровень аутофагии в клетках С2С12 после трансдукции вирусными конструкциями, несущими различные варианты DES. Базовый уровень аутофагии в нетрансдуцированных клетках С2С12, а также в клетках, трансдуцированных десмином дикого типа и клетках, несущих мутацию DES L345P, проводили двумя методами: иммуноцитохимии и Вестерн-блот-анализа. В клетках, трансдуцированных вирусным вектором (несущим как DES дикого типа, так и мутантный вариант DES L345P), наблюдали тенденцию к большему количеству изоформы LC3-II, чем в нетрансдуцированных клетках, что свидетельствует о повышении базового уровня аутофагии вследствие проведения вирусной трансдукции (рис. 5 I). Применение метода иммуноцитохимии показало, что в клетках С2С12 с мутацией DES L345P до индукции аутофагии количество аутофагосом отмечается значимо больше, чем в нетрансдуцированных клетках (рис. 5 IIа–г).
Рис. 5.
Сравнение базовых уровней аутофагии в клетках С2С12, не подвергшихся стимуляции аутофагии, по данным Вестерн-блот-анализа (I) и в клетках, трансдуцированных различными формами гена десмина по данным иммуноцитохимии (IIг). II: а–в – Окраска только нерастворимой фракции белков, содержащей LC3-II, анти-LС3-антителами в нетрансдуцированных клетках (НТК, a), в клетках, трансдуцированных DES дикого типа (ДТ, б) и его мутантом DES L345P (L345P, в); ядра окрашены DAPI (синий цвет), аутофагомомы – анти-LC3-антителами (красный цвет), об.: 100×. На графиках базовые уровни показаны относительно НТК, даны средние значения и среднеквадратичные отклонения (вертикальные отрезки); достоверность различий Р оценивали при помощи непараметрического критерия Манна–Уитни.
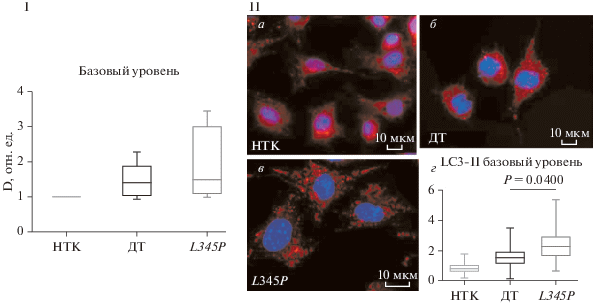
Важно отметить, что трансдукция клеток конструкцией с DES дикого типа также приводила к незначимому повышению базового количества аутофагосом по сравнению с нетрансдуцированными клетками. Таким образом, иммуноцитохимическое окрашивание показало, что трансдукция клеток С2С12 мутацией DES L345P приводит к повышению базового содержания аутофагосом и нерастворимой изоформы белка LC3-II в клетках.
ОБСУЖДЕНИЕ
Известно, что мутации в гене DES связаны с развитием нервно-мышечной патологии и патологии миокарда (Brodehl, Gaertner-Rommel et al., 2018). В основе развития данной патологии на клеточном уровне лежит дезинтеграция мышечных волокон и образование нерастворимых внутриклеточных белковых агрегатов (Bar, Kostareva et al., 2006). Существует предположение, что в патогенезе формирования белковых агрегатов имеет значение нарушение эффективности процесса аутофагии (Bhuiyan, Pattison et al., 2013). Помимо структурной основы Z-диска, десмин также представляет собой одну из основных транспортных магистралей для созревания аутофагосом и дальнейшего их слияния с лизосомами (Tsoupri, Capetanaki, 2013). Таким образом, нарушение структуры десмина вследствие мутаций в кодирующем его гене может приводить как к нарушению в структуре клеточного цитоскелета, так и к нарушению процесса аутофагии, внося, тем самым, вклад в прогрессирующее формирование внутриклеточных белковых агрегатов.
Наблюдаемое в нашей работе снижение содержания изоформы LC3-I по мере увеличения времени бессывороточного голодания отражает закономерную динамику процесса аутофагии в мышечных клетках в нормальных условиях. В то же время в исследуемых клетках лини С2С12 стимуляция аутофагии не приводит к значительному накоплению изоформы LC3-II, что характерно для других типов клеток, например, клеток линии НеLa, MEF или HEK293 (Klionsky, Abdelmohsen et al., 2016). Такая динамика может являться отражением интенсивного процесса деградации аутофагосом путем быстрого слияния их с лизосомами в мышечных клетках и быть направлена на поддержание эффективного протеостаза в активно сокращающихся и подверженных механическому воздействию миоцитах. Более того, в клетках с мутацией DES L345P не происходит даже незначительного накопления изоформы LC3-II в течение 2 ч, что может являться отражением еще более быстрого процесса деградации аутофагосом в клетках, несущих мутацию DES L345P.
