

Экология, 2021, № 3, стр. 179-192
Анализ экологических сетей многокомпонентных сообществ микроорганизмов: возможности, ограничения, потенциальные ошибки
В. С. Микрюков a, *, О. В. Дуля a, Г. А. Лиходеевский b, Е. Л. Воробейчик a
a Институт экологии растений и животных УрО РАН
620144 Екатеринбург, ул. 8 Марта, 202, Россия
b Уральский федеральный аграрный научно-исследовательский центр УрО РАН
620142 Екатеринбург, ул. Белинского, 112a, Россия
* E-mail: vmikryukov@gmail.com
Поступила в редакцию 20.08.2020
После доработки 13.11.2020
Принята к публикации 20.11.2020
Аннотация
Благодаря высокой разрешающей способности и производительности секвенирования ДНК исследователям открывается чрезвычайно высокое разнообразие сообществ бактерий, грибов, протистов и микробеспозвоночных почвы, древесины, филлосферы и других природных сред. Для изучения свойств таких сообществ необходимы мощные инструменты анализа многокомпонентных систем. Один из них – анализ экологических сетей, позволяющий решить широкий круг задач экологии. В обзоре кратко описаны возможности анализа сетей, его базовые понятия и метрики топологии сетей. Указаны ограничения, связанные с особенностями секвенирования ДНК (композиционность и разреженность данных), а также потенциальные источники ошибок при интерпретации результатов (реликтовая ДНК, артефактные последовательности ДНК, артефактные связи). Основное внимание уделено сообществам микроорганизмов, но обсуждаемые вопросы релевантны большинству других групп биоты.
Взаимосвязи компонентов экологических сообществ во многом определяют их функционирование и устойчивость к внешним воздействиям, что обусловливает необходимость их изучения и охраны [1–4]. Однако межвидовые связи в сообществах важнейших организмов планеты – микроорганизмов – исследованы недостаточно [5], так как до начала эры молекулярной экологии полная таксономическая инвентаризация их природных сообществ была недостижима [6]. В настоящее время ситуация кардинально изменилась: с помощью молекулярно-генетических методов можно с высоким таксономическим разрешением описать микробоценоз любой среды. С недавних пор идентификация микроорганизмов на основе методов секвенирования следующего поколения (NGS, next-generation sequencing) стала настолько доступной и высокопроизводительной [7], что исследования микробиомов в региональном, континентальном и даже глобальном масштабах, выполненные в рамках одной работы, уже не вызывают удивления [8–11]. Несмотря на ряд технических проблем (наличие реликтовой ДНК, недостаточная специфичность или универсальность праймеров, неполнота референтных баз данных и др.), решением которых активно занимается исследовательское сообщество, NGS уже сейчас обеспечило возможность не только инвентаризации таксономического разнообразия сообществ, но и изучения взаимосвязей между видами [12, 13].
Анализ структуры взаимосвязей в многокомпонентных системах, в том числе их наглядную визуализацию, часто реализуют в рамках анализа сетей – одного из разделов математической теории графов [5, 14]. Уже несколько десятилетий внутри этой области развивается отдельное направление – анализ экологических сетей. Круг его приложений очень широк: от выявления факторов формирования сообществ и изучения связи их сложности с устойчивостью до описания и моделирования экологических ниш отдельных видов. Несколько примеров, демонстрирующих успешное решение разнообразных задач, приведено в табл. 1.
Таблица 1.
Примеры задач, решаемых с помощью анализа экологических сетей
| Группа биоты | Местообитание | Задача исследования | Публикация |
|---|---|---|---|
| Грибы и прокариоты | Почва | Разделение сообществ ризосферы и дрилосферы | [15] |
| Грибы и бактерии | Легкие человека | Оценка роли грибов в сложности и устойчивости структуры микробиома | [16] |
| Нематоды и бактерии-нитрификаторы | Почва | Анализ пространственной ниши на основе модулярности | [17] |
| Грибы | Ткани листьев | Анализ пространственной ниши на основе модулярности | [18] |
| Прокариоты | Почва | Анализ пространственной ниши на основе модулярности | [19] |
| Прокариоты | Разные слои океанической толщи | Анализ пространственной ниши на основе модулярности | [20] |
| Прокариоты | Глобальные сборы из множества местообитаний (по материалам базы данных Greengenes) | Исследование связи между филогенетическим родством и пространственной нишей | [21] |
| Прокариоты | Коралловые рифы | Поиск ключевых видов, ответственных за развитие черноленточной болезни коралловых полипов (black band disease) | [22] |
| Прокариоты | Биопленка на поверхности океанической воды | Поиск ключевых компонентов сообщества биопленки | [23] |
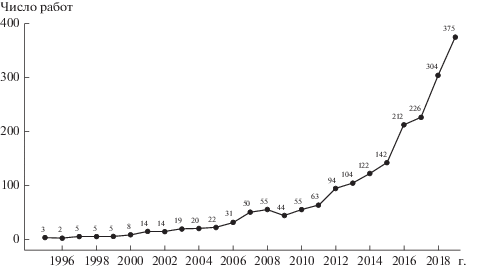
В последнее время наблюдается всплеск интереса к анализу экологических сетей: в библиографической базе Scopus число публикаций возросло от единиц в год в середине 2000-х гг. до сотен к концу 2010-х гг. (рис. 1). К сожалению, анализ сетей пока не оценен по достоинству в отечественной экологии: российские исследователи были авторами лишь 25 из 1994 работ, представленных в базе Scopus на конец октября 2020 г.
Рис. 1.
Динамика числа работ, посвященных изучению взаимосвязей в биотических сообществах с помощью анализа экологических сетей по результатам поискового запроса в базе Scopus на 24.10.2020: TITLE-ABS-KEY ((“network analysis” OR “co-occurrence”) AND “community”) AND SUBJAREA (“AGRI” OR “IMMU”) AND EXACTKEYWORD (“Actinobacteria” OR “Animal*” OR “Bacteria (microorganisms)” OR “Bacteri*” OR “Bacterial Community” OR “Bacteroidetes” OR “Biodiversity” OR “Bioinformatics” OR “Community Composition” OR “Community Structure” OR “DNA Sequence” OR “Ecology” OR “Ecosystem*” OR “Firmicutes” OR “Food Web” OR “Fung*” OR “High-throughput Sequencing” OR “Metagenomics” OR “Microbial Activity” OR “Microbial Communit*” OR “Microbial Diversity” OR “Microbiology” OR “Microbiome” OR “Microbiota” OR “Microflora” OR “Microorganisms” OR “Phylogeny” OR “RNA 16S” OR “RNA, Ribosomal, 16S” OR “Soil Microbiology” OR “Soil*” OR “Species Diversity”).
Наш обзор посвящен не столько описанию возможностей анализа сетей в экологических приложениях, сколько его ограничениям и потенциальным ошибкам в интерпретации результатов, а также способам их устранения. Наличие обзоров, например [5, 24, 25], монографий [26] и учебных пособий [14, 27–29] по анализу сетей избавило нас от необходимости подробного введения в проблему и рассмотрения вычислительных алгоритмов.
