

Электрохимия, 2019, T. 55, № 6, стр. 687-695
Влияние микроструктуры на электрохимические свойства композитного катода LiFePO4/C/восстановленный оксид графена
Г. Кучинскис a, *, Г. Баярс a, **, К. Бикова a, К. Капранс a, Ю. Клеперис a
a Институт физики твердого тела, Латвийский Университет
LV-1063 Рига, ул. Кенгарага, 8, Латвия
* E-mail: gints.kucinskis@cfi.lu.lv
** E-mail: gunars.bajars@gmail.com
Поступила в редакцию 11.10.2018
После доработки 30.11.2018
Принята к публикации 22.01.2019
Аннотация
Синтезированы композиты LiFePO4/C/восстановленный оксид графена с различной морфологией, что позволило оценить электрохимическое поведение в зависимости от морфологии образца. Частицы LiFePO4, закрепившиеся на листках восстановленного оксида графена, а также листки восстановленного оксида графена, обернутые вокруг агломератов LiFePO4, – это две наиболее хорошо выраженные наблюдаемые структуры. Из них именно частицы LiFePO4, закрепившиеся на листках восстановленного оксида графена, оказались оптимальной структурой и являются причиной повышенной емкости и хорошей работоспособности электрода при высоких токах заряда.
ВВЕДЕНИЕ
Существует ограниченное число катодных материалов для внедрения лития, которые по своей емкости и стабильности удовлетворяют существующим требованиям в производстве литий-ионных аккумуляторов. Обычно используемые классы материалов – это слоистые оксиды переходных металлов (включая LiCoO2 [1], NCM, или LiNi0.33Co0.33 Mn0.33O2 [2, 3], катоды, стабилизированные Li2MnO3 [4], и другие варианты этой структуры), LiMn2O4 типа шпинели [5, 6] и LiFePO4 [7]. Среди них LiFePO4, теоретическая емкость которого составляет 170 мА ч г–1, демонстрирует высокую химическую устойчивость и хорошую циклируемость, сырье для его производства недорого, и он относительно экологичен [8].
Несмотря на то, что величина гравиметрической зарядной емкости фиксирована, все же желательно улучшить работоспособность электродов при высоких токах заряда и их циклируемость. Средства для достижения этой цели – это электронопроводящие добавки [9–11] и оптимизация “архитектуры” электрода, включая смешивание материалов [12], пористость [13] и толщину [14]. Обычно электронопроводящие добавки – это материалы на основе углерода, благодаря их высокой электронной проводимости, устойчивости и дешевизне [15]. Хорошо исследованы углеродные покрытия [16, 17], углеволокно [18], нанотрубки [19, 20] и различные другие пористые структуры. Эти электронопроводящие добавки образуют внутри электрода сетчатую структуру, обеспечивая электронопроводящие пути между токоподводом и практически каждым зерном в электроде. Это и приводит к улучшению как работоспособности электродов при высоких токах заряда, так и устойчивости при циклировании.
После открытия графена с его выдающимися свойствами и электронной проводимостью [21], делались попытки использовать его в литий-ионных аккумуляторах (подробный литературный обзор читатель найдет в статье [22]). Восстановленный оксид графена, “близкий родственник” графена, – это монослой графита, состоящий из sp2-гибридизованных атомов углерода, образующих кристаллическую решетку в форме пчелиных сот [22]. Восстановленный оксид графена содержит больше дефектов решетки, а часто также и функциональных групп, поскольку он получается восстановлением оксида графена, материала с графеноподобной решеткой в форме пчелиных сот, декорированной функциональными гидроксильными и эпокси-группами, а также карбонильными группами (карбоксильная, карбонильная, сложноэфирная), расположенными по краям листков оксида графена [23, 24]. Часто листки восстановленного оксида графена состоят из нескольких слоев. Однако, поскольку оксид графена более гидрофилен, чем восстановленный оксид графена, у него имеется преимущество более легкого смешивания с наночастицами катодного материала [25].
