

Цитология, 2020, T. 62, № 9, стр. 672-678
Влияние тяжелой гипобарической гипоксии на содержание маркера аутофагии LC3 в гиппокампе крыс
А. В. Чурилова 1, *, Т. Г. Зачепило 1, М. Ю. Зенько 1
1 Федеральное Государственное Бюджетное Учреждение Науки Институт физиологии им. И.П. Павлова РАН
199034 Санкт-Петербург, Россия
* E-mail: annch05@mail.ru
Поступила в редакцию 02.06.2020
После доработки 11.06.2020
Принята к публикации 14.06.2020
Аннотация
Аутофагия – внутриклеточный механизм деградации цитоплазматических молекул и органелл в аутофагосомах, необходимый для поддержания клеточного гомеостаза и нормального функционирования нейронов, в том числе при действии экстремальных факторов. Несмотря на это, роль аутофагии в механизмах адаптивных и патологических реакций мозга до сих пор изучена слабо и представляет собой актуальную проблему для исследований. Изучение процессов аутофагии в нейронах в условиях повреждающих воздействий, имеет фундаментальное значение и одновременно может быть полезным с точки зрения разработки специфичных препаратов, направленных на звенья аутофагического каскада. Поскольку одним из наиболее распространенных повреждающих факторов является гипоксия, цель настоящей работы заключалась в оценке активности процесса аутофагии в гиппокампе крыс после действия тяжелой гипоксии. Использовали иммуногистохимический метод. Было выявлено, что тяжелая гипобарическая гипоксия (180 мм рт. ст., 3 ч) приводит к увеличению активности аутофагии в нейронах полей гиппокампа, которая проявляется снижением содержания маркера LC3 через 1 сут после воздействия. Вместе с тем, через 3 сут после гипоксии уровень LC3 восстанавливается до контрольных значений. Таким образом, полученные в модели in vivo данные свидетельствуют об усилении процессов аутофагической деградации в гиппокампе крыс в ответ на действие гипобарической гипоксии.
Аутофагия – внутриклеточный регулируемый механизм деградации в аутофагосомах белков с нарушенной структурой и поврежденных органелл (Klionsky, Emr, 2000; Levine, Klionsky, 2004; Mizushima et al., 2004; Komatsu et al., 2006). Механизмы аутофагии осуществляют контроль качества внутриклеточных компонентов и направлены на поддержание клеточного гомеостаза, необходимого для дифференциации, развития и нормального функционирования клеток (Kuma et al., 2004). Особенно большое значение аутофагическая деградация поврежденных клеточных органелл и белков приобретает при различных экстремальных воздействиях, в том числе окислительном стрессе, за счет своевременной утилизации поврежденных компонентов (Kiffin et al., 2004).
Гипоксия – один из наиболее распространенных повреждающих факторов, ведущий компонент патогенеза многих заболеваний, в том числе ишемической болезни сердца, инсульта головного мозга и многих неврологических нарушений. В экспериментальных моделях на животных показано, что гипоксия, вызванная снижением атмосферного давления (тяжелая гипобарическая гипоксия (ТГГ), 180 мм рт. ст. в течение 3 ч) вызывает структурные повреждения нейронов наиболее уязвимых областей мозга (неокортекса, гиппокампа) (Рыбникова и др., 2004). Возможно, одним из важнейших компонентов ответной реакции нейронов мозга на действие гипоксии является изменение активности аутофагии.
Нейроны являются высоко дифференцированными клетками с протяженными отростками, и поэтому их функционирование в значительной мере зависит от механизмов аутофагии (Hara et al., 2006; Komatsu et al., 2006). Показано, что активация аутофагии отмечается после ишемии мозга (Zhu et al., 2005; Rami et al., 2008), а также в нейронах, подверженных эксайтотоксическому стрессу (Wang et al., 2008). Дальнейшее детальное изучение роли аутофагии в механизмах повреждающего действия гипоксии на нейроны мозга представляет собой актуальную проблему, исследование которой имеет не только фундаментальное, но и практическое значение, поскольку создает основу для разработки специфических препаратов, оказывающих таргетное действие на отдельные звенья каскада аутофагии.
