

Доклады Российской академии наук. Химия, науки о материалах , 2020, T. 494, № 1, стр. 27-31
Новый метод синтеза бис-фосфороксидов c 1,3-бутадиеновым спейсером из 1-фосфинил-2-арилацетиленов с использованием системы Cp2ZrCl2–EtAlCl2–Mg
И. Р. Рамазанов 1, *, Р. Н. Кадикова 1, А. К. Амирова 1, член-корреспондент РАН У. М. Джемилев 1
1 Институт Нефтехимии и Катализа Уфимского федерального исследовательского центра Российской академии наук
Республика Башкортостан, Уфа, Россия
* E-mail: ilfir.ramazanov@gmail.com
Поступила в редакцию 09.06.2020
После доработки 12.08.2020
Принята к публикации 02.09.2020
Аннотация
Реакция 1-фосфинил-2-арилацетиленов с Cp2ZrCl2, Mg и EtAlCl2 в ТГФ при комнатной температуре приводит к образованию после гидролиза и последующего окисления [(1E,3E)-2,3-диарилбута-1,3-диен-1,4-диил]бис(дифенилфосфороксидов) с хорошим выходом.
Циклометаллирование ацетиленов с использованием низковалентных комплексов металлов является удобным методом конструирования металлациклопентадиеновых систем [1–3]. Создание новых эффективных методов конструирования функционально замещенных металлациклопентадиенов на основе фосфорсодержащих ацетиленов открывает перспективы использования в области супрамолекулярной химии для разработки современных материалов [4]. Ранее мы пытались использовать реакцию Zr-катализируемого циклоалюминирования для получения алюминациклопентадиенов из 1-алкинилфосфинов [5]. Однако использование системы Et3Al/[Cp2ZrCl2]/гексан приводило только к образованию фосфорзамещенных металлациклопентенов. Тот же результат был получен при взаимодействии 1-алкинилфосфина с системой Et2Zn/[Ti(O-iPr)4‒EtMgBr]/Et2O [6]. Ранее Такахаши (Takahashi) обнаружил, что 1‑алкинилфосфины реагируют с диэтилцирконоценом с образованием соответствующего цирконациклопентена [7]. Опираясь на эти результаты, мы выдвинули идею о возможности синтеза замещенных металлациклопентадиенов циклометаллированием фосфорсодержащих ацетиленов по реакции Джемилева [8] с использованием в качестве катализатора комплексов циркония, проявляющих наибольшую активность и селективность действия в подобных реакциях. Наиболее перспективной из них представлялась Cp2ZrCl2-катализируемая реакция ацетиленов с EtAlCl2 в присутствии Mg.
При проведении предварительных экспериментов мы установили, что при взаимодействии дифенил(фенилэтинил)фосфина с 0.5 эквивалентами этилалюминийдихлорида (EtAlCl2), 0.6 эквивалентами порошкообразного магния в присутствии 10 мол. % цирконоцендихлорида (Cp2ZrCl2) в ТГФ при температуре 23°С в течение 18 ч после дейтеролиза реакционной массы и окисления системой Н2O2/NaOH наблюдалось образование следовых количеств 1,3-диенового бис-фосфороксида. Необходимость обработки реакционной массы системой Н2O2/NaOH вызвана тем, что образующиеся после гидролиза или дейтеролиза соответствующие фосфины частично окисляются в процессе выделения методом колоночной хроматографии. Наличие в продуктах реакции дейтерированных непредельных фосфороксидов, построенных из двух молекул исходных ацетиленов, а также присутствие двух атомов дейтерия в 1,4-положении 1,3-диеновой системы, свидетельствует о возможном образовании в ходе реакции интермедиатных циркона- или алюминациклопентадиенов, как показано на схеме 1. Все наши попытки повысить степень конверсии исходных ацетиленов путем увеличения времени и температуры прохождения реакции, а также количества EtAlCl2, не дали положительных результатов. Однако при подборе оптимальных условий реакции мы обнаружили, что с увеличением концентрации Cp2ZrCl2 значительно повышается выход 1,3-диенового бис-фосфороксида. Так, например, при взаимодействии 1-фосфинил-2-арилацетиленов – дифенил(фенилэтинил)фосфина или p-толилэтинилдифенилфосфина – с 0.5 экв. EtAlCl2 в растворе ТГФ (23°C, 18 ч) после гидролиза или дейтеролиза с последующим окислением полученных соединений системой Н2O2/NaOH были выделены соответствующие 1,3-диеновые бис-фосфороксиды 1а,b и 2а с выходами более 65%. В отличие от 1-фосфинил-2-арилацетиленов, использование 1-фосфинил-2-алкилацетиленов в аналогичных условиях приводит к образованию сложной смеси продуктов димеризации и олигомеризации исходных ацетиленов.