Несмотря на многочисленные исследования графена, как электронопроводящей добавки для литий-ионных аккумуляторов [25–52 ], не существует согласия по вопросу о том, каков оптимальный процесс приготовления или смешивания обоих соединений – графена и LiFePO4. Часто в разных работах используют различные методы приготовления LiFePO4, которые дают разный размер зерна, меняющееся количество углеродного покрытия и других добавок в электроде. Более того, сложность заключается в том, чтобы дать оценку восстановленному оксиду графена, использованному в каждой из этих работ, так как различия в концентрации дефектов, размере и толщине листков могут привести к изменению электронной проводимости восстановленного оксида графена [53]. Это затрудняет сравнение различных работ, поскольку речь идет о влиянии морфологии на электрохимические свойства. В настоящей работе мы смешивали оксид графена или его прекурсор с LiFePO4 или его прекурсором на различных стадиях синтеза, сохраняя при этом постоянным как массовое отношение реагентов C : восстановленный оксид графена : LiFePO4, так и путь синтеза. Мы приготовили различные морфологии и структуры зерна, в то время, как другие параметры материала оставались практически одинаковыми. Это позволило нам лучше оценить электрохимические свойства, в особенности, работоспособности при высоких токах заряжения, как функцию морфологии композита LiFePO4/C/восстановленный оксид графена.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
LiFePO4/C готовили синтезом в растворе. Для этого LiH2PO4 (Aldrich, ≥99%), FeC2O4 ∙ 2H2O (Aldrich, ≥99%) и лимонную кислоту (C6H8O7, Sigma-Aldrich, ≥99.5%), как источник углерода, смешивали в деионизованной воде; затем воду выпаривали на горячей плите (Thotplate = 100°C). Молярное отношение LiH2PO4 и FeC2O4 ∙ 2H2O равнялось 1 : 1. Масса лимонной кислоты составляла 16% от общей массы реагентов. После высушивания смесь реагентов измельчали и нагревали в течение 3 ч в смеси Ar/5 об. % H2 при 350°C. После этого прекурсор вновь измельчали и прокаливали в течение 3.5 ч в смеси Ar/5 об. % H2 при 700°C.
Композиты LiFePO4/C/восстановленный оксид графена готовили, добавляя листки восстановленного оксида графена (суспензию оксида графена в деионизованной воде Bluestone, размер хлопьев 1–20 мкм) на различных стадиях синтеза:
LiFePO4/C/восстановленный оксид графена, (образец 1) оксид графена добавляли во время приготовления прекурсора;
LiFePO4/C/восстановленный оксид графена, (образец 2) оксид графена добавляли перед стадией прокаливания;
LiFePO4/C/восстановленный оксид графена, (образец 3) оксид графена добавляли к LiFePO4/C после синтеза, после чего выполняли дополнительно стадию восстановления оксида графена, прогревая образец в течение 3.5 ч в смеси Ar/5 об. % H2 при 700°C.
Это добавление оксида графена выполняли, смешивая оксид графена с остальными реагентами в 40 мл деионизованной воды, после чего воду выпаривали на горячей плите. Количество добавляемого оксида графена выбирали таким образом, чтобы содержание восстановленного оксида графена в окончательном образце составляло 3 вес. %. Термообработка, следовавшая за добавлением оксида графена, удаляла основную часть функциональных групп и таким образом превращала оксид графена в восстановленный оксид графена [54].
Измерения методом порошковой рентгеновской дифракции были проведены на дифрактометре Philips X’Pert Pro MPD с CuKα-излучением. Рамановские спектры снимали на спектрометре SciAps Advantage с лазерным излучением 532 нм. Образцы исследовали методом сканирующей электронной микроскопии (SEM) на приборе Tescan Lyra. Содержание углерода в пленках определяли методами термогравиметрического анализа и дифференциального термического анализа на воздухе с помощью дифференциального термогравиметрического анализатора Shimadzu DTG-60. Удельную площадь поверхности и пористость определяли методом адсорбции газообразного азота: данные, полученные на приборе Costech Kelvin 1042, анализировали по теории Брунауэра–Эмметта–Теллера (БЭТ) [55].