Основное внимание мы уделили сообществам микроорганизмов – пожалуй, наиболее сложному объекту синэкологии, но обсуждаемые вопросы в полной мере относятся и к другим группам биоты. Поскольку в анализе сетей особенно актуален принцип GIGO (“Garbage In, Garbage Out”, т.е. качество результатов в первую очередь определяется качеством исходных данных), мы отдельно остановились на специфике информационных массивов, получаемых в ходе ДНК-метабаркодирования – основного метода изучения микробиомов природных сред. Представленная информация, на наш взгляд, может быть полезна при планировании применения ДНК-метабаркодирования для исследования сообществ бактерий, грибов, протистов и микробеспозвоночных почвы, древесины, филлосферы, донных отложений и других аналогичных сред.
ИССЛЕДОВАНИЯ СООБЩЕСТВ МИКРООРГАНИЗМОВ С ПОМОЩЬЮ СЕКВЕНИРОВАНИЯ ДНК
С каждым годом разрешающая способность, точность и производительность методов инвентаризации биоты с помощью анализа состава нуклеиновых кислот и белков увеличиваются, а стоимость снижается. Поэтому в последнее десятилетие ДНК-метабаркодирование стало основным инструментом анализа разнообразия микроорганизмов природных сред [10, 11, 30–32]. Переход от традиционной “чашечной микробиологии” к метабаркодированию вскрыл неожиданно высокое биоразнообразие даже в очень небольших объемах природных субстратов. Например, число OTU уровня вида, выявленных в образце почвы или мертвой древесины массой 250–500 мг, может достигать сотен для грибов и микроартропод и тысяч для прокариот [10, 33–36].
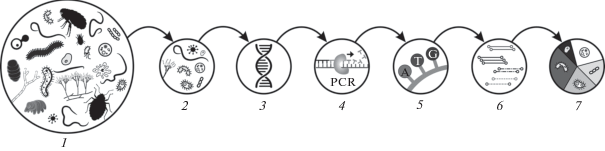
Хотя специфика исследуемой группы или среды может потребовать дополнительных манипуляций на начальных этапах работы, последовательность действий по молекулярной идентификации представителей любого таксона в образце любой природной среды однотипна (рис. 2). Вначале из образца выделяют тотальную ДНК, т.е. смесь ДНК всех организмов (environmental DNA или eDNA). Далее на основе полимеразной цепной реакции (ПЦР) с использованием таксон-специфичных праймеров в экстракте увеличивают концентрацию ДНК-маркеров интересующей группы. ДНК-маркеры – это короткие участки генома, изменчивость последовательности нуклеотидов которых позволяет различать более мелкие таксономические единицы (например, виды) в пределах более крупного таксона (например, класса). В качестве ДНК-маркеров для грибов наиболее часто используют участки внутренних транскрибируемых спейсеров рРНК (internal transcribed spacer, ITS) [37], для прокариот – фрагменты генов малой субъединицы рРНК 16S, для большинства микроэукариот – 18S [38], для беспозвоночных – ген цитохромоксидазы [39].
Рис. 2.
Последовательность анализа состава биоты природных сред с помощью ДНК-метабаркодирования: 1 – биота природной среды; 2 – отбор репрезентативных образцов; 3 – экстракция ДНК; 4 – ПЦР, в ходе которой увеличивают концентрацию ДНК-маркера для инвентаризации целевой группы организмов, а также проводят подготовку ДНК к секвенированию; 5 – секвенирование ДНК; 6 – биоинформационный анализ последовательностей ДНК; 7 – список OTU с таксономическими аннотациями и относительными обилиями.
В результате секвенирования получают прочтения, т.е. нуклеотидные последовательности ДНК-маркеров. В ходе последующего биоинформационного анализа сходные последовательности объединяют в “оперативные (операционные, операциональные) таксономические единицы” (operational taxonomic unit, OTU) или в “варианты последовательностей ампликонов” (amplicon sequence variant, ASV) : OTU – набор последовательностей, которые могут отличаться друг от друга в границах выбранного порога сходства, например из-за генетической изменчивости или ошибок ПЦР; ASV – набор строго идентичных последовательностей (иногда его обозначают как zero-radius OTU); чаще используют OTU. Таксономическое разрешение зависит от задач исследования и регулируется выбором ДНК-маркера и порога сходства последовательностей [40]. Для идентификации принадлежности OTU к определенным таксонам их последовательности сопоставляют с последовательностями ДНК известных таксонов, депонированных в различных базах данных: UNITE [41], SILVA [42], PR2 [43], BOLD [44] и др. В итоге формируется список OTU с таксономическими аннотациями и относительными обилиями в виде числа прочтений ДНК в каждом образце.
Из-за ограниченной производительности секвенаторов оценка абсолютного обилия OTU напрямую невозможна, поэтому по результатам секвенирования каждая OTU представлена долей прочтений ее ДНК от суммарного количества прочтений. Важное свойство таких данных, существенно осложняющее анализ, – композиционность, т.е. взаимозависимость относительных обилий OTU: поскольку в сумме все доли равны единице, увеличение обилия любой OTU приводит к снижению обилия остальных OTU и наоборот. Композиционность накладывает ограничения на выбор методов математического анализа данных [45, 46] и вызывает сложности при интерпретации результатов.
Еще одно “малоприятное” свойство результатов секвенирования ДНК – разреженность данных (data sparsity), т.е. большое число нулевых значений обилия. В значительной степени оно обусловлено спецификой микробных сообществ, для многих видов которых характерна низкая встречаемость. Важно, что такая разреженность может быть следствием не только редкости видов, действительно отсутствующих в большинстве образцов (так называемые “настоящие нули”, essential zeros). Проблема заключается в том, что в ходе секвенирования на разные образцы, как правило, приходится разное количество прочтений, т.е. разное выборочное усилие. Поэтому в образцах с малым количеством прочтений вероятность попадания в выборку ДНК видов с низким обилием может быть настолько небольшой, что в массиве данных появляются “ложные нули” (иначе их называют “округленные нули”, rounded zeros). При возможности образцы с малым количеством прочтений секвенируют повторно или же прибегают к различным приемам замены ложных нулей ожидаемыми значениями (zero imputation) [47].
ЭКОЛОГИЧЕСКИЕ СЕТИ
Визуализация взаимоотношений множества объектов с помощью графов давно используется в социологии, нейробиологии, теории эволюции, техническом проектировании, химии и многих других дисциплинах [14, 48]. Первые и наиболее известные примеры отображения с помощью графов экологических взаимодействий – пищевые сети и цепи – относятся к концу XIX в. [4, 28]. Сегодня анализу биологических и экологических сетей посвящено много обобщающих обзоров, например [5, 24, 26–29].
Важно подчеркнуть ключевую особенность экологических сетей: в отличие от нейронных, химических или технических систем, связи в которых чаще всего можно наблюдать непосредственно, экологические сети строятся на основе информации о совместной встречаемости видов (presence–absence data) или сопряженности их обилий (abundance data) в выборках образцов, наблюдений, учетов, описаний и пр. Статистически значимую положительную связь между обилием или встречаемостью пары видов принято интерпретировать как их симбиотические взаимоотношения или сходные экологические предпочтения, а отрицательную – как антагонизм или разделение экологических ниш. К сожалению, отражение экологических процессов в сетях может быть искажено как по объективным причинам, так и из-за методических ограничений, причины которых мы обсудим далее.