Резкое снижение количества изоформы LC3-II в образцах с мутацией DES L345P за 4 ч бессывороточного голодания может указывать как на снижение скорости образования аутофагосом, так и на повышенную скорость деградации за счет лизосомальных ферментов. В то же время, сравнение скорости перехода изоформы LC3-I в изоформу LC3-II в течение всего времени голодания в клетках дикого типа и клетках с мутацией DES L345P выявило, что скорость расходования изоформы LC3-I являлась одинаковой. Таким образом, можно констатировать отсутствие значимого влияния мутаций DES L345P на процесс созревания аутофагосом, и предположить, что в основе наблюдаемого эффекта лежит повышение скорости их деградации для своевременной утилизации мутантных форм белка.
Анализ результатов иммуноцитохимического исследования также выявил более высокий базальный уровень изоформы LC3-II в клетках с мутацией по сравнению с клетками дикого типа. Вопрос о природе повышения базального уровня аутофагии в клетках с мутацией остается открытым. Так как мутация DES L345P приводит к формированию внутриклеточных белковых агрегатов, а ее присутствие в клетках ведет к нарушению сборки десминовых филаментов, можно предположить, что процесс аутофагии в клетках С2С12 имеет большую интенсивность еще до стимуляции бессывороточным голоданием для поддержания протеостаза мышечных клеток (Ulbricht, Gehlert et al., 2015). Таким образом, присутствие в мутантных клетках большего количества аутофагосом может быть связано с повышением базового уровня аутофагии для утилизации белков с нарушенной конформацией (King, 2012). Важно также отметить, что в клетках линии С2С12 вирусная трансдукция с использованием лентивирусных плазмид сама по себе приводит к активации процесса аутофагии.
Механизм запуска процесса аутофагии различается в зависимости от тканевой принадлежности клеток. Известно, что одним из основных сигналов к запуску процесса аутофагии в мышечных клетках является наличие белковых агрегатов, которые необходимо удалить из клетки для обеспечения поддержания гомеостаза, а также для поддержания структурной и функциональной целостности клетки (Bravo-San Pedro, Kroemer et al., 2017). В нашей работе мы использовали мутацию гена DES L345P, которая ассоциирована с наиболее клинически выраженными фенотипическими проявлениями десминовой кардиомиопати и миопатии (Capetanaki, Papathanasiou et al., 2015). Наличие в организме этой мутации препятствует правильной сборке десминовых филаментов, что ведет к образованию в клетке миофибриллярных агрегатов. Можно предполагать, что интенсификация процесса аутофагии в мутантных мышечных клетках линии С2С12 предотвращает накопление образовавшихся агрегатов, защищает клетку от нарушения функциональной целостности и способствует сохранению гомеостаза. В связи с этим процесс аутофагии в настоящее время активно рассматривается в качестве одной из мишеней для терапевтического воздействия при лечении миофибриллярных миопатий, в частности, путем применения активаторов и блокаторов аутофагии (Grumati, Coletto et al., 2011; Perrone, Squillaro et al., 2019).
Итак, в настоящей работе нами было показано, что трансдукция клеток линии С2С12 лентивируснымии конструкциями, несущими мутантую форму DES L345P, проявляется в повышении базального уровня образования аутофагосом. В клетках линии С2С12 динамика уменьшения изоформы LC3-I при переходе в изоформу LC3-II с накоплением последней за 2 ч бессывороточного голодания и последующей деградацией при увеличении времени голодания отражает нормальный процесс аутофагии в клетках, трансдуциированных геном DES дикого типа. Мутация DES L345P приводит к индукции процесса аутофагии с клетках С2С12, что отражается в повышенном базальном содержании аутофагосом и ускорении утилизации изоформы LC3-II по сравнению с клетками, трансдуцированными DES дикого типа. В продолжение данной работы планируется исследовать действие ингибиторов и активаторов аутофагии, таких как хлорокин, вортманин, пепстатин, рапамицин.
Список литературы
Сухарева К.С., Смолина Н.А., Головкин А.С., Худяков А.А., Князева А.А., Мишанин А.И., Костарева А.А. 2016. Методические подходы к детекции процесса аутофагии в мышечных клетках. Трансляционная медицина. Т. 3. С. 129. (Sukhareva K.S., Smolina N.A., Golovkin A.S., Khudiakov A.A., Knyazeva A.A., Mishanin A.B., Kostareva A.A. 2016. Methodic approaches to detect autophagy in muscle cells. Translat.Med. (Russ.). V. 3. P. 129.)
Bar H., Kostareva A., Sjoberg G., T. Sejersen T., Katus H. A., Herrmann H. 2006. Forced expression of desmin and desmin mutants in cultured cells: impact of myopathic missense mutations in the central coiled-coil domain on network formation. Exp. Cell Res. V. 312. P. 1554.
Bhuiyan M. S., Pattison J. S., Osinska H., James J., Gulick J., McLendon P.M., Hill J.A., Sadoshima J., Robbins J. 2013. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. V. 123. P. 5284.