Электроды готовили, смешивая активный материал с ацетиленовой сажей (Alfa Aesar, ≥99.9%) и поливинилиденфторидом (Sigma-Aldrich, ≥98%) в массовом отношении 75 : 15 : 10 в N-метилпирролидоне (Sigma-Aldrich, ≥99.9%) в шаровой мельнице (Retch MM200) в течение 20 мин. Полученную кашицу наносили на алюминиевую фольгу. Двухэлектродные электрохимические ячейки с быстроразъемным соединением (диаметр 10 мм) собирали в перчаточном боксе в атмосфере Ar. Использовали стекловолоконный сепаратор Whatman GF/F и 1 M раствор электролита LiPF6 в смеси этиленкарбонат–диметилкарбонат с объемным соотношением 1 : 1 (Sigma-Aldrich, ≥98%). Электрохимические измерения проводили на приборе Voltalab PGZ-301. Вычисляя удельные зарядную и разрядную емкости, использовали значение массы LiFePO4 без добавок.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
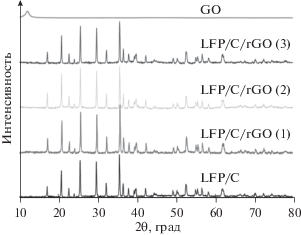
Данные рентгеновской дифракции (рис. 1) показали наличие чистого LiFePO4 с орторомбической структурой (пространственная группа Pnma). Каких-либо примесей не обнаружено. Для оксида графена наблюдался слабый пик при 2θ ≈ 13°, который исчез после термического восстановления и не наблюдался в композитах LiFePO4/C/восстановленный оксид графена.
Рис. 1.
Рентгеновские дифрактограммы оксида графена (GO), LiFePO4/C и композитов LiFePO4/C/восстановленный оксид графена (rGO).
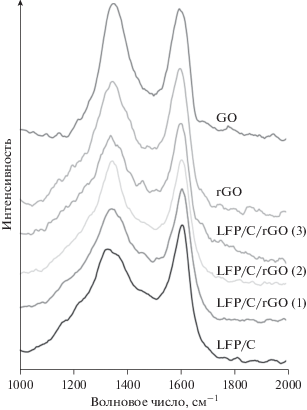
В Рамановских спектрах всех приготовленных материалов наблюдались полосы углерода D и G при 1350 и 1600 см–1 (рис. 2). Полоса D указывает на колебания sp3-гибридизованной связи, что характерно для разупорядоченных структур углерода и дефектного графена. Пик в полосе G связан с колебаниями sp2-гибридизованных углеродных связей, что характерно для чистых графита и графена. При восстановлении оксида графена интенсивность полосы G возрастала. Это приводит к изменению отношения интенсивностей сигналов полос G и D: значение IG/ID выросло с 0.97 в случае оксида графена до 1.10 для восстановленного оксида графена. Не отмечено сколько-нибудь значительных различий между образцами LiFePO4/C и LiFePO4/C/восстановленный оксид графена, скорее всего потому, что сигнал исходит главным образом от углеродного покрытия, а не от графена.
Рис. 2.
Рамановские спектры оксида графена (GO), восстановленного оксида графена (rGO) и композитов LiFePO4/C и LiFePO4/C/восстановленный оксид графена.
Для более глубокого исследования содержания углерода и количеств обоих типов аморфного углерода – образовавшегося в результате разложения лимонной кислоты и восстановленного оксида графена – мы провели термогравиметрический анализ и дифференциальный термический анализ (рис. 3). Ранее детальное исследование термического разложения LiFePO4 показало, что при окислении LiFePO4 при 700°C масса образца возрастает на 5.1 мас. % в результате образования Li3Fe2(PO4)3 и Fe2O3 [56]. С другой стороны, наблюдается потеря массы, связанная с потерей углерода: при температуре выше 450°C углерод превращается в CO2. Было показано, что приготовленные порошки содержали 1.1 вес. % углерода, образовавшегося из лимонной кислоты, и FeC2O4 ∙ 2H2O.