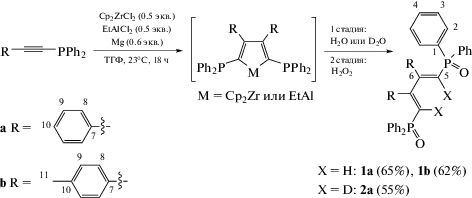
Идентификация полученных диеновых соединений была проведена методами одномерной и двумерной ЯМР-спектроскопии, а также ESI-MS спектрометрии высокого разрешения. Наличие в спектре 1H ЯМР соединения 1a дублетного сигнала при 6.38 м. д. с J = 19.3 Гц обусловлено значительной константой спин-спинового взаимодействия (КССВ) 2JPH между атомом фосфора и атомом водорода при двойной связи. Аналогичные высокие КССВ 2JPH наблюдались нами в случае ранее синтезированных 1-алкенилфосфороксидов и 1-алкенилфосфорсульфидов [5]. Сигналы фенильной группы при атоме фосфора смещены в область слабого поля относительно сигналов фенильной группы при двойной связи. Кросс-пик между этими сигналами в спектре NOESY соединения 1а указывает на E-ориентацию двойной связи. На этот же факт косвенно указывает отсутствие в спектре сигналов, указывающих на взаимодействие между атомом водорода и фенильной группой при двойной связи. Положение атомов дейтерия в соединении 2а было установлено на основании сравнения спектров ЯМР со спектральными параметрами недейтерированного соединения 1а. Сигналы атомов углерода C(5) отсутствуют, что характерно для дейтерозамещенных олефинов. Массы протонированных аддуктов молекулярных ионов, которые наблюдались в спектре MS-ESI соединения 1a, хорошо согласуются с расчетными.
Установлено, что реакция идет только в присутствии EtAlCl2. Введение в исследуемую реакцию вместо EtAlCl2 других кислот Льюиса на основе соединений магния или цинка (EtMgBr, Et2Zn, ZnCl2) не приводило к селективному образованию продуктов димеризации. Использование Et3Al, Et2AlCl и AlCl3 приводило к более низким выходам целевых продуктов по сравнению с EtAlCl2. Существенное влияние на реакцию оказывает природа растворителя. В неэфирных растворителях (толуол, гексан) образовывались продукты олигомеризации и полимеризации алкинов, а в диэтиловом эфире и диизопропиловом эфире реакция протекала с меньшей скоростью, что, может быть, связано с плохой растворимостью в них компонентов металлоорганической системы по сравнению с ТГФ. Кроме того, влияние на скорость реакции может оказывать эффект большей стабилизации металлоорганического соединения в растворе ТГФ вследствие большей компактности молекулы ТГФ по сравнению с молекулами диэтилового и диизопропилового эфира, а, следовательно, лучшей сольватации.
Мы предположили, что реакция проходит через стадию промежуточного образования металла-циклопентадиена. Из-за высокой реакционной способности алюминийорганических соединений в реакционной смеси по отношению к кислороду выделение и идентификация этого металлациклопентадиена затруднена. Нами была высказана гипотеза, заключающаяся в том, что в отсутствие сильной кислоты Льюиса в результате взаимодействия атомов циркония и фосфора образуется цирконоценовый комплекс с 1-фосфинил-2-арилацетиленом A (схема 2). Образование комплексов такого рода было зафиксировано ранее [9]. Добавление же органического галогенида алюминия приводит к образованию комплекса 1-фосфинил-2-арилацетилена с EtAlCl2B. Таким образом, галогенид алюминия выступает в качестве своеобразной защиты для фосфиновой функции группы.