Цель настоящей работы состояла в оценке активности аутофагии в структурах мозга крыс (гиппокампе) после воздействия ТГГ.
Поскольку аутофагия является динамическим процессом, зависящим от скорости образования аутофагосом и скорости их деградации, то под активностью аутофагии (autophagic flux) подразумевается то, насколько интенсивно происходит оборот аутофагосом в клетке. Для оценки активности аутофагии в настоящей работе использовали хлорохин – неселективный ингибитор аутофагии поздней стадии, действие которого связано с изменением рН лизосом, в результате чего блокируется завершение процесса аутофагии и происходит накопление аутофагосом в клетке (Solomon, Lee, 2009). Количество аутофагосом и активность аутофагии пропорциональна содержанию специфических белков-маркеров аутофагии и может быть выявлена иммуногистохимически. Одним из широко используемых маркеров аутофагии является LC3, который специфически локализуется в аутофагических структурах на протяжении всего процесса от формирования аутофагосомы до деградации в лизосомах (Mizushima et al, 2010). В работе были использованы антитела к маркеру LC3 (microtubule-associated proteins 1A and 1B, MAP1LC3), по изменению содержания которого в клетках гиппокампа в ответ на воздействие ТГГ судили об интенсивности процесса аутофагии.
МАТЕРИАЛ И МЕТОДИКА
Работа выполнена на взрослых крысах-самцах линии Вистар (200–220 г), полученных из ЦКП Биоколлекция Института физиологии РАН.
Модель ТГГ и схема эксперимента. ТГГ создавали в барокамере проточного типа при поддержании в ней атмосферного давления 180 мм рт. ст. в течение 3 ч. В эксперименте участвовали следующие группы животных: 1) контрольная; 2) группа Хлорохин – животные, которым производили инъекцию хлорохина (ингибитора завершения процесса аутофагии); 3) группа ТГГ – животные, подвергнутые действию ТГГ; 4) группа ТГГ + Хлорохин – животные, подвергнутые действию ТГГ, которым сразу после окончания ТГГ производили инъекцию хлорохина. Крысам групп 1 и 3 внутрибрюшинно водили 200 мкл 0.9%-ного NaCl, а групп 2 и 4 – 200 мкл раствора хлорохина, приготовленного на 0.9%-ном NaCl, в дозе 3 мг на 1 кг массы животного. Во всех группах было по 5–6 животных. Животных наркотизировали с помощью внутрибрюшинной инъекции двухкомпонентного раствора (60 мг золетила и 10 мг 2%-ного ксилазина на 1 кг веса), выполняли транскардиально перфузию, после чего извлекали головной мозг, выделяли область, содержащую париетальный неокортекс и гиппокамп, и фиксировали в течение 1 сут в 4%-ном параформальдегиде (0.01 М фосфатный буфер, рН 7.4).
Гистологическая обработка ткани. Фиксированные препараты отмывали в проточной воде в течение 2 ч. Препараты дегидратировали в спирте восходящей концентрации по следующей схеме: изопропанол 50% (1 ч), изопропанол 70% (1 ч), изопропанол 80% (ночь), изопропанол 100% (1 ч), изопропанол 100% (5 ч). После этого образцы помещали в парафин (58°С) на 3 ч (3 смены парафина по 1 ч каждая) и заливали в парафиновые блоки. Изготавливали серии парафиновых фронтальных срезов мозга толщиной 4 мкм на уровне −2.8 мм от брегмы, монтировали их на предметные стекла.