Рис. 3.
Результаты термогравиметрического анализа (TGA) и дифференциального термического анализа (DTA) композитов LiFePO4/C и LiFePO4/C/восстановленный оксид графена (rGO); данные TGA для LiFePO4 из работы [56].
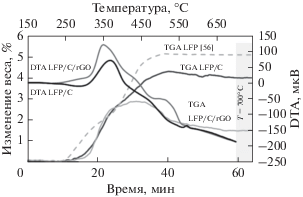
При исследовании образца LiFePO4/C/восстановленный оксид графена (1), также содержащего восстановленный оксид графена, полная потеря массы составила 3.6 вес. %; это означает, что содержание восстановленного оксида графена составляет 2.5 вес. %. Здесь дифференциальный термический анализ выявил дополнительную экзотермическую реакцию, которая начинается при температуре выше 500°C. Мы относим ее к окислению восстановленного оксида графена, поскольку сообщалось, что пик при 500°C относится к окислению монослоя графена [57].
Следует отметить, что полученное содержание восстановленного оксида графена (2.5 вес. %) меньше ожидаемых 3 вес. %. Это можно объяснить потерей массы при исчезновении функциональных групп из оксида графена – предположительно, главным образом, гидроксильных и эпокси-групп, а также некоторых карбонильных (карбоксильная, карбонильная, сложноэфирная), расположенных по краям листков оксида графена [23, 24]. Было также показано, что при поглощении тепловой энергии в ходе термообработки атомы углерода могут не только реорганизовываться [58], но также газифицироваться, так что в сетке восстановленного оксида графена образуются отверстия [59]. Таким образом может теряться около 10% всех атомов углерода [60].
Площадь поверхности, определенная методом БЭТ, оказалась в пределах ошибки одинаковой для образцов LiFePO4/C и LiFePO4/C/восстановленный оксид графена. Среднее значение этой площади равно 30 м2 г–1. Средняя площадь поверхности, определенная по уравнению Ленгмюра, равна 41 м2 г–1, а средний объем пор – 56 мм3 г–1.
По визуальной оценке SEM-микрофотографий образцов LiFePO4/C, размер зерна лежит в пределах 100–700 нм (рис. 4). Наблюдались также агломераты большего размера, состоящие из нескольких зерен. Хотя и не вполне надежный, рентгенодифракционный анализ с использованием формулы Шеррера дал близкое значение размера зерна: 100 нм, что согласуется с данными SEM-анализа. Содержащие восстановленный оксид графена образцы композита LiFePO4/C/восстановленный оксид графена имеют почти одинаковый размер зерна.
Рис. 4.
SEM-микрофотографии: (a) LiFePO4/C; (б) LiFePO4/C/восстановленный оксид графена (1) с группами, закрепленными на листках восстановленного оксида графена; (в) LiFePO4/C/восстановленный оксид графена (2) с упакованными в листки агломератами LiFePO4; (г) LiFePO4/C/восстановленный оксид графена (3) с агломератами LiFePO4, покрытыми листками восстановленного оксида графена.

Листки восстановленного оксида графена равномерно распределены в приготовленных композитах LiFePO4/C/восстановленный оксид графена. Однако, согласно SEM-исследованиям (рис. 4), есть разница в морфологии полученных композитов с восстановленным оксидом графена. Эти образцы можно сгруппировать по их морфологии (см., например, анализ, проделанный в работе [61]). В образце LiFePO4/C/восстановленный оксид графена (1) мы видим больше групп LiFePO4, закрепленных на листках восстановленного оксида графена, а также агломераты LiFePO4, упакованные в листки восстановленного оксида графена. В образце LiFePO4/C/восстановленный оксид графена (2) больше листков восстановленного оксида графена с упакованными в них агломератами LiFePO4, и почти нет групп LiFePO4, закрепленных на листках. В образце LiFePO4/C/восстановленный оксид графена (3) также нет таких закрепленных групп. Вместо этого, в листки восстановленного оксида графена упакованы один или несколько агломератов LiFePO4.