Базовые понятия
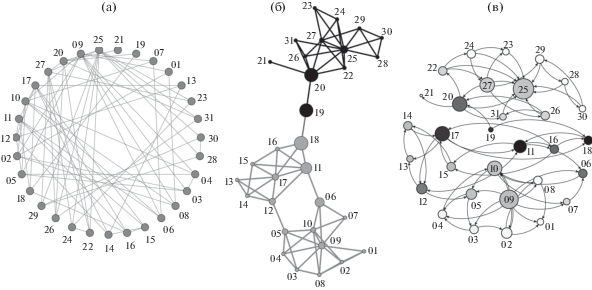
В большинстве случаев узлы экологической сети – это виды, а ребра – связи между парами видов в контексте исследуемого процесса (рис. 3). Базовые понятия при работе с сетями – путь и степень узла. Путь (иногда его называют геодезический или кратчайший путь, path) – это наименьшее число связей, соединяющих два узла, а степень узла (node degree) – число принадлежащих узлу связей. Например, на рис. 3б степень узлов 07 и 15 равна трем и четырем соответственно, а путь между этими узлами составляет три ребра, несмотря на то, что от узла 07 до 15 можно пройти по большему числу ребер, например по шести (через узлы 10, 06, 11, 12 и 17).
Рис. 3.
Примеры визуализации одной и той же сети (искусственные данные): а – круговая раскладка с узлами, упорядоченными по их степени; б – раскладка NEATO, размер узла пропорционален центральности по промежуточности, разным цветом обозначены модули сети; в – раскладка Фрухтермана-Рейнгольда, показана направленность связей, размер узла пропорционален центральности по степени, насыщенность заливки – центральности по близости.
Поскольку построение экологических сетей основано на оценках связи между встречаемостью или обилием пар видов, то в качестве меры силы связи используют стандартные коэффициенты корреляции (Спирмена или Пирсона). Однако их применение напрямую к данным ДНК-метабаркодирования некорректно вследствие композиционности величин, поэтому оно возможно только после трансформации обилий OTU. Для этого чаще всего используют CLR-трансформацию (centered log ratio transformation) – логарифм отношения обилия к среднему геометрическому обилий всех OTU в образце [45].
Расчет среднего геометрического при наличии нулевых значений невозможен, поэтому нули предварительно заменяют произвольно выбранными малыми величинами (pseudocounts) или ожидаемыми значениями [47]. Однако при высокой разреженности данных, о которой мы упомянули выше, замена большого количества нулей может существенно повлиять на конечный результат. В такой ситуации целесообразно использовать альтернативные способы трансформации без замены нулей: ALR (additive log-ratio), IQLR (inter-quartile log ratio) [49] или mCLR (modified CLR) [50].
Алгоритмы построения сетей
Разработано множество алгоритмов построения сетей, в том числе учитывающих композиционность и разреженность данных (табл. 2). Помимо мер корреляции, в них используют альтернативные метрики для измерения силы связи: индексы несходства пар видов по наборам образцов, в которых они обнаружены, а также меры условной зависимости (conditional dependence), исключающие из сети опосредованные связи. Некоторые методы, например CoNet, позволяют одновременно использовать разные метрики и строить консенсусные сети [12, 51].
Таблица 2.
Методы построения экологических сетей на основе встречаемости или обилия видов
| Пакет программ | Метод | Метрика силы связи | Источник информации |
|---|---|---|---|
| SparCC | Sparse correlations for compositional data | Корреляция | [49] |
| MENAP | Molecular ecological network analysis pipeline | Корреляция | [25] |
| CCREPE | Compositionality corrected by permutation and renormalization | Корреляция | [50] |
| MINE | Maximal information-based nonparametric exploration | Максимальная информация | [51] |
| LSA | Local similarity analysis | Сходство | [52] |
| CCLasso | Correlation inference for compositional data through lasso | Корреляция | [53] |
| propr | Proportionality between vectors of compositional data | Пропорциональность | [54] |
| REBACCA | Regularized estimation of the basis covariance based on compositional data | Корреляция | [55] |
| CoNet; CoNetinR | Co-occurrence network inference | Корреляция, несходство | [56] |
| SPIEC-EASI | Sparse and compositionally robust inference of microbial ecological networks | Условная зависимость | [47] |
| SPRING | Semi-parametric rank-based approach for inference in graphical model | Условная зависимость | [36] |
При выборе подходящего алгоритма построения сетей в первую очередь необходимо учитывать характер данных. Высокая чувствительность некоторых алгоритмов к композиционности и разреженности данных может проявляться в увеличении долей выявленных ложных либо не выявленных истинных связей. В ряде работ [12, 24, 50, 52] сравнивают эффективность разных алгоритмов и обсуждают стратегии их выбора.
Итог работы представленных в табл. 2 пакетов программ – это матрицы взаимосвязей пар видов. Для визуализации взаимосвязей в виде графов и различных манипуляций с графами разработаны специальные программы, из которых наиболее известны igraph [53], Gephi [54] и Cytoscape [55].
Топология сетей
Хотя сеть и может служить средством визуализации взаимосвязей объектов, ее облик сам по себе вряд ли может объективно характеризовать сообщество, поэтому попытки напрямую интерпретировать изображения сетей многовидовых микробиомов часто обречены на неудачу. Более того, разнообразные приемы визуализации могут ввести неподготовленного исследователя в заблуждение, акцентируя внимание на одних свойствах сети или объектов и отвлекая от других (см. рис. 3).
Для описания взаимосвязей в сообществе и роли в нем отдельных видов более продуктивно и надежно использовать формализованные параметры топологии сети. Таких параметров разработано чрезвычайно много [5, 14, 27, 64]. Мы остановимся только на тех из них, которые, с нашей точки зрения, информативны в контексте экологических процессов в сообществах микроорганизмов (табл. 3). Параметры топологии, используемые в анализе экологических сетей (в основном в трофических сетях многоклеточных организмов), хорошо описаны в [64], а в [27] даны учебные примеры их расчетов.
Таблица 3.
Параметры топологии сетей, информативные для экологических приложений
| Свойство | Метрика | Способ расчета | Интерпретация |
|---|---|---|---|
| Интегральные параметры топологии сети | |||
| Сложность сети | Число связей (edge number) | Абсолютное число связей | Чем больше метрика, тем сложнее сеть. Можно использовать только для сравнения сетей со сходным числом узлов |
| Плотность связей (linkage density) | Число связей, нормированное на число узлов сети | ||
| Связность (connectance) | Число связей, нормированное на число потенциальных связей | Чем больше метрика, тем сложнее сеть. Можно сравнивать сети с разным числом узлов | |
| Модулярность сети | Коэффициент модулярности (modularity index) | Степень подразделенности сети на подсети по сравнению со случайной сетью с таким же числом узлов и связей | Чем больше метрика, тем более обособлены подсети внутри сети |
| Число модулей | Абсолютное число подсетей | Чем больше метрика, тем больше обособленных подсетей внутри сети | |
| Ассортативность сети | Коэффициент ассортативности (assortativity index) | Мера сопряженности связей между узлами со сходством свойств узлов; приведена к интервалу от –1 до +1 | Чем ближе значение к 1, тем вероятнее формирование связей между сходными объектами (ассортативная сеть); чем ближе значение к –1, тем вероятнее формирование связи между несходными объектами (диссассортативная сеть). Близкие к нулю значения указывают на преимущественно случайное формирование связей |
| Параметры топологии отдельных узлов | |||
| Центральность узла | Центральность по степени узла (node degree centrality) | Число связей узла | Чем больше метрика, тем с большим числом узлов связан узел и тем более он важен. Важно учитывать направленность связей |
| Центральность по промежуточности (betweenness centrality) | Доля путей, проходящих через узел | Чем больше метрика, тем чаще узел встречается на путях между парами узлов в сети. Важно учитывать направленность связей | |
| Центральность по близости (closeness centrality) | Обратная величина к сумме путей от узла до всех других узлов сети | Чем больше метрика, тем ближе узел к остальным узлам сети. Важно учитывать направленность связей | |
| Локальная ассортативность | Коэффициент локальной ассортативности (local assortativity) | Мера аналогична ассортативности сети, но для окружения узла, а не всей сети | Интерпретация аналогична таковой для ассортативности сети, но в пределах окружения узла |
Меры сложности сети. В качестве наиболее простого показателя сложности может выступать абсолютное число связей, но оно, как правило, сильно зависит от числа узлов сети, поскольку чем больше объектов, тем больше возможностей для формирования связей. Число связей, отнесенное к числу узлов – плотность связей, также зависит от числа объектов, но слабее. Оба показателя больше подходят для сравнения сложности сообществ со сходным числом видов. Более информативна связность сети – доля реализованных связей от всех потенциально возможных [65]. При определении числа потенциальных связей можно учитывать особенности сообществ: например, каннибализм некоторых видов (т.е. связь узла “на себя”), возможность связи только между видами, принадлежащими разным трофическим уровням, а не между всеми, и пр. [66]. В многокомпонентных сообществах, как правило, реализовано менее 20% потенциальных связей [65, 67, 68].
Показатели плотности связей и связности сети нашли применение в исследованиях зависимостей между сложностью и устойчивостью сообществ (complexity–stability relationship). В этом активно разрабатываемом направлении синэкологии сталкиваются две противоположные позиции: с экологической точки зрения чем больше путей переноса энергии в экосистеме, тем она устойчивей [69], тогда как с математической верно обратное [70]. В реальности форма связи между сложностью и устойчивостью сообществ зависит от его размера, состава и силы отдельных взаимодействий [71].
Меры центральности узлов используют для оценки важности объектов в сети, в частности видов в сообществе. Разработано более 250 мер центральности [72], из которых широко известны импакт-фактор научных журналов, а также PageRank, на которой ранее был основан поиск информации в поисковой системе Google. Релевантные сообществам микроорганизмов меры – центральность по степени узла (число связей узла), промежуточности (доля путей, проходящих через узел) и близости (обратная величина к сумме путей от узла до всех остальных узлов). На основе мер центральности можно выявлять так называемые хабы (hubs) – узлы с высокой интегрированностью в сеть, т.е. играющие ключевую роль в ее формировании.
Интерпретация мер центральности неодинакова для ориентированных (directed networks) и неориентированных (undirected) сетей: в первом случае направленность связей между узлами известна, во втором – нет. В ориентированных сетях хабы можно интерпретировать как ключевые виды (keystone species), которые настолько важны для функционирования сообщества, что их исчезновение влечет за собой его кардинальную перестройку. На рис. 3в узел 09 с учетом большого числа исходящих из него связей представляет собой хаб. Узлы с высокой центральностью по степени и близости могут быть интерпретированы как эдификаторы – виды с сильным прямым и опосредованным влиянием на все остальные виды в сообществе.
В большинстве сетей меры центральности тесно коррелируют друг с другом [64, 73], поэтому наличие отклонений от такой корреляции – это дополнительный источник информации. Известны примеры, когда высокая центральность по промежуточности в сочетании с низкой степенью узла указывает на важную регуляторную роль видов [22], которые могут соединять большие подсети (как, например, узел 19 на рис. 3б).
В неориентированных сетях на основе центральности узла нельзя сделать вывод о роли вида, поскольку большое число связей может иметь узел, зависящий от множества других видов, но не влияющий на них [74, 75]. Например, на рис. 3б вывод о важности узла 25, который можно сделать с учетом его высокой степени, меняется на противоположный, если принять во внимание направленность связей, показанную на рис. 3в.
Направленность связей можно определить с помощью анализа временной динамики сообщества или на основе знания биологии видов, а ключевую роль хабов проверить экспериментально, сравнивая последствия их удаления из сообщества с последствиями удаления случайных узлов (targeted vs. random node removal) [75, 76]. Однако из-за высокой микромасштабной изменчивости повторный отбор многокомпонентных природных сообществ, как и экспериментальное манипулирование с их составом практически невозможны. На наш взгляд, выявление хабов более перспективно осуществлять на основе информации о синтрофии (syntrophy, cross-feeding) или цепях трансформации органического вещества. При таком подходе направленность связей можно определить с помощью предсказанного функционального потенциала вида по разнообразию семейств генов (например, на основе баз данных KEGG и EC [77]), кодирующих ферменты, ответственные за разложение тех или иных веществ. Узлом, интерпретируемым как хаб, может быть вид, осуществляющий разложение трудноразлагаемых субстратов или представляющий уникальное звено в цепях их превращений.
Модулярность. С помощью индекса модулярности оценивают степень подразделенности наблюдаемой сети на подсети (сообщества, группы или модули) по сравнению со случайной сетью той же размерности [78]. Анализ модулярности, пожалуй, один из наиболее перспективных инструментов в исследованиях экологических сетей. В зависимости от целей работы группы объектов могут быть выбраны на основе разных критериев (например, по таксономической или функциональной принадлежности). Еще больший интерес представляет выявление самих модулей, т.е. подсетей тесно связанных видов, а также сравнение сетей по числу модулей и поиск детерминантов, обусловливающих их существование. Из-за важности выявления обособленных частей сообществ в эпидемиологии, физиологии, социологии и технической инженерии разработано множество алгоритмов поиска модулей (community detection algorithms) [78, 79], которые можно использовать и при анализе экологических сетей.
Ассортативность, т.е. предпочтение объектов формировать связи с себе подобными, – хорошо известный феномен в социологии, психологии и популяционной генетике [78]. Анализ сетей позволяет оценить ассортативность количественно. В отличие от модулярности сходство объектов можно оценивать не только в номинальных, но и непрерывных шкалах (например, степень сходства по фенотипу, генотипу или числу связей).
Считается, что большинство биологических сетей дисассортативны по степени, т.е. большинство связей формируется между узлами с разной степенью, что среди прочего обусловливает неустойчивость сети к удалению хабов [29, 78, 80]. Дисассортативность по функциональным признакам может проявляться в пищевых сетях, в которых связаны представители разных трофических групп.
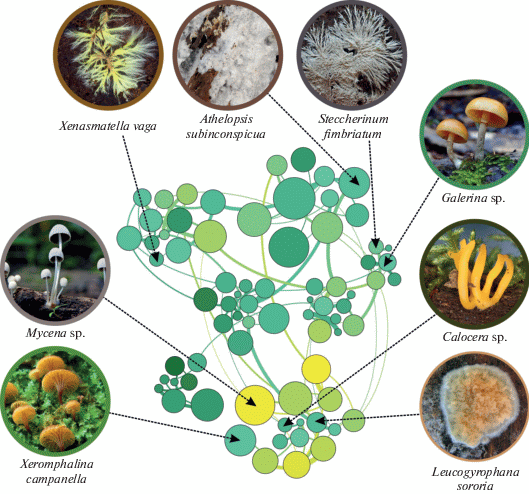
Важно помнить, что сети, построенные на оценках совместной встречаемости (даже при учете обилий видов), могут отражать не только непосредственно взаимоотношения между видами, но также сходство их экологических предпочтений [20, 21, 24]. Анализ модулярности и ассортативности позволяет выявлять группы видов, объединенных пространственной нишей. Примером может служить сеть, построенная на наших данных ДНК-метабаркодирования сообществ грибов мертвой древесины в южной тайге (рис. 4). На ней выделенные в ходе анализа модули состоят из OTU со сходными экологическими предпочтениями, что, помимо визуального результата, подтверждается высокими и статистически значимыми коэффициентами ассортативности по экологическим предпочтениям OTU.
Рис. 4.
Сеть положительных взаимодействий между OTU грибных сообществ мертвой древесины. Параметры топологии сети: число узлов равно 74, число связей – 97, плотность связей – 1.31 связей/узел, связность сети – 3.59%, индекс модулярности – 0.63, число модулей – 7, коэффициент ассортативности по экологическим предпочтениям – 0.64. Толщина линии пропорциональна силе связи, размер узла – относительному обилию OTU. Узлы сгруппированы по принадлежности к модулям. Цветом обозначены предпочтения OTU: от темно-зеленого к желтому увеличиваются встречаемость и обилие OTU в более мягкой, кислой и влажной древесине. Показана таксономическая принадлежность некоторых узлов (фотографии). Построение и расчет интегральных характеристик сети выполнены в R v.4.0.2 [82] с использованием пакетов SPRING v.1.0.2 [50] и igraph v.1.2.5 [53], визуализация – в Gephi v.0.9.2 [54].
Если задача состоит в анализе трофической ниши видов, то можно заранее избавиться от влияния пространственной ниши на структуру сети, добиваясь максимальной однородности выборок по условиям среды [24]. При работе с большинством природных сообществ микроорганизмов полная однородность серии образцов практически невозможна, поэтому полезным инструментом для анализа трофических ниш могут быть меры локальной ассортативности [81].
Поиск мотивов
С исследованием трофических взаимоотношений также связана задача поиска элементарных функциональных единиц сообщества. К ее решению можно подойти с помощью выявления мотивов сети (network motifs) – часто повторяющихся небольших подграфов, которые логично интерпретировать как ее “строительные блоки” [83]. В пищевых сетях макроорганизмов мотивы могут представлять, например, группы хищник–жертва–ресурс или пары конкурирующих видов [84, 85]. Из-за сложности интерпретации громоздких функциональных единиц искомые мотивы обычно включают не более четырех узлов. В отношении сообществ микроорганизмов поиск мотивов перспективен с помощью характеристики трофической ниши по разнообразию генов, кодирующих ответственные за превращения тех или иных веществ ферменты.
ИСТОЧНИКИ ОШИБОК В АНАЛИЗЕ ЭКОЛОГИЧЕСКИХ СЕТЕЙ
Условием корректного анализа сетей должен быть учет ограничений, обусловленных как многокомпонентностью и пространственно-временной изменчивостью объектов исследования, так и особенностями методов получения данных. Некоторые из таких ограничений рассмотрены ниже.
Наличие реликтовой ДНК. Доля находящейся в свободном виде или в мертвых клетках ДНК (extracellular DNA, relic DNA) иногда достигает 90% от всей ДНК в исследуемой среде [86]. В макроэкологических исследованиях, выполняемых в масштабе регионов и континентов, а также при работе с сообществами со сглаженной динамикой состава наличие реликтовой ДНК может лишь незначительно искажать (как занижать, так и завышать) оценки разнообразия микроорганизмов [87, 88]. Однако при работе в микромасштабе – а именно этот масштаб в пространстве и во времени чаще всего интересен при исследовании взаимоотношений микроорганизмов – реликтовая ДНК из-за возможного смещения обилия видов может стать причиной как ложноположительных, так и ложноотрицательных связей в сетях [87]. Избавиться от ДНК, находящейся на поверхности почвенных частиц и в поврежденных клетках, позволяют некоторые технические приемы, в частности предварительная обработка образцов моноазидом этидия [89]. Кроме того, наличие реликтовой ДНК не препятствует анализу разнообразия актуальной (физиологически активной) части микробного сообщества с помощью секвенирования РНК [87, 90].
Артефактные последовательности ДНК. Помимо упомянутой выше проблемы ложных нулей (из-за малой глубины секвенирования), существует опасность возникновения артефактных последовательностей ДНК. Причины этого – ошибки ПЦР, контаминация реагентов или помещений, а также обмен индексами, идентифицирующими образцы в процессе секвенирования (index switching или cross-talk) [91]. Поскольку алгоритмы поиска контаминантов пока несовершенны [92], исходные данные принято предварительно очищать от редких OTU, устанавливая порог фильтрации по встречаемости [93]. К сожалению, выбор величины такого порога произволен, поэтому при фильтрации всегда есть риск исключения из выборки действительно редких видов. Пока решение этой проблемы, как и при любом другом методе описания сообществ, сводится к увеличению размера выборки и/или выборочного усилия (т.е. глубины секвенирования).
Артефактные связи. Исследователи большинства микробоценозов природных сред сталкиваются с тем, что количество образцов несопоставимо мало по сравнению с видовым богатством сообществ (эту проблему обозначают large p, small n problem). Это может приводить к появлению артефактных связей по причинам инфляции уровня значимости из-за множественного тестирования статистических гипотез, наличия опосредованных взаимодействий между видами, сосуществования видов в результате разделения их трофических ниш. Для минимизации риска появления артефактных связей, помимо “универсального средства” – увеличения объема выборки, необходимы поправки на множественное тестирование гипотез (например, контроль доли ложных гипотез, false discovery rate, FDR) [94]. Для устранения влияния опосредованных связей между видами можно использовать методы, основанные на показателях частной или условной корреляций [5].
Ошибочные интерпретации. На протяжении почти всей истории экологии исследователи редко могли устоять перед соблазном интерпретировать совместную встречаемость видов (co-occurrence) в контексте межвидовых отношений [95]. Однако такая интерпретация полна артефактами. Простейший из них – трактовка высокой совместной встречаемости видов исключительно как свидетельство их симбиоза. Добавление информации об обилии видов может указать на их антагонистические взаимоотношения, при которых обилие одного вида снижается при увеличении другого. В обзорных работах приведены многочисленные “подводные камни” этого подхода [52, 75, 95, 96].
При обсуждении параметров топологии мы уже упоминали еще один источник потенциальных ошибок в анализе экологических сетей – интерпретацию хабов как важных для функционирования сообщества видов при отсутствии информации о направленности связей.
ЗАКЛЮЧЕНИЕ
Анализ сетей – это стремительно развивающееся направление, завоевывающее все большую популярность в самых разных предметных областях, в первую очередь связанных с изучением многокомпонентных систем. Благодаря этому в распоряжении экологов оказался мощный инструментарий для описания структуры многовидовых сообществ и выявления их ключевых свойств. Поэтому первоочередной становится задача не “изобретения” новых показателей и методов, а выбора информативных метрик и алгоритмов построения сетей из богатого арсенала уже существующих. Такие метрики и алгоритмы должны не только учитывать специфику экологических сетей, но и быть содержательно интерпретируемы в контексте изучаемых экологических процессов.
При выборе конкретного метода анализа сетей важно убедиться в соответствии исходных данных его допущениям, поскольку результаты зависят и от выбранной метрики связи, и от порога фильтрации по ее силе, и от собственно алгоритма построения сети [97]. Впрочем, устойчивые ассоциации и большинство интегральных параметров топологии обычно сохраняются при использовании разных методов построения сети [24].