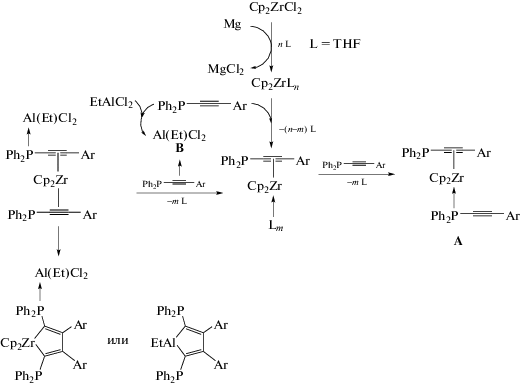
Следует отметить, что ранее сообщалось о двухстадийной процедуре получения дифосфиноцирконациклопентадиена Cp2Zr[2,5-(Ph2P)2-3,4-Ph2C4] с выходом 49% из дифенил(фенилэтинил)фосфина [8]. На первом этапе цирконациклопропеновый комплекс готовили с использованием реагента Розенталя. Последующая реакция моноалкинового комплекса с 1-фосфинил-2-фенилацетиленом приводила к образованию цирконациклопентадиена. Разработанный нами метод впервые позволил проводить превращение 1-фосфинил-2-арилацетиленов в 1,3-диеновые бис-фосфороксиды в одну препаративную стадию. Данная реакция имеет общий характер и позволяет синтезировать ранее труднодоступные и практически важные непредельные бис-фосфороксиды, соединенные сопряженным углеводородным спейсером. Первые положительные результаты открывают путь для синтеза новых фосфорсодержащих соединений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для проведения экспериментов использовались коммерчески доступные реагенты. 1-Алкинилфосфины синтезировали каталитической реакцией 1-алкинов с Ph2PCl [10]. Реакции с алюминийорганическими соединениями проводили в атмосфере осушенного аргона. Гексан перегоняли над P2O5. Спектры 1Н и 13C ЯМР регистрировались на спектрометре Bruker Avance 400 (100.62 МГц для 13С и 400.13 МГц для 1H) (Германия). При съемке спектров 1H и 13С ЯМР в качестве внутреннего стандарта использовали SiMe4 и CDCl3 соответственно. Масс-спектры снимали с помощью прибора “Finnigan 4021” (США) с энергией ионизирующих электронов 70 эВ и температурой камеры ионизации 200°С. Элементный анализ образцов определяли на элементном анализаторе фирмы Carlo Erba, модель 1106 (Италия). Тонкослойную хроматографию проводили на пластинах Silufol UV-254 (Чехия) в системе гексан : EtOAc : MeOH = 5 : 1 : 0.5.
[(1E,3E)-2,3-Дифенилбута-1,3-диен-1,4-диил]бис(дифенилфосфороксид) 1a.
К магниевому порошку (0.288 мг, 1.2 ммоль) в тетрагидрофуране (2 мл) в атмосфере аргона добавляли EtAlCl2 (0.14 мл, 1 ммоль), дифенил(фенилэтинил)фосфин (0.572 г, 2 ммоль) и Cp2ZrCl2 (0.292 г, 1 ммоль) при 0°C. После перемешивания в течение 18 ч при комнатной температуре реакционную массу охладили на ледяной бане и добавили Et2O (5 мл) и 25%-й раствор KOH (3 мл). Водный слой экстрагировали диэтиловым эфиром (3 × 5 мл). Объединенные органические слои промыли водой (10 мл), высушили над безводным MgSO4. Растворитель отогнали на роторном испарителе. Продукт реакции растворили в 5 мл хлороформа и к полученному раствору при интенсивном перемешивании медленно по каплям добавили 30%-й водный раствор H2O2 (0.35 мл, 3 ммоль). Реакционную массу перемешивали в течение 1 ч, затем промыли водой (3 × 5 мл). Органический слой сушили над безводным MgSO4, отфильтровали и раствор упарили на роторном испарителе. Продукт реакции в виде желтого масла очистили методом колоночной хроматографией на силикагеле (гексан : EtOAc : MeOH = 5 : 1 : 0.5) и получили 0.79 г продукта в виде вязкого желтоватого масла (выход 65%), Rf 0.16. 1H ЯМР (δ, м. д.): 6.38 (д, J = 19.3 Гц, 2Н, С(5)Н), 7.05–7.85 (м, 30Н, Ph). 13C ЯМР (δ, м. д.): 127.83, 128.20, 128.25, 128.30, 128.64, 129.96, 130.55, 130.59, 130.63, 131.17, 134.14. ESI-MS: m/z 607.1945 [M+H]+; выч. для C40H32O2P2: 607.1950.
Найдено, %: C, 79.02; H, 5.38.
Вычислено для C40H32O2P2, %: C, 79.20; H, 5.32.
[(1E,3E)-2,3-Дифенилбута-1,3-диен-1,4-диил-1,4-d2]бис(дифенилфосфороксид) 2a.