Можно сделать вывод о том, что добавка восстановленного оксида графена к LiFePO4 на различных стадиях синтеза ведет к изменениям морфологии конечного композита. Мы показали, что добавка восстановленного оксида графена на самой ранней стадии синтеза в растворе увеличивает количество LiFePO4, закрепленного на листках восстановленного оксида графена. Поскольку прекурсор LiFePO4 впервые нагревался только после смешивания с оксидом графена, это дало возможность частицам LiFePO4 образоваться и расти поверх листков восстановленного оксида графена, и поэтому возможно образование их хорошего электрического контакта с этими листками.
Электрохимические свойства композитов LiFePO4 с восстановленным оксидом графена рассмотрены в ряде работ [25–34, 45–52]. В большинстве их сообщается об улучшении емкости и способности работать при больших скоростях заряжения–разряда по сравнению с образцами сравнения, не содержавшими восстановленного оксида графена. Однако, часто трудно распознать вклад других факторов, таких как размер зерна, пористость и методика приготовления электрода – в особенности, при сравнении результатов, полученных разными исследовательскими группами [22]. Сравнение композитов LiFePO4/C/восстановленный оксид графена с различной структурой зерен, при сохранении, насколько это возможно, одинаковыми размера зерен и пористости позволило нам непосредственно оценить электрохимические свойства материала LiFePO4/C/восстановленный оксид графена, как функцию его специфической морфологии.
На рис. 5 приведены кривые заряжения–разряда образцов LiFePO4 и проведено сравнение их работоспособности при больших токах. Зарядная емкость LiFePO4/C, т.е. контрольного образца, не содержащего восстановленного оксида графена, равна 144 мА ч г–1 при скорости заряжения 0.1 C. При скорости 1 C емкость равняется 117 мА ч г–1, а при 10 C – 71 мА ч г–1.
Рис. 5.
Гальваностатические кривые заряжения–разряда образцов (a) LiFePO4; (б) LiFePO4/C/восстановленный оксид графена (1); (в) LiFePO4/C/восстановленный оксид графена (2); (г) LiFePO4/C/восстановленный оксид графена (3); (д) циклируемость образцов.
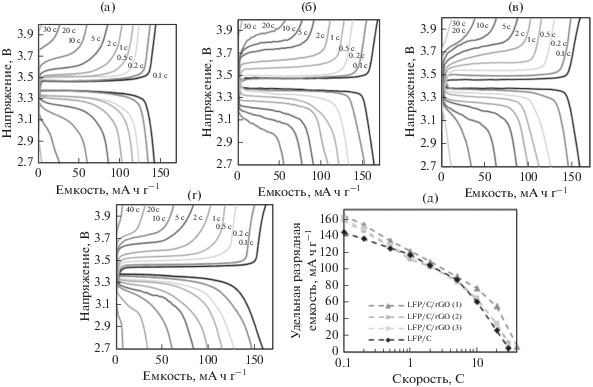
Емкость композитов LiFePO4/C/восстановленный оксид графена выше: при скорости заряжения 0.1 C – до 163 мА ч г–1 в случае композита LiFePO4/C/восстановленный оксид графена (1). Возможно, благодаря улучшенной электронной проводимости сетчатой структуры в композите LiFePO4/C/восстановленный оксид графена больше частиц LiFePO4 образуют хороший электрический контакт с токоподводом, чем в случае композита LiFePO4/C. Тот факт, что емкость LiFePO4 увеличивается при добавке восстановленного оксида графена даже при медленном заряжении (малые значения C), не является чем-то необычным, поскольку этот эффект наблюдали в многочисленных работах [26, 27, 30, 45, 47, 48, 51, 52]. Максимальное увеличение емкости (почти на 40% при скорости заряжения 0.1 C) по сравнению с образцом сравнения наблюдали в работе [27]. Емкости образцов LiFePO4/C/восстановленный оксид графена подобны емкостям LiFePO4/C при скоростях выше, чем 0.5 C. Образец LiFePO4/C/восстановленный оксид графена (1) демонстрирует повышенную емкость при скоростях 10 C и 20 C, хотя при скорости 40 C количество лития, внедренного в этот композит и экстрагированного из него, начинает стремиться к нулю.