С одной стороны, широкое внедрение в практику экологических исследований методов анализа сетей может способствовать развитию понятийного аппарата и наработке полезного опыта. Однако, с другой стороны, некритичное заимствование подходов из смежных областей чревато не только ошибочными выводами, но и в определенной степени может компрометировать саму идею использования анализа сетей в экологических приложениях. В отечественной биологии, начиная с классических работ А.А. Любищева, ошибки применения количественных методов делят на две группы – связанные с “недостатком осведомленности” и “избытком энтузиазма”. Мы надеемся, что представленный обзор современных подходов к анализу экологических сетей и его ограничений в определенной степени будет препятствовать распространению и тех, и других.
Авторы признательны М.В. Модорову, О.Е. Лиходеевской, И.А. Шадрину и Е.А. Бельскому за ценные комментарии к рукописи. Сбор материала, построение сетей, биоинформационный и статистический анализ данных выполнены при поддержке РФФИ (проекты № 18-29-05042 и № 19-04-00921), подготовка рукописи завершена в рамках государственного задания Института экологии растений и животных УрО РАН. При проведении биоинформационного анализа был использован суперкомпьютер “Уран” Института математики и механики УрО РАН. Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Ives A.R., Carpenter S.R. Stability and diversity of ecosystems // Science. 2007. V. 317. № 5834. P. 58–62. https://doi.org/10.1126/science.1133258
Pimm S.L. The complexity and stability of ecosystems // Nature. 1984. V. 307. № 5949. P. 321–326. https://doi.org/10.1038/307321a0
Tylianakis J.M., Didham R.K., Bascompte J., Wardle D.A. Global change and species interactions in terrestrial ecosystems // Ecology Letters. 2008. V. 11. № 12. P. 1351–1363. https://doi.org/10.1111/j.1461-0248.2008.01250.x
Бигон М., Харпер Д., Таунсенд К. Экология. Особи, популяции и сообщества. М.: Мир, 1989. 667 с.
Layeghifard M., Hwang D.M., Guttman D.S. Disentangling interactions in the microbiome: A network perspective // Trends in Microbiology. 2017. V. 25. № 3. P. 217–228. https://doi.org/10.1016/j.tim.2016.11.008
Nannipieri P., Ascher J., Ceccherini M.T. et al. Microbial diversity and soil functions // European Journal of Soil Science. 2003. V. 54. № 4. P. 655–670. https://doi.org/10.1046/j.1351-0754.2003.0556.x
Metzker M.L. Sequencing technologies – the next generation // Nature Reviews Genetics. 2010. V. 11. № 1. P. 31–46. https://doi.org/10.1038/nrg2626
Bahram M., Netherway T., Hildebrand F. et al. Plant nutrient-acquisition strategies drive topsoil microbiome structure and function // New Phytologist. 2020. V. 227. № 4. P. 1189–1199. https://doi.org/10.1111/nph.16598
Davison J., Moora M., Öpik M. et al. Global assessment of arbuscular mycorrhizal fungus diversity reveals very low endemism // Science. 2015. V. 349. № 6251. P. 970–973. https://doi.org/10.1126/science.aab1161
Tedersoo L., Bahram M., Põlme S. et al. Global diversity and geography of soil fungi // Science. 2014. V. 346. № 6213. 1256688. https://doi.org/10.1126/Science.1256688
Tisthammer K.H., Cobian G.M., Amend A.S. Global biogeography of marine fungi is shaped by the environment // Fungal Ecology. 2016. V. 19. P. 39–46. https://doi.org/10.1016/j.funeco.2015.09.003
eiss S., van Treuren W., Lozupone C. et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision // The ISME Journal. 2016. V. 10. № 7. P. 1669–1681. 10.1038/ismej.2015.23
Faust K., Raes J. Microbial interactions: from networks to models // Nature Reviews Microbiology. 2012. V. 10. № 8. P. 538–550. https://doi.org/10.1038/nrmicro2832
Newman M. Networks: An Introduction. Oxford: Oxford University Press, 2010. 772 p. https://doi.org/10.1093/acprof:oso/9780199206650. 001.0001
Jacquiod S., Puga-Freitas R., Spor A. et al. A core microbiota of the plant-earthworm interaction conserved across soils // Soil Biology and Biochemistry. 2020. V. 144. 107754. https://doi.org/10.1016/j.soilbio.2020.107754
Tipton L., Muller C.L., Kurtz Z.D. et al. Fungi stabilize connectivity in the lung and skin microbial ecosystems // Microbiome. 2018. V. 6. № 1. 12. https://doi.org/10.1186/s40168-017-0393-0
Jiang Y.J., Sun B., Li H.X. et al. Aggregate-related changes in network patterns of nematodes and ammonia oxidizers in an acidic soil // Soil Biology and Biochemistry. 2015. V. 88. P. 101–109. https://doi.org/10.1016/j.soilbio.2015.05.013
Darcy J.L., Swift S.O.I., Cobian G.M. et al. Fungal communities living within leaves of native Hawaiian dicots are structured by landscape-scale variables as well as by host plants // Molecular Ecology. 2020. V. 29. P. 3102–3115. https://doi.org/10.1111/mec.15544
Banerjee S., Thrall P.H., Bissett A. et al. Linking microbial co-occurrences to soil ecological processes across a woodland-grassland ecotone // Ecology and Evolution. 2018. V. 8. № 16. P. 8217–8230. https://doi.org/10.1002/ece3.4346
Cram J.A., Xia L.C., Needham D.M. et al. Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes // The ISME Journal. 2015. V. 9. № 12. P. 2573–2586. https://doi.org/10.1038/ismej.2015.76
Chaffron S., Rehrauer H., Pernthaler J., von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data // Genome Research. 2010. V. 20. № 7. P. 947–959. https://doi.org/10.1101/gr.104521.109
Meyer J.L., Gunasekera S.P., Scott R.M. et al. Microbiome shifts and the inhibition of quorum sensing by Black Band Disease cyanobacteria // The ISME Journal. 2016. V. 10. № 5. P. 1204–1216. https://doi.org/10.1038/ismej.2015.184
Pollet T., Berdjeb L., Garnier C. et al. Prokaryotic community successions and interactions in marine biofilms: the key role of Flavobacteriia // FEMS Microbiology Ecology. 2018. V. 94. № 6. https://doi.org/10.1093/femsec/fiy083
Rottjers L., Faust K. From hairballs to hypotheses-biological insights from microbial networks // FEMS Microbiology Reviews. 2018. V. 42. № 6. P. 761–780. https://doi.org/10.1093/femsre/fuy030
Deng Y., Jiang Y.-H., Yang Y. et al. Molecular ecological network analyses // BMC Bioinformatics. 2012. V. 13. № 1. P. 113. https://doi.org/10.1186/1471-2105-13-113
Dáttilo W., Rico-Gray V. Ecological networks in the tropics: An integrative overview of species interactions from some of the most species-rich habitats on earth. Cham: Springer, 2018. 202 p. https://doi.org/10.1007/978-3-319-68228-0
Junker B.H., Schreiber F. Analysis of biological networks. Hoboken, N.J.: Wiley-Interscience, 2008. 346 p.
Képès F. Biological networks. Singapore: World Scientific, 2007. 516 p. https://doi.org/10.1142/6459
Woodward G. Advances in ecological research: Ecological networks. Amsterdam: Elsevier, 2010. 454 p.