Процедура получения соединения 2а аналогична вышеописанной процедуре для соединения 1a с учетом обработки реакционной массы D2O вместо H2O. 1H ЯМР (δ, м. д.): 7.0–7.85 (м, 30Н, Ph). 13C ЯМР (δ, м. д.): 128.31, 128.34, 128.69, 130.04, 130.60, 130.66, 130.69, 131.21, 134.19. ESI‑MS: m/z 609.2075 [M+H]+; выч. для C40H30D2O2P2: 609.2079.
[(1E,3E)-2,3-Ди-p-толилбута-1,3-диен-1,4-диил)бис(дифенилфосфороксид) 1b.
Процедура получения аналогична процедуре, описанной для получения соединения 1a, но вместо дифенил(фенилэтинил)фосфина использовали p-толилэтинилдифенилфосфин (0.60 г, 2 ммоль). В результате было выделено 0.78 г вязкого желтоватого масла (выход 61%), Rf 0,19. 1H ЯМР (δ, м. д.): 2.25 (с, 3H, C(11)H3), 6.27 (д, J = 19.3 Гц, 2Н, С(5)Н), 6.9‒7.6 (м, 28Н, арил). 13C ЯМР (δ, м. д.): 21.25 [C(11)], 127.60, 128.10, 128.15, 128.20, 128.46, 129.87, 130.63, 130.67, 130.71, 131.03, 134.32. ESI-MS: m/z 635.2261[M+H]+; выч. для C42H36O2P2: 635.2263.
Найдено, %: C, 79.51; H, 5.56.
Вычислено для C42H36O2P2, %: C, 79.48; H, 5.72.
Список литературы
Negishi E.I., Takahashi T. Alkene and alkyne complexes of zirconocene. Their Preparation, Structure, and Novel Transformations // Bull. Chem. Soc. Jpn. 1998. V. 71. P. 755–769. https://doi.org/10.1246/bcsj.71.755
Broene R.D., Buchwald S.L. Zirconocene complexes of unsaturated organic molecules: new vehicles for organic synthesis // Science. 1993. V. 261. P. 1696–1701. https://doi.org/10.1126/science.8378769
Buchwald S.L., Nielsen R.B. Group 4 metal complexes of benzynes, cycloalkynes, acyclic alkynes, and alkenes // Chem. Rev. 1988. V. 88. P. 1047–1058. https://doi.org/10.1021/cr00089a004
Gessner V.H., Tannaci J.F., Miller A.D., Tilley T.D. Assembly of macrocycles by zirconocene-mediated, reversible carbon-carbon bond formation // Acc. Chem. Res. 2011. V. 44. P. 435–446. https://doi.org/10.1021/ar100148g
Ramazanov I.R., Kadikova R.N., Saitova Z.R., Dzhemilev U.M. A Route to 1-alkenylphosphine derivatives via the Zr-catalyzed reaction of 1‑alkynylphosphines with triethylaluminum // Asian J. Org. Chem. 2015. V. 4. P. 1301–1307. https://doi.org/10.1002/ajoc.201500274
Kadikova R.N., Ramazanov I.R., Mozgovoi O.S., Gabdullin A.M., Dzhemilev U.M. 2-Zincoethylzincation of 2-alkynylamines and 1-alkynylphosphines catalyzed by titanium(iv) isopropoxide and ethylmagnesium bromide // Synlett. 2019. V. 30. P. 311–314. https://doi.org/10.1055/s-0037-1612009
Xi Z., Zhang W., Takahashi T. Selective preparation of 1,3-butadienyl phosphines, 1-iodo- and 1,4-diiodo-butadienyl phosphine oxides via zirconocene-mediated cross-coupling of alkynylphosphines // Tetrahedron Lett. 2004. V. 45. P. 2427–2429. https://doi.org/10.1016/j.tetlet.2004.01.098
Dyakonov V.A. Dzhemilev reactions in organic and organometallic synthesis. New York: Nova Science Publishers, Inc., 2010. 96 p.
Miller A.D., Johnson S.A., Tupper K.A., McBee J.L., Tilley T.D. Unsymmetrical zirconacyclopentadienes from isolated zirconacyclopropenes with 1-alkynylphosphine ligands // Organometallics. 2009. V. 28. P. 1252–1262. https://doi.org/10.1021/om801040t
Beletskaya I.P., Afanasiev V.V., Kazankova M.A., Efimova I.V. New approach to phosphinoalkynes based on Pd- and Ni-catalyzed cross-coupling of terminal alkynes with chlorophosphanes // Org. Lett. 2003. V. 5. P. 4309–4311. https://doi.org/10.1021/ol035562c
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах