

Электрохимия, 2019, T. 55, № 4, стр. 508-512
Электрокаталитические свойства Rh/C- и Pt–Rh/C-катализаторов, полученных методом электрохимического диспергирования
Н. А. Фаддеев a, А. Б. Куриганова a, *, И. Н. Леонтьев b, Н. В. Смирнова a, **
a Южно-Российский государственный политехнический университет (НПИ) имени М.И. Платова
346428 Ростовская обл., Новочеркасск, ул. Просвещения, 132, Россия
b Южный федеральный университет
344090 Ростов-на-Дону, ул. Зорге, 5, Россия
* E-mail: kuriganova_@mail.ru
** E-mail: smirnova_nv@mail.ru
Поступила в редакцию 02.05.2017
После доработки 31.10.2017
Принята к публикации 21.11.2018
Аннотация
Методом электрохимического диспергирования металлов в Na+-содержащих водных электролитах были получены Pt/C-, Rh/C- и Pt–Rh/C-каталитические системы, с равномерным распределением металлической фазы по поверхности углеродного носителя и средним размером наночастиц Pt и Rh 7.3 и 6.5 нм соответственно. Показано, что для определения электрохимически активной площади поверхности Pt/C-, Rh/C- и Pt–Rh/C-катализаторов методом окислительной десорбции СО оптимальным потенциалом адсорбции СО является потенциал двойнослойной области E = 300 мВ (о. в. э.). Введение Rh в состав Pt/C-катализатора позволило увеличить удельную активность и снизить потенциал начала окисления метанола.
ВВЕДЕНИЕ
Низкотемпературные топливные элементы (НТЭ) с прямым окислением жидкого топлива являются альтернативой водородным НТЭ. Применение жидкого топлива (метанол, этанол) имеет ряд преимуществ перед водородом: простота транспортировки, хранения и использования, низкая стоимость. Однако, промежуточные продукты окисления спиртов, в частности CO, адсорбируясь, могут блокировать активные центры Pt и снижать активность Pt/C-катализатора, что приводит к снижению мощности НТЭ [1].
Окисление органических соединений на Pt-содержащих катализаторах определяется степенью заполнения адсорбированных органических (COадс) и кислородсодержащих частиц (OHадс или Oадс) [2]. Термодинамически рассчитанные потенциалы окисления спиртов близки к 0 В (о. в. э.), тогда как адсорбция кислородсодержащих частиц на Pt начинается только при E > 0.6 В, что и обуславливает возникновение перенапряжения анодной реакции окисления спирта [1, 2]. Для снижения перенапряжения в состав Pt-содержащих катализаторов вводят ряд металлов (Ru, Rh, Sn, Ni), адсорбция кислородсодержащих частиц на которых протекает легче, чем на Pt [3, 4]. Наилучшей кислородадсорбирующей способностью обладает Ru. Однако Ru (как и Ni или Sn) в большей степени подвержен выщелачиванию, чем Rh.
Для получения Rh/C- и Pt–Rh/C-каталитических систем был использован метод электрохимического диспергирования Pt и Rh электродов в импульсном режиме электролиза, который уже был применен к синтезу Pt-содержащих катализаторов [5, 6]. Ранее нами были продемонстрированы преимущества метода электрохимического диспергирования для получения Pt-содержащих катализаторов перед традиционным методом химического восстановления прекурсоров платины [5], такие как сокращение числа технологических процедур в процессе синтеза, отсутствие необходимости использования органических растворителей и стабилизаторов, улучшенные электрохимические характеристики Pt/C-катализаторов по сравнению с коммерческим аналогом, а также возможность получения Pt-содержащих катализаторов на основе углеродных носителей с различной морфологией, но одинаковой микроструктурой платиновых наночастиц [6]. В данной работе мы продемонстрировали применимость метода электрохимического диспергирования металлов к синтезу наноструктур на основе Rh‑наночастиц, которые являются сокатализатором в Pt-содержащих каталитических системах в анодных процессах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез Rh/C- и Pt/C-каталитических систем осуществляли по методике, представленной в [5]. Получение Pt–Rh/C-каталитических систем осуществляли аналогичным способом, путем последовательным диспергирования Rh, а затем Pt электродов в суспензии углеродного носителя (УН) в 2 M NaOH. Содержание металлической фазы в катализаторах (табл. 1) регулировалось продолжительностью синтеза по заранее известным скоростям диспергирования Pt и Rh.
Таблица 1.
Характеристики Pt/C-, Rh/C- и Pt-Rh/C-катализаторов
| Образец | Загрузка металла, % | a, Å | D111, нм | Eпик СО, мВ | Sуд, м2 г–1 | ${{j}_{{{\text{п и к }}\,\,{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}},$ мА см–2 | ||
|---|---|---|---|---|---|---|---|---|
| Eадс = 100 мВ | Eадс = 300 мВ | Eадс= 500 мВ | ||||||
| Pt/C | 25 | 3.9178 | 7.3 | 756 | 6 ± 1.0 | 11 ± 1.0 | 8 ± 1.0 | 27.10 |
| Rh/C | 25 | 3.8022 | 6.5 | 668 | 11 ± 1.0 | 16 ± 1.5 | 15 ± 1.5 | – |
| Pt–Rh/C | 16.6% – Pt 8.4% – Rh |
– | – | 772 | 11 ± 1.0 | 15 ± 1.5 | 11 ± 1.0 | 34.85 |
Рентгеноструктурные исследования были проведены на Швейцарско-Норвежской линии Европейского центра синхротронного излучения (SNBL ESRF). Размер наночастиц рассчитывали, используя формулу Шеррера [7]. При определении размеров частиц (D111) использовались результаты аппроксимации рефлекса (111). Параметры элементарной ячейки (a) определяли при помощи UnitCell [8], используя все рефлексы рентгенограммы. Микроскопические исследования образцов катализаторов проводились с помощью просвечивающей электронной микроскопии (ПЭМ) (Zeiss LEO 912AB).
Электрохимически активную (ЭХАП) и удельную (Sуд) площадь поверхности катализаторов определяли методом окислительной десорбции CO [9]. Исследование электрокаталитических свойств осуществляли в растворах 0.5 M CH3OH + + 0.5 M H2SO4 методом циклической вольтамперометрии (ЦВА) в интервале потенциалов 0.05–1.3 В (о. в. э.).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
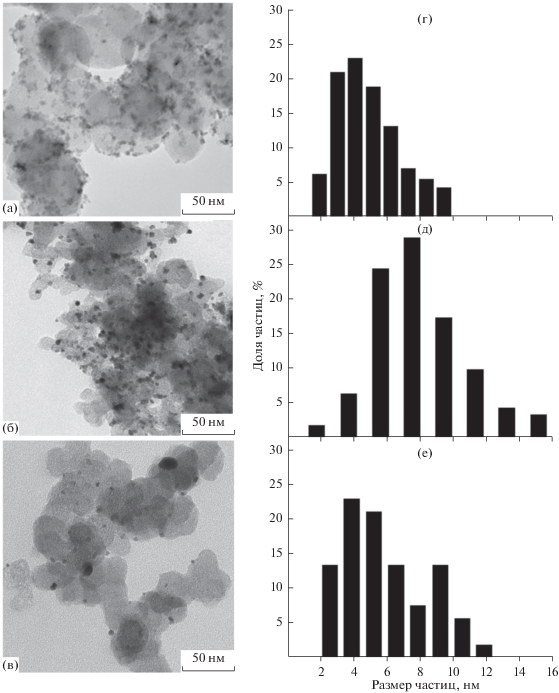
Рассчитанный на основании данных рентгенофазового анализа средний размер кристаллитов Pt и Rh в полученных Pt/C- и Rh/C-катализаторах составил 7.3 и 6.5 нм соответственно (рис. 1, табл. 1). Данные электронной микроскопии также свидетельствуют о преобладании частиц Rh и Pt размером 4–5 и 6–8 нм соответственно (рис. 2г, 2д), для смешанного образца Pt–Rh/C было характерно бимодальное распределение наночастиц по размерам (рис. 2е). Все синтезированные образцы катализаторов характеризовались равномерным распределением наночастиц металлов по поверхности УН (рис. 2а–2в).
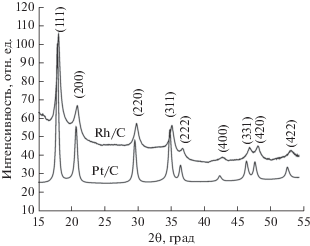
Рис. 2.
ПЭМ-изображения Rh/C- (a), Pt/C- (б), PtRh/C- (в) катализаторов и распределение частиц Rh (г), Pt (д) и Pt + Rh (e) по размерам.