Иммуногистохимическое окрашивание. Препараты депарафинизировали в ксилоле (2 смены по 5 мин каждая) и регидратировали в этаноле нисходящей концентрации, после этого помещали в фосфатно-солевой буферный раствор (PBS). Демаскировку антигена проводили в цитратном буфере (0.01 М, pH 6.0) при 100°С под давлением в течение 1 мин. Препараты оставляли на 30 мин остывать в цитратном буфере, после чего отмывали 2 раза в PBS по 5 мин. Далее образец ткани обводили гидрофобным маркером и наносили на него блокирующую сыворотку, препараты оставляли для инкубации во влажной камере при комнатной температуре на 30 мин. Затем аккуратно снимали излишки блокирующей сыворотки и наносили на стекла первичные антитела к LC3, оставляли для инкубации на ночь во влажной камере при 4°С. Промывали препараты в PBS 2 раза по 5 мин. Далее последовательно инкубировали препараты со вторичными биотинилированными антителами и авидин-биотиновым комплексом согласно рекомендации производителя. Между инкубациями препараты отмывали в PBS 2 раза по 5 мин. Визуализацию реакции проводили с использованием набора для визуализации на основе диаминобензидина. Препараты дегидратировали и заключали в монтирующую среду.
Обработка данных. Анализ препаратов проводили с использованием светового микроскопа Olympus CX31 (Olympus, Япония), цифровой камеры Progres CT1 (Jenoptic, Германия) и персонального компьютера. Количественный анализ препаратов выполняли в программе ImageJ (https://imagej.nih.gov/ij/) с использованием плагина SDA (Nurzynska et al., 2017). Производили подсчет интегральной яркости иммунореактивного к LC3 вещества и его площади окрашивания; полученные значения пересчитывали и выражали в усл. ед. оптической плотности. Таким образом, расчетные значения оптической плотности отражали содержание белка LC3 в клетках. Результаты статистически обрабатывали с помощью пакетов анализа данных Microsoft Excel 2010, STATISTICA 7.0, использовали критерий Манна–Уитни. Различия между выборками считали статистически значимыми при Р < 0.05. Данные на рисунках представлены в виде средних значений и ошибок среднего.
Реактивы. В работе использовали первичные антитела к LC3 (Sigma Aldrich, USA); блокирующую сыворотку, вторичные биотинилированные антитела, авидин-биотиновый комплекс в составе набора VECTASTAIN ABC-Peroxidase Kit (Vector Laboratories PK-4001, Inc, США); набор для визуализации DAB peroxidase substrate kit (Vector Laboratories, Inc, США); монтирующую среду Bio-Mount (Bio-Optica, Италия); хлорохин дифосфат (Sigma Aldrich, США); параформальдегид (ЛенРеактив, Россия); ксилавет 2% (Alpha-Vet Veterinary Ltd, Венгрия); золетил (Valdepharm, Франция).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
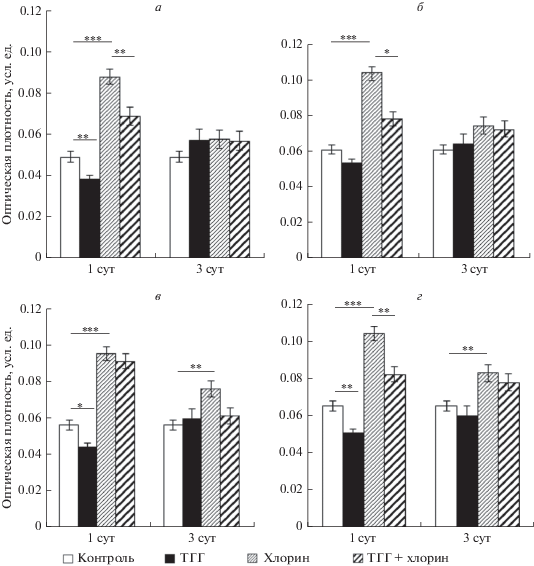
В исследуемых препаратах локализация LC3 была цитоплазматической. Известно, что изоформы белка LC3 имеют преимущественно цитоплазматическую локализацию (Koukourakis et al., 2015). В исследованных полях гиппокампа (СА1, СА3, СА4) и в зубчатой извилине контрольной группы иммунореактивное к LC3 вещество было сосредоточено как в соме клеток, так и в клеточных отростках. В частности, в поле СА1 гиппокампа интенсивно окрашивались фрагменты дендритов (рис. 1а, черные стрелки), в поле СА3 – нейропиль. У крыс, которым вводили хлорохин, через 1 сут после инъекции уровень иммунореактивности к LC3 в нейронах исследованных полей гиппокампа (CA1, СА3, СА4, зубчатая извилина) был выше относительно контроля, что проявлялось увеличением оптической плотности иммунореактивного вещества (рис. 1а, б; рис. 2). Через 3 сут содержание LC3 не отличалось от контрольных значений в полях СА1 и СА3, но было выше контроля в поле СА4 (Р = 0.005616) и зубчатой извилине (Р = = 0.008352) гиппокампа (рис. 2).