Потрясающе, что образцы композитов LiFePO4/C/восстановленный оксид графена (2) и (3) при больших скоростях заряжения–разряда не демонстрируют улучшения циклируемости по сравнению с композитом LiFePO4/C. Поскольку и использованные реагенты, и общий путь синтеза одинаковы для всех образцов, это указывает на то, что важнейшим параметром для улучшения работы при больших скоростях заряжения–разряда является получение подходящей структуры электрода LiFePO4/C/восстановленный оксид графена на уровне морфологии. Скажем аккуратнее: хорошая способность образца LiFePO4/C/восстановленный оксид графена (1) работать при больших скоростях скорее всего есть следствие особенностей его морфологии (а именно, наличия частиц LiFePO4, закрепленных на листках восстановленного оксида графена), полученной путем смешивания LiFePO4 с восстановленным оксидом графена на самой ранней стадии синтеза. Причиной улучшения работы при больших скоростях заряжения–разряда при такой морфологии может быть улучшенный контакт частиц LiFePO4 с восстановленным оксидом графена. К тому же частицы LiFePO4, закрепленные на восстановленном оксиде графена, по-видимому, меньше среднего размера, а было показано, что уменьшение размера частиц LiFePO4 обычно приводит к улучшению работы при больших скоростях заряжения из-за укорачивания пути диффузии лития в LiFePO4 [62]. Однако, трудно судить о том, достаточно ли одного только большого числа частиц LiFePO4, закрепленных на восстановленном оксиде графена, для того, чтобы вызвать такой эффект.
Рассмотрим вкратце и другие явления, которые могут быть альтернативным объяснением описанного нами эффекта. Прежде всего, можно предположить, что дополнительные стадии в нашем методе приготовления композитов LiFePO4/C/восстановленный оксид графена (2) и (3) могут быть причиной появления рентгеноаморфных примесей, которые снижают емкость. Они не определяются рентгенодифракционным методом, но, тем не менее, могут стать причиной уменьшения емкости. Однако, многие из возможных примесей (например, LiFeO2 [63] или Li3Fe2(PO4)3 [64, 65]) способны запасать литий при напряжении 2.7–4.0 В, поэтому на кривых заряжения–разряда должны наблюдаться дополнительные плато или наклонные участки. Хотя возможность появления примесей существует всегда, мы не наблюдали ничего подобного ни на рентгеновских дифрактограммах, ни на кривых заряжения–разряда. Более того, емкости при скорости 0.1 C были практически одинаковы для всех образцов, содержащих восстановленный оксид графена.
Далее, в некоторых работах сообщалось об избыточной теоретической емкости LiFePO4 при использовании восстановленного оксида графена в качестве добавки. В работе [66] полученную емкость 208 мА ч г–1 композита LiFePO4/восстановленный оксид графена объяснили избыточным литием, “запасенным в обратимой редокс-реакции между ионами лития из электролита и отслоившимися чешуйками графена”. В другом исследовании инкапсулированный в углероде композит LiFePO4 (с наружной углеродной оболочкой) также показал емкость 192 мА ч г–1 – выше теоретической емкости, равной 170 мА ч г–1 [67]. В работе [68] сообщалось о емкостях вплоть до 208 мА ч г–1, как о результате появления дополнительных “мест” для запасания Li на поверхности LiFePO4. На основе нашей работы нельзя ни исключить гипотезу о создании дополнительных мест для запасания лития, ни поддержать ее. Однако, хотя об эффектах проводимости по границе раздела и о запасании лития на границах раздела сообщалось и ранее [69–72], все же, из-за ограниченного числа исследований композитов на основе восстановленного оксида графена и недостаточного понимания этих явлений, заявления об избыточной теоретической емкости композитов LiFePO4–углерод мы воспринимаем с осторожностью. К тому же в другом исследовании мы изучали восстановленный оксид графена в качестве анода и обнаружили, что у него пренебрежимо малая емкость по запасанию лития в интервале напряжений 2.7–4.0 В [73, 74].