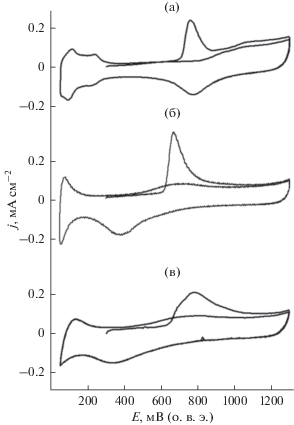
На рисунках 3а–3в представлены ЦВА Pt/C, Rh/C и Pt–Rh/C в 0.5 M H2SO4 в присутствии адсорбированного СО. Потенциал пика окисления СО на Rh/C смещен в катодную область более чем на 80 мВ по сравнению с Pt/C и Pt–Rh/C, что обусловлено более высокой кислородадсорбирующей способностью Rh в результате формирования поверхностных оксидов Rh [10]. При этом, при адсорбции СО в водородной области потенциалов (E = 100 мВ) (табл. 1) значения Sуд поверхности катализаторов существенно занижены, что может быть связано с частичным гидрированием СО [11]. При адсорбции СО в области более анодных потенциалов (E = 500 мВ) также наблюдаются заниженные значения Sуд, что обусловлено наличием в этой области потенциалов адсорбированных кислородсодержащих частиц, как на Pt, так и на Rh [12], обеспечивающих частичное окисление СО в процессе его адсорбции. Поэтому для измерения ЭХАП и Sуд Pt/C, Rh/C и Pt–Rh/C адсорбцию СО проводили при потенциалах двойнослойной области (E = 300 мВ) в отсутствии адсорбированных кислородсодержащих частиц или водорода. Наибольшей Sуд обладает Rh/C-катализатор (табл. 1), что обусловлено сравнительно низким размером частиц Rh по сравнению с Pt и отсутствием частиц размером более 10 нм (рис. 2а, 2г).
Рис. 3.
ЦВА Pt/C- (a), Rh/C- (б) и Pt–Rh/C- (в) катализаторов в 0.5 М H2SO4 в присутствии CO, адсорбированного при E = 300 мВ.
На рис. 4 представлены ЦВА Pt/C-, Rh/C- и Pt–Rh/C-катализаторов в растворе 0.5 М CH3OH + + 0.5 M H2SO4. При анодной развертке потенциала скорость окисления метанола на Pt–Rh/C на 30% превышает скорость окисления метанола на Pt/C, при этом процесс начинается при более чем на 100 мВ более катодных потенциалах.
Рис. 4.
ЦВА Pt/C, Rh/C и Pt–Rh/C в растворе 0.5 М CH3OH + 0.5 M H2SO4, скорость развертки потенциала 50 мВ с–1.
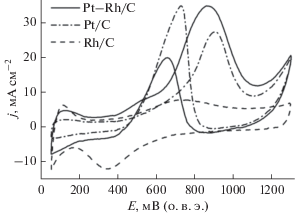
Аналогичный каталитический эффект наблюдается как для сплавов Pt и Rh [13], так и для смешанных Pt + Rh-катализаторов [14] и обусловлен бифункциональным механизмом окисления адсорбированных интермедиатов органической природы [15].Низкая удельная электрокаталитическая активность Rh/C-катализатора в реакции электроокисления метанола обусловлена низкой скоростью процесса дегидрирования молекулы метанола на наночастицах Rh по сравнению с Pt в кислой среде [16, 17].
ЗАКЛЮЧЕНИЕ
В работе предложен способ получения Rh/C- и Pt–Rh/C-катализаторов путем электрохимического диспергирования Pt- и Rh-электродов под действием переменного импульсного тока. Показано, что присутствие Rh- в Pt/C-катализаторе снижает перенапряжение окисления как СО, так и метанола, что обусловлено бифункциональным механизмом окисления адсорбированных органических частиц.
Список литературы
Pedersen, C.M., Escudero-Escribano, M., Velázquez-Palenzuela, A., Christensen, L.H., Chorkendorff, I., and Stephens, I.E.L., Benchmarking Pt-based electrocatalysts for low temperature fuel cell reactions with the rotating disk electrode: oxygen reduction and hydrogen oxidation in the presence of CO (review article), Electrochim. Acta, 2015, vol. 179, p. 647.
Gomez-Marin, A.M. and HernáNdez-Ortiz, J.P., Langmuir–Hinshelwood Mechanism Including Lateral Interactions and Species Diffusion for CO Electro-Oxidation on Metallic Surfaces, J. Phys. Chem. C, 2014, vol. 118, p. 2475.
Михайлова, А.А., Пасынский, А.А., Гринберг, В.А., Великодный, Ю.А., Хазова, О.А. Окисление СО и метанола на платино-оловянных электродах. Электрохимия. 2010. Т. 46. С. 29. [Mikhailova, A.A., Pasynskii, A.A., Grinberg, V.A., Velikodnyi, Y.A., and Khazova, O.A., CO and methanol oxidation at platinum-tin electrodes, Russ. J. Electrochem., 2010, vol. 46, p. 26.]