Рис. 1.
Гистологический препарат фронтального среза мозга, иллюстрирующий иммунореактивность к LC3 в поле СА1 гиппокампа крыс: а – контроль; б – через 1 сут после инъекции хлорохина (3мг/кг); в – через 1 сут после тяжелой гипобарической гипоксии (ТГГ, 180 мм рт. ст., 3 ч); г – через 1 сут после ТГГ и инъекции хлорохина по окончании ТТГ. Иммунопероксидазный метод с окраской диаминобензидином. Стрелками на фотографии контрольного препарата указаны отростки нейронов, иммунореактивные к LC3.
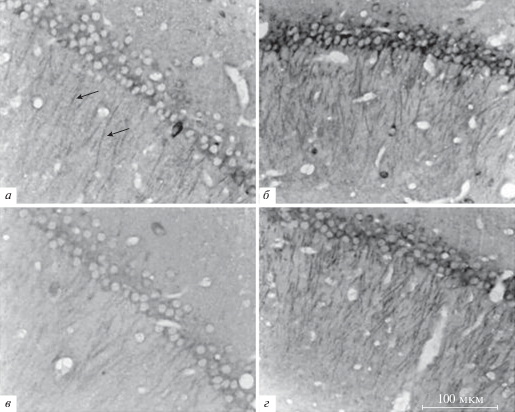
Рис. 2.
Изменения иммунореактивности к LC3 в полях СА1 (а), СА3 (б), СА4 (в) и зубчатой извилине гиппокампа (г) после ТГГ (180 мм рт. ст., 3 ч), а также на фоне введения хлорохина (3 мг/кг). По вертикали – оптическая плотность иммунореактивных к LC3 объектов, усл. ед. Данные представлены в виде средних значений и их ошибок. Различия между группами по критерию Манна–Уитни достоверны при *P ≤ 0.05, **P ≤ 0.01 или ***P ≤ 0.001.
Как показано в модели in vitro, в нейронах поддерживается достаточно высокий базальный уровень активности аутофагии, при этом количество аутофагосом невелико за счет их быстрой деградации (Boland et al., 2008). Полученные нами результаты in vivo согласуются с данным утверждением и свидетельствуют об интенсивном протекании процесса аутофагии в клетках гиппокампа крыс. Несмотря на сравнительно низкий уровень иммунореактивности к LC3 в нейронах гиппокампа контрольной группы животных, через 1 сут после введения хлорохина уровень LC3 возрастал значительно, т.е. происходило накопление данного белка вследствие блокирования его деградации в лизосомах.
Достаточно большое количество работ свидетельствуют об исключительно важной роли базального уровня активности аутофагии для нормального функционирования нейронов (Boland et al., 2008; Tooze, Schiavo, 2008). В частности, показано, что у животных с нокаутом по генам atg5 или atg7 (генам сборки аутофагосом) увеличивается число белковых включений в нейронах и развивается нейродегенерация (Hara et al., 2006; Komatsu et al., 2006). Дефекты на различных этапах процесса аутофагии связаны с прогрессированием таких заболеваний, как болезни Паркинсона, Хантингтона, Альцгеймера (см. обзор: Ghavami et al., 2014). Поэтому изучение процессов аутофагии, как одного из основных механизмов поддержания гомеостаза в нейронах, представляется важным, особенно в условиях действия различного рода экстремальных факторов, в том числе гипоксии.