Bravo-San Pedro J.M., Kroemer G., Galluzzi L. 2017. Autophagy and mitophagy in cardiovascular disease. Circ. Res. V. 120. P. 1812.
Brodehl A., Gaertner-Rommel A., Milting H. 2018. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. V. 10. P. 983.
Capetanaki Y., Bloch R.J., Kouloumenta A., Mavroidis M., Psarras S. 2007. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. V. 313. P. 2063.
Capetanaki Y., Papathanasiou S.A., Diokmetzidou A., Vatsellas G., Tsikitis M. 2015. Desmin related disease: a matter of cell survival failure. Curr. Opin. Cell. Biol. V. 32. P. 113.
Dalakas M.C., M.D., Park K.-Y., Ph.D., Semino-Mora C., M.D., Ph.D., Lee H.S., M.D., Sivakumar K., M.D., Goldfarb L.G., M.D. 2000. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. The New Engl. J. Med. V. 342. P. 770.
Elliott P., Andersson B., Arbustini E., Bilinska Z., Cecchi F., Charron P., Dubourg O., Kuhl U., Maisch B., McKenna W.J., Monserrat L., Pankuweit S., Rapezzi C., Seferovic P., Tavazzi L., Keren A. 2008. Classification of the cardiomyoies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. V. 29. P. 270.
Goldfarb L.G., Park K.Y., Cervenakova L., Gorokhova S., Lee H.S., Vasconcelos O., Nagle J.W., Semino-Mora C., Sivakumar K., Dalakas M.C. 1998. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. V. 19. P. 402.
Grumati P., Coletto L., Sandri M., Bonaldo P. 2011. Autophagy induction rescues muscular dystrophy. Autophagy. V. 7. P. 426.
Herrmann H., Aebi U. 2016. Intermediate filaments: structure and assembly. Cold Spring Harb. Perspect. Biol. V. 8. pii: a018242. https://doi.org/10.1101/cshperspect.a018242
Jerabkova B., Marek J., Buckova H., Kopeckova L., Vesely K., Valickova J., Fajkus L., Fajkusova J. 2010. Keratin mutations in patients with epidermolysis bullosa simplex: correlations between phenotype severity and disturbance of intermediate filament molecular structure. Br. J. Dermatol. V. 162. P. 1004.
Jia B., Xue Y., Yan X., Li J., Wu Y., Guo R., Zhang J., Zhang L., Li Y., Liu Y., Sun L. 2018. Autophagy inhibitor chloroquine induces apoptosis of cholangiocarcinoma cells via endoplasmic reticulum stress. Oncol. Lett. V. 16. P. 3509.
King J.S. 2012. Mechanical stress meets autophagy: potential implications for physiology and pathology. Trends Mol. Med. V. 18. P. 583.
Kley R.A., van der Ven P.F., Olive M., Hohfeld J., Goldfarb L.G., Furst D.O., Vorgerd M. 2013. Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy. V. 9. P. 422.
Klionsky D.J., Abdelmohsen K., Abe A, Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams CM., Adams P.D., Adeli K., Adhihetty P.J., Adler S.G., Agam G., Agarwal R., Aghi M.K. et al. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. V. 12. P. 1.
Nair U., Klionsky D.J. 2011. Activation of autophagy is required for muscle homeostsis during physical exercise. Autophagy. V. 7. P. 1405.
Omary M.B. 2009. “IF-pathies”: A broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. V. 119. P. 1756.
Paulin D., Li Z. 2004. Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. V. 301. P. 1.
Perrone L., Squillaro T., Napolitano F., Terracciano C., Sampaolo S., Melone M.A.B. 2019. The autophagy signaling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients. V. 11. P. 1881.
Sanghvi-Shah R., Weber G.F. 2017. Intermediate filaments at the junction of mechanotransduction, migration, and development. Front. Cell Dev. Biol. V. 5. P. 81.
Sjoberg G., Saavedra-Matiz C.A., Rosen D.R., Wijsman E.M., Borg K., Horowitz S.H., Sejersen T. 1999. A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum. Mol. Genet. V. 8. P. 2191.
Tang G., Yue Z., Talloczy Z., Hagemann T., Cho W., Messing A., Sulzer D.L., Goldman J.E. 2008. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. V. 17. P. 1540.
Tsoupri E., Capetanaki Y. 2013. Muyospryn: a multifunctional desmin-associated protein. Histochem. Cell Biol. V. 140. P. 55.
Ulbricht, A., Gehlert S., Leciejewski B., Schiffer T., Bloch W., Hohfeld J. 2015. Induction and adaptation of chaperone-assisted selective autophagy CASA in res ponse to resistance exercise in human skeletal muscle. Autophagy. V. 11. P. 538.
Worman H.J., Courvalin J.C. 2004. How do mutations in lamins A and C cause disease? J. Clin. Invest. V. 113. P. 349.
Дополнительные материалы отсутствуют.