Семенов М.В. Метабаркодинг и метагеномика в почвенно-экологических исследованиях: успехи, проблемы и возможности // Журн. общ. биол. 2019. Т. 80. № 6. С. 403–417. https://doi.org/10.1134/S004445961906006X
Salazar G., Cornejo-Castillo F.M., Benitez-Barrios V. et al. Global diversity and biogeography of deep-sea pelagic prokaryotes // The ISME Journal. 2016. V. 10. № 3. P. 596–608. https://doi.org/10.1038/ismej.2015.137
Pawlowski J., Audic S., Adl S. et al. CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms // PLoS Biology. 2012. V. 10. № 11. e1001419. https://doi.org/10.1371/journal.pbio.1001419
Mikryukov V.S., Dulya O.V., Modorov M.V. Phylogenetic signature of fungal response to long-term chemical pollution // Soil Biology and Biochemistry. 2020. V. 140. P. 107 644. https://doi.org/10.1016/j.soilbio.2019.107644
Porter T.M., Morris D.M., Basiliko N. et al. Variations in terrestrial arthropod DNA metabarcoding methods recovers robust beta diversity but variable richness and site indicators // Scientific Reports. 2019. V. 9. 18218. https://doi.org/10.1038/s41598-019-54532-0
Чернов Т.И., Железова А.Д., Кутовая О.В. и др. Сравнительная оценка структуры микробиомов погребенных и современных почв при помощи анализа микробной ДНК // Микробиология. 2018. Т. 87. № 6. С. 737–746. https://doi.org/10.1134/S0026365618060071
Walters K.E., Martiny J.B.H. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats // PLoS One. 2020. V. 15. № 9. e0233872. https://doi.org/10.1371/journal.pone.0233872
Schoch C.L., Seifert K.A., Huhndorf S. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi // Proceedings of the National Academy of Sciences. 2012. V. 109. № 16. P. 6241–6246. https://doi.org/10.1073/pnas.1117018109
Pace N.R. A molecular view of microbial diversity and the biosphere // Science. 1997. V. 276. № 5313. P. 734–740. https://doi.org/10.1126/science.276.5313.734
Liu M., Clarke L.J., Baker S.C. et al. A practical guide to DNA metabarcoding for entomological ecologists // Ecological Entomology. 2020. V. 45. № 3. P. 373–385. https://doi.org/10.1111/een.12831
Callahan B.J., McMurdie P.J., Holmes S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis // The ISME Journal. 2017. V. 11. № 12. P. 2639–2643. https://doi.org/10.1038/ismej.2017.119
Nilsson R.H., Larsson K.H., Taylor A.F.S. et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications // Nucleic Acids Research. 2019. V. 47. № D1. P. D259–D264. https://doi.org/10.1093/nar/gky1022
Pruesse E., Quast C., Knittel K. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB // Nucleic Acids Research. 2007. V. 35. № 21. P. 7188–7196. https://doi.org/10.1093/nar/gkm864
Guillou L., Bachar D., Audic S. et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy // Nucleic Acids Research. 2013. V. 41. № D1. P. 597–604. https://doi.org/10.1093/nar/gks1160
Ratnasingham S., Hebert P.D. BOLD: The barcode of life data system (http://www.barcodinglife.org) // Molecular Ecology Notes. 2007. V. 7. № 3. P. 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J. Microbiome datasets are composItional: And this is not optional // Frontiers in Microbiology. 2017. V. 8. 2224. https://doi.org/10.3389/fmicb.2017.02224
Morton J.T., Marotz C., Washburne A. et al. Establishing microbial composition measurement standards with reference frames // Nature Communications. 2019. V. 10. № 1. 2719. https://doi.org/10.1038/s41467-019-10656-5
Martín-Fernández J.-A., Hron K., Templ M. et al. Bayesian-multiplicative treatment of count zeros in compositional data sets // Statistical Modelling. 2015. V. 15. № 2. P. 134–158. https://doi.org/10.1177/1471082X14535524
Freeman L.C. The development of social network analysis: A study in the sociology of science. Vancouver BC: Empirical Press, 2004. 218 p.
Fernandes A.D., Reid J.N., Macklaim J.M. et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis // Microbiome. 2014. V. 2. 15. https://doi.org/10.1186/2049-2618-2-15
Yoon G., Gaynanova I., Muller C.L. Microbial networks in SPRING – Semi-parametric rank-based correlation and partial correlation estimation for quantitative microbiome data // Frontiers in Genetics. 2019. V. 10. P. 516. https://doi.org/10.3389/fgene.2019.00516
Faust K., Sathirapongsasuti J.F., Izard J. et al. Microbial co-occurrence relationships in the human microbiome // PLoS Computational Biology. 2012. V. 8. № 7. e1002606. https://doi.org/10.1371/journal.pcbi.1002606
Hirano H., Takemoto K. Difficulty in inferring microbial community structure based on co-occurrence network approaches // BMC Bioinformatics. 2019. V. 20. 329. https://doi.org/10.1186/s12859-019-2915-1
Csardi G., Nepusz T. The igraph software package for complex network research // InterJournal, complex systems. 2006. V. 1695. № 5. P. 1–9.
Bastian M., Heymann S., Jacomy M. Gephi: An open source software for exploring and manipulating networks // Proceedings of the third international AAAI conference on weblogs and social media. 2009. V. 8. P. 361–362. https://doi.org/10.13140/2.1.1341.1520
Shannon P., Markiel A., Ozier O. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks // Genome Research. 2003. V. 13. № 11. P. 2498–2504. https://doi.org/10.1101/gr.1239303
Friedman J., Alm E.J. Inferring correlation networks from genomic survey data // PLoS Computational Biology. 2012. V. 8. № 9. e1002687. https://doi.org/10.1371/journal.pcbi.1002687
Schwager E., Weingart G., Bielski C. CCREPE: Compositionality corrected by permutation and renormalization / URL: https://www.bioconductor.org/packages/devel/bioc/html/ccrepe.html
Reshef D.N., Reshef Y.A., Finucane H.K. et al. Detecting novel associations in large datasets // Science. 2011. V. 334. № 6062. P. 1518–1524. https://doi.org/10.1126/science.1205438
Ruan Q., Dutta D., Schwalbach M.S. et al. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors // Bioinformatics. 2006. V. 22. № 20. P. 2532–2538. https://doi.org/10.1093/bioinformatics/btl417
Fang H., Huang C., Zhao H., Deng M. CCLasso: correlation inference for compositional data through Lasso // Bioinformatics. 2015. V. 31. № 19. P. 3172–3180. https://doi.org/10.1093/bioinformatics/btv349
Quinn T.P., Richardson M.F., Lovell D., Crowley T.M. propr: An R-package for identifying proportionally abundant features using compositional data analysis // Scientific Reports. 2017. V. 7. https://doi.org/10.1038/s41598-017-16520-0
Ban Y., An L., Jiang H. Investigating microbial co-occurrence patterns based on metagenomic compositional data // Bioinformatics. 2015. V. 31. № 20. P. 3322–3329. https://doi.org/10.1093/bioinformatics/btv364
Faust K., Raes J. CoNet app: inference of biological association networks using Cytoscape // F1000Research. 2016. V. 5. https://doi.org/10.12688/f1000research.9050.2
Antoniazzi R., Dáttilo W., Rico-Gray V. A useful guide of main indices and software used for ecological networks studies // Ecological networks in the tropics / Eds. Dáttilo W., Rico-Gray V. Cham, 2018. P. 185–196. https://doi.org/10.1007/978-3-319-68228-0_13
Dunne J.A., Williams R.J., Martinez N.D. Food-web structure and network theory: The role of connectance and size // Proceedings of the National Academy of Sciences of the United States of America. 2002. V. 99. № 20. P. 12917–12922. https://doi.org/10.1073/pnas.192407699
Delmas E., Besson M., Brice M.H. et al. Analysing ecological networks of species interactions // Biological Reviews. 2019. V. 94. № 1. P. 16–36. https://doi.org/10.1111/brv.12433
Lupatini M., Suleiman A.K.A., Jacques R.J.S. et al. Network topology reveals high connectance levels and few key microbial genera within soils // Frontiers in Environmental Science. 2014. V. 2. https://doi.org/10.3389/fenvs.2014.00010
Mikhailov I.S., Zakharova Y.R., Bukin Y.S. et al. Co-occurrence networks among bacteria and microbial eukaryotes of lake Baikal during a spring phytoplankton bloom // Microbial Ecology. 2019. V. 77. № 1. P. 96–109. https://doi.org/10.1007/s00248-018-1212-2
MacArthur R. Fluctuations of animal populations and a measure of community stability // Ecology. 1955. V. 36. № 3. P. 533–536. https://doi.org/10.2307/1929601
May R.M. Will a large complex system be stable? // Nature. 1972. V. 238. № 5364. P. 413–414. https://doi.org/10.1038/238413a0
Landi P., Minoarivelo H.O., Brannstrom A. et al. Complexity and stability of ecological networks: a review of the theory // Population Ecology. 2018. V. 60. № 4. P. 319–345. https://doi.org/10.1007/s10144-018-0628-3
Jalili M., Salehzadeh-Yazdi A., Asgari Y. et al. CentiServer: A comprehensive resource, web-based application and R package for centrality analysis // PLoS One. 2015. V. 10. № 11. e0143111. https://doi.org/10.1371/journal.pone.0143111
Lau M.K., Borrett S.R., Baiser B. et al. Ecological network metrics: opportunities for synthesis // Ecosphere. 2017. V. 8. № 8. e01900. https://doi.org/10.1002/ecs2.1900
Faust K., Bauchinger F., Laroche B. et al. Signatures of ecological processes in microbial community time series // Microbiome. 2018. V. 6. 120. https://doi.org/10.1186/s40168-018-0496-2
Rottjers L., Faust K. Can we predict keystones? // Nature Reviews Microbiology. 2019. V. 17. № 3. P. 193–193. https://doi.org/10.1038/s41579-018-0132-y
Agler M.T., Ruhe J., Kroll S. et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation // PLoS Biology. 2016. V. 14. № 1. P. 1–31. https://doi.org/10.1371/journal.pbio.1002352
Douglas G.M., Maffei V.J., Zaneveld J.R. et al. PICRUSt2 for prediction of metagenome functions // Nature Biotechnology. 2020. V. 38. № 6. P. 685–688. https://doi.org/10.1038/s41587-020-0548-6
Newman M.E.J. Assortative mixing in networks // Physical Review Letters. 2002. V. 89. № 20. 208701. https://doi.org/10.1103/PhysRevLett.89.208701
Yang Z., Algesheimer R., Tessone C.J. A comparative analysis of community detection algorithms on artificial networks // Scientific Reports. 2016. V. 6. 30 750. https://doi.org/10.1038/srep30750
Piraveenan M., Uddin S., Chung K.S.K. Measuring topological robustness of networks under sustained targeted attacks // 2012 IEEE/ACM international conference on advances in social networks analysis and mining (Asonam). 2012. P. 38–45. https://doi.org/10.1109/Asonam.2012.17
Peel L., Delvenne J.C., Lambiotte R. Multiscale mixing patterns in networks // Proceedings of the National Academy of Sciences of the United States of America. 2018. V. 115. № 16. P. 4057–4062. https://doi.org/10.1073/pnas.1713019115
R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. URL: https://www.R-project.org/
Milo R., Shen-Orr S., Itzkovitz S. et al. Network motifs: Simple building blocks of complex networks // Science. 2002. V. 298. № 5594. P. 824–827. https://doi.org/10.1126/science.298.5594.824
Giling D.P., Ebeling A., Eisenhauer N. et al. Plant diversity alters the representation of motifs in food webs // Nature Communications. 2019. V. 10. № 1. P. 1226. https://doi.org/10.1038/s41467-019-08856-0
Stouffer D.B., Camacho J., Jiang W., Amaral L.A.N. Evidence for the existence of a robust pattern of prey selection in food webs // Proceedings of the Royal Society B-Biological Sciences. 2007. V. 274. № 1621. P. 1931–1940. https://doi.org/10.1098/rspb.2007.0571
Dell’Anno A., Danovaro R. Extracellular DNA plays a key role in deep-sea ecosystem functioning // Science. 2005. V. 309. № 5744. P. 2179–2179. https://doi.org/10.1126/science.1117475
Carini P., Delgado-Baquerizo M., Hinckley E.L.S. et al. Effects of spatial variability and relic DNA removal on the detection of temporal dynamics in soil microbial communities // Mbio. 2020. V. 11. № 1. e02776–19. https://doi.org/10.1128/mBio.02776-19
Lennon J.T., Muscarella M.E., Placella S.A., Lehmkuhl B.K. How, when, and where relic DNA affects microbial diversity // Mbio. 2018. V. 9. № 3. e00637–18. https://doi.org/10.1128/mBio.00637-18
Wagner A.O., Malin C., Knapp B.A., Illmer P. Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide // Applied and Environmental Microbiology. 2008. V. 74. № 8. P. 2537–2539. https://doi.org/10.1128/Aem.02288-07
Baldrian P., Kolařík M., Štursová M. et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition // The ISME Journal. 2012. V. 6. № 2. P. 248–258. https://doi.org/10.1038/ismej.2011.95
Edgar R.C. UNCROSS: Filtering of high-frequency cross-talk in 16S amplicon reads // bioRxiv. 2016. P. 088 666. https://doi.org/10.1101/088666
Davis N.M., Proctor D.M., Holmes S.P. et al. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data // Microbiome. 2018. V. 6. 226. https://doi.org/10.1186/s40168-018-0605-2
Lagkouvardos I., Fischer S., Kumar N., Clavel T. Rhea: a transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons // Peerj. 2017. V. 5. e2836. https://doi.org/10.7717/peerj.2836
Verhoeven K.J.F., Simonsen K.L., McIntyre L.M. Implementing false discovery rate control: Increasing your power // Oikos. 2005. V. 108. P. 643–647. https://doi.org/10.1111/j.0030-1299.2005.13727.x
Blanchet F.G., Cazelles K., Gravel D. Co-occurrence is not evidence of ecological interactions // Ecology Letters. 2020. V. 23. № 7. P. 1050–1063. https://doi.org/10.1111/ele.13525
Berry D., Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks // Frontiers in Microbiology. 2014. V. 5. 219. https://doi.org/10.3389/fmicb.2014.00219
Connor N., Barberan A., Clauset A. Using null models to infer microbial co-occurrence networks // PLoS One. 2017. V. 12. № 5. e0176751. https://doi.org/10.1371/journal.pone.0176751
Дополнительные материалы отсутствуют.