В настоящей работе мы обнаружили, что через 1 сут после ТГГ уровень иммунореактивности к LC3 был снижен относительно контроля в полях CA1, СА4 и зубчатой извилине гиппокампа (рис. 1а, в; рис. 2а, в, г). В поле СА3 действие ТГГ не влияло на уровень иммунореактивности к LC3 (рис. 2б). Поскольку в контрольной группе уровень иммунореактивности к LC3 достаточно низкий, то судить о его уменьшении после действия ТГГ затруднительно. Поэтому мы использовали в эксперименте дополнительную группу животных, которым после воздействия ТГГ вводили хлорохин. Было выявлено уменьшение уровня иммунореактивности к LC3 в полях СА1, СА3 и зубчатой извилине гиппокампа крыс в группе ТГГ + Хлорохин относительно группы сравнения (Хлорохин) через 1 сут после ТГГ (рис. 1в, г; рис. 2а, б, г).
Обнаруженная разница в уровне иммунореактивности к LC3 между группами Хлорохин и ТГГ + Хлорохин в данных областях гиппокампа, как мы полагаем, обусловлена тем, что усиление процессов аутофагической деградации начинается на раннем сроке после начала воздействия ТГГ – тогда, когда животные находятся в барокамере. Кроме того, действие препарата начиналось не сразу, а через некоторое время после инъекции, согласно фармакокинетике препарата (Adelusi, Salako, 1982). Таким образом, к моменту, когда хлорохин попадал в клетки и блокировал лизосомальную деградацию, интенсивность аутофагии была повышена под действием ТГГ и, следовательно, увеличивалась скорость распада LC3, а его содержание снижалось. В поле СА4 гиппокампа значимых изменений иммунореактивности к LC3 на сроке 1 сут после ТГГ выявлено не было (рис. 2в).
Обнаруженное нами снижение уровня LC3 в нейронах гиппокампа через 1 сут после ТГГ как в группе ТГГ, так и в группе ТГГ + Хлорохин возможно связано с быстрой деградацией белка при активации аутофагии. Скорость распада белка увеличивается и не компенсируется на этом сроке синтезом его de novo. Имеются данные литературы, свидетельствующие о том, что количество LC3 обратно пропорционально активности аутофагии, т.е. снижение уровня LC3 является признаком усиления процесса аутофагии на более поздних сроках после начала активации аутофагии (Mizushima et al., 2010).
Через 3 сут после ТГГ в наших экспериментах уровень иммунореактивности к LC3 в группе ТГГ не отличался от такового в контрольной группе, а в группе ТГГ + Хлорохин – от такового в группе Хлорохин (рис. 2). Возможно, что через 3 сут после ТГГ интенсивность процесса аутофагии снижается, или происходит восстановление баланса между уровнем деградации и синтеза белка.
Таким образом, анализ полученных данных позволяет сделать заключение о том, что процесс аутофагической деградации молекул усиливается действием ТГГ в нейронах гиппокампа крыс на раннем сроке (1 сут) и стабилизируется через 3 сут после воздействия. Возможно, что подобная динамика активности аутофагии в ответ на ТГГ характеризует адаптивные механизмы, активируемые в нейронах и направленные на утилизацию поврежденных органелл, белков и других токсических продуктов. Эта точка зрения подкрепляется рядом исследований. В частности, показано, что аутофагия является одним их важных механизмов регуляции активности транкрипционного фактора HIF-1 (за счет его избирательной деградации) – одного из основных посредников, индуцирующих клеточный ответ на гипоксию (Hubbi et al., 2013). Аутофагической деградации может подвергаться каспаза-3 (Dohi et al., 2012). Кроме того, показано, что при активации аутофагии происходит отсоединение антиапоптотического фактора Bcl-2 от белка Ambra1, в результате чего Bcl-2 становится функционально активным (Toozel, Codogno, 2011). Важная роль аутофагии принадлежит в антиоксидантной защите организма (Giordano et al., 2013).
Тем не менее, в некоторых случаях активацию аутофагии связывают с клеточной гибелью, особенно при острых воздействиях, таких как ишемия (Adhami et al., 2006; Koike et al., 2008; Rami et al., 2008; Puyal, Clarke, 2009). Вопрос о том, является ли увеличение активности аутофагии в ответ на экстремальные воздействия механизмом клеточной гибели или частью адаптивной стратегии, остается дискуссионным (Зубова, 2019). Возможно, это во многом зависит от параметров используемой экспериментальной модели. Известно, что при гипоксическом (ишемическом) воздействии основными механизмами клеточной гибели являются некроз, апоптоз или смешанный тип гибели (Liu et al., 2004; Rybnikova et al., 2006). В ряде работ показано, что механизмы апоптоза и аутофагии взаимосвязаны, и при блокировании апоптоза клетка погибает по механизму, опосредованному активацией аутофагии (Shimizu et al., 2004; Yu et al., 2004; Boya et al., 2005; Yousefi et al., 2006).
Таким образом, учитывая большое значение аутофагии в функционировании нейронов, выяснение ее роли в компенсаторных или про-апоптотических механизмах, индуцируемых в нейронах действием ТГГ, является важной фундаментальной задачей и станет предметом дальнейших исследований. Вместе с тем, обоснованная исследованиями в этой области разработка препаратов, специфичных к компонентам аутофагического каскада, возможно, послужит эффективной стратегией для профилактики или коррекции заболеваний, имеющих гипоксию в качестве основного фактора патогенеза.
Список литературы
Рыбникова Е.А., Хожай Л.И., Тюлькова Е.И., Глущенко Т.С., Ситник Н.А., Отеллин В.А., Самойлов М.О. 2004. Влияние гипобарической гипоксии на экспрессию белков ранних генов и структурные изменения нейронов мозга: корректирующий эффект прекондиционирования. Морфология. Т. 125. № 2. С. 10. (Rybnikova E.A., Khozhai L.I., Tyul’kova E.I., Glushchenko T.S., Sitnik N.A., Pelto-Huikko M., Otellin V.A., Samoilov M.O. 2005. Expression of early gene proteins, structural changes in brain neurons in hypobaric hypoxia and the correcting effects of preconditioning. Neurosc. Behav. Physiol. V. 35. № 4. P. 383.)
Зубова С.Г. 2019. Многоликость аутофагии и ее неоднозначная роль в биологических процессах. Цитология. Т. 61. № 12. С. 941. (Zubova S.G. 2019. The diversity of autophagy and its controversial role in biological processes. Tsitologya. V. 6. № 12. P. 941.)
Adelusi S.A., Salako L.A. 1982. Kinetics of the distribution and elimination of chloroquine in the rat. Gen. Pharmacol. V. 13. P. 433.
Adhami F., Liao G., Morozov Y.M., Schloemer A., Schmithorst V.J., Lorenz J.N., Dunn R.S., Vorhees C.V., Wills-Karp M., Degen J.L., Davis R.J., Mizushima N., Rakic P., Dardzinski B.J., Holland S.K. et al. 2006. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. V. 169. P. 566.
Boland B., Kumar A., Lee S., Platt F.M., Wegiel J., Yu W.H., Nixon R.A. 2008. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. V. 28. P. 6926.
Boya P., Gonzalez-Polo R.A., Casares N., Perfettini J.L., Dessen P., Larochette N., Metivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. 2005. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. V. 25. P. 1025.
Dohi E., Tanaka S., Seki T., Miyagi T., Hide I., Takahashi T., Matsumoto M., Sakai N. 2012. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. V. 60. P. 431.
Giordano S., Darley-Usmar V., Zhang J. 2013. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. V. 2. P. 82.