Кузов, А.В., Тарасевич, М.Р., Богдановская, В.А. Катализаторы анодного окисления этанола для этанольно-воздушного топливного элемента с протонпроводящим полимерным электролитом. Электрохимия. 2010. Т. 46. С. 444. [Kuzov, A.V., Tarasevich, M.R., and Bogdanovskaya, V.A., Catalysts of ethanol anodic oxidation for ethanol-air fuel cell with a proton-conducting polymer electrolyte, Russ. J. Electrochem., 2010, vol. 46, p. 422.]
Leontyev, I., Kuriganova, A., Kudryavtsev, Y., Dkhil, B., and Smirnova, N., New life of a forgotten method: Electrochemical route toward highly efficient Pt/C catalysts for low-temperature fuel cells, Appl. Catal. A, 2012, vol. 431–432, p. 120.
Смирнова, Н.В., Куриганова, А.Б., Новикова, К.С., Герасимова, Е.В. О роли морфолгии углеродного носителя в формировании каталитического слоя твердополимерного топливного элемента. Электрохимия. 2014. Т. 50. С. 999. [Smirnova, N.V., Kuriganova, A.B., Novikova, K.S., and Gerasimova, E.V., The role of carbon support morphology in the formation of catalytic layer of solid-polymer fuel cell, Russ. J. Electrochem., 2014, vol. 50, p. 899.]
Roisnel, T. and Rodríquez-Carvajal, J., WinPLOTR: A windows tool for powder diffraction pattern analysis J. Rodrigues–Carvajal. Proceedings of the European Powder Diffraction Conference (EPDIC7), 2001, vol. 378–381, p. 118.
Holland, T.J.B. and Redfern, S.A.T., UNITCELL: a nonlinear least-squares program for cell-parameter refinement and implementing regression and deletion diagnostics, J. Appl. Crystallogr., 1997, vol. 30, p. 84.
Watt-Smith, M.J., Friedrich, J.M., Rigby, S.P., Ralph, T.R., and Walsh, F.C., Determination of the electrochemically active surface area of Pt/C PEM fuel cell electrodes using different adsorbates, J. Phys. D: Appl. Phys., 2008, vol. 41, p. 174004.
Delpeuch, A.B., Maillard, F., Chatenet, M., Soudant, P., and Cremers, C., Ethanol oxidation reaction (EOR) investigation on Pt/C, Rh/C, and Pt-based bi- and tri-metallic electrocatalysts: A DEMS and in situ FTIR study, Appl. Catal. B: Environ., 2016, vol. 181, p. 672.
Дамаскин, Б.Б., Петрий, О.А., Батраков, В.В. Адсорбция органических соединений на электродах из платиновых металлов. M.: Наука, 1968. 246 p.
Zhang, Y., Gao, X., and Weaver, M.J., Nature of surface bonding on voltammetrically oxidized noble metals in aqueous media as probed by real-time surface-enhanced Raman spectroscopy, J. Phys. Chem., 1993, vol. 97, p. 8656.
Lima, F.H.B., Profeti, D., Lizcano-Valbuena, W.H., Ticianelli, E.A., and Gonzalez, E.R., Carbon-dispersed Pt–Rh nanoparticles for ethanol electro-oxidation. Effect of the crystallite size and of temperature, J. Electroanal. Chem., 2008, vol. 617, p. 121.
Fang, L., Vidal-Iglesias, F.J., Huxter, S.E., Attard, G.A., and Wells, P.B., RhPt/graphite catalysts for CO electrooxidation: Performance of mixed metal and alloyed surfaces, Surf. Sci., 2015, vol. 631, p. 258.
Hammer, B. and Nørskov, J.K., Theoretical surface science and catalysis–calculations and concepts, Academic Press, 2000. p. 71.
Lin, X.-F., Ren, B., and Tian, Z.-Q., Electrochemical and Surface-Enhanced Raman Spectroscopic Studies on the Adsorption and Electrooxidation of C1 Molecules on a Roughened Rh Electrode, J. Phys. Chem. B, 2004, vol. 108, p. 981.
De Souza, J.P.I., Queiroz, S.L., Bergamaski, K., Gonzalez, E.R., and Nart, F.C., Electro-Oxidation of Ethanol on Pt, Rh, and PtRh Electrodes. A Study Using DEMS and in-situ FTIR Techniques, J. Phys. Chem. B, 2002, vol. 106, p. 9825.
Дополнительные материалы отсутствуют.
Инструменты
Электрохимия