Ghavami S., Shojaei S., Yeganeh B., Andee S.R., Jangamreddy J.R., Mehrpour M., Christoffersson J., Chaabane W., Moghadam A.R., Kashani H.H., Hashemi M., Owji A.A., Łos M.J. 2014. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Progress Neurobiol. V. 112. P. 24.
Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. V. 441. P. 885.
Hubbi M.E., Hu H., Kshitiz, Ahmed I., Levchenko A., Semenza G.L. 2013. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J. Biol. Chem. V. 288. P. 10703.
Kiffin R., Christian C., Knecht E., Cuervo A.M. 2004. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell. V. 15. P. 4829.
Klionsky D.J., Emr S.D. 2000. Autophagy as a regulated pathway of cellular degradation. Science. V. 290. P. 1717.
Koike M., Shibata M., Tadakoshi M., Gotoh K., Komatsu M., Waguri S., Kawahara N., Kuida K., Nagata S., Kominami E., Tanaka K., Uchiyama Y. 2008. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic/ischemic injury. Am. J. Pathol. V. 172. P. 454.
Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. V. 441. P. 880.
Koukourakis M.I., Kalamida D., Giatromanolaki A., Zois C. E., Sivridis E., Pouliliou S., Mitrakas A., Gatter K.C., Harris A.L. 2015. Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One. V. 10. e0137675. https://doi.org/10.1371/journal.pone.0137675
Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature. V. 432. P. 1032.
Levine B., Klionsky D.J. 2004. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. V. 6. P. 463.
Liu C.L., Siesjö B.K., Hu B.R. 2004. Pathogenesis of hippocampal neuronal death after hypoxia-ischemia changes during brain development. Neurosci. V. 127. P. 113.
Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. 2004. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. V. 15. P. 1101.
Mizushima N., Yoshimori T., Levine B. 2010. Methods in mammalian autophagy research. Cell. V. 140. P. 313.
Nurzynska K., Mikhalkin A., Piorkowski A. 2017. CAS: Cell annotation software – research on neuronal tissue has never been so transparent. Neuroinformatics. V. 15. P. 365.
Puyal J., Clarke P.G.H. 2009. Targeting autophagy to prevent neonatal stroke damage. Autophagy. V. 5. P. 1060.
Rami A., Langhagen A., Steiger S. 2008. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. V. 29. P. 132.
Rybnikova E., Sitnik N., Gluschenko T., Tjulkova E., Samoilov M.O. 2006. The preconditioning modified neuronal expression of apoptosis-related proteins of Bcl-2 superfamily following severe hypobaric hypoxia in rats. Brain Res. V. 1089. P. 195.
Shimizu S., Kanaseki T., Mizushima N., Mizuta T., Arakawa-Kobayashi S., Thompson C.B., Tsujimoto Y. 2004. Role of Bcl-2 family proteins in a nonapoptotic programmed cell death dependent on autophagy genes. Nat. Cell. Biol. V. 6. P. 1221.
Solomon V.R., Lee H. 2009. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. V. 625. P. 220.
Tooze S.A., Schiavo G. 2008. Liaisons dangereuses: Autophagy, neuronal survival and neurodegeneration. Curr. Opin. Neurobiol. V. 18. P. 504.
Toozel S.A., Codogno P. 2011. Compartmentalized regulation of autophagy regulators: fine-tuning AMBRA1 by Bcl-2. EMBO J. V. 30. P. 1185.
Wang Y., Han R., Liang Z.Q.,Wu J.C., Zhang X.D., Gu Z.L., Qin Z.H. 2008. An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy. V. 4. P. 214.
Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., Brunner T., Simon H.U. 2006. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell. Biol. V. 8. P. 1124.
Yu L., Alva A., Su H., Dutt P., Freundt E., Welsh S., Baehrecke E.H., Lenardo M.J. 2004. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. V. 304. P. 1500.
Zhu C., Wang X., Xu F., Bahr B.A., Shibata M., Uchiyama Y., Hagberg H., Blomgren K. 2005. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia–ischemia. Cell Death Differ. V. 12. P. 162.
Дополнительные материалы отсутствуют.