Наконец, следует отметить, что восстановленный оксид графена может замедлять кинетику лития на электроде [75], предположительно, замедляя ионный транспорт. Это потрясающее заключение означает, что, вопреки широко распространенному мнению, некоторые типы морфологии композитных электродов с восстановленным оксидом графена могут ухудшать поведение таких электродов, вместо того чтобы улучшать его. Этот эффект должен быть выражен сильнее при относительно больших токах заряжения или разряда. Очень может быть, что такие эффекты дают вклад в поведение и наших материалов, потому что работоспособность при высоких токах заряжения образцов LiFePO4/C/восстановленный оксид графена (2) и (3) оказалась практически такой же, как у композита LiFePO4/C.
ЗАКЛЮЧЕНИЕ
Мы показали, что можно приготовить композиты LiFePO4/C/восстановленный оксид графена с различной морфологией путем добавления оксида графена к LiFePO4 на различных этапах синтеза из раствора и восстанавления его при нагревании до восстановленного оксида графена. Добавляя оксид графена к прекурсору LiFePO4, можно получить смесь зерен LiFePO4, закрепленных на листках восстановленного оксида графена, и агломератов LiFePO4, упакованных в листки восстановленного оксида графена, в то время, как добавление оксида графена на поздних этапах синтеза дает главным образом листки восстановленного оксида графена, в которые упакованы агломераты LiFePO4.
Мы исследовали электрохимические свойства композита LiFePO4/C/восстановленный оксид графена, как функцию его морфологии, и нашли, что наиболее благоприятная для получения высокой емкости и улучшения работы электрода при больших скоростях заряжения–разряда морфология – это частицы LiFePO4, закрепленные на листках восстановленного оксида графена. Мы предполагаем, что причины такого поведения – это улучшенный электронный контакт между частицами LiFePO4 и некоторое уменьшение самих частиц LiFePO4, следовательно, уменьшение пути диффузии из объема LiFePO4 в электрод.
Список литературы
Mizushima, K., Jones, P.C., Wiseman, P.J., and Goodenough, J.B., Mater. Res. Bull., 1980, vol. 15, p. 783.
Liu, Z., Yu, A. and Lee, J.Y., J. Power Sources, 1999, vols. 81–82, p. 416.
Ohzuku, T., and Makimura, Y., Chem. Lett., 2001, vol., p. 744.
Thackeray, M.M., Kang, S.-H., Johnson, C.S., et al., J. Mater. Chem., 2007, vol. 17, p. 3112.
Thackeray, M.M., Johnson, P.J., de Picciotto, L.A., et al., Mater. Res. Bull., 1984, vol. 19, p. 179.
Ohzuku, T., Kitagawa, M., and Hirai, T., J. Electrochem. Soc., 1990, vol. 137, p. 769.
Padhi, A.K., Nanjundaswarny, K., and Goodenough, J., J. Electrochem. Soc., 1997, vol. 144, p. 1188.
Mekonnen, Y., Sundararajan, A., and Sarwat, A.I., SoutheastCon., 2016. IEEE, p. 1.
Park, M., Zhang, X., Chung, M., et al., J. Power Sources, 2010, vol. 195, p. 7904.
Nishizawa, M., Koshika, H., Itoh, T., et al., Electrochem. Commun., 1999, vol. 1, p. 375.
Zaghib, K., Shim, J., Guerfi, A., et al., Electrochem. Solid-State Lett., 2005, vol. 8, p. A207.
Liu, D., Chen, L.-C., Liu, T.-J., et al., Adv. Chem. Eng. Sci., 2014, vol. 04, p. 515.
Indrikova, M., Grunwald, S., Golks, F., et al., J. Electrochem. Soc., 2015, vol. 162, p. A2021.
Zheng, H., Li, J., Song, X., et al., Electrochim. Acta, 2012, vol. 71, p. 258.
Li, L., Wu, L., Wu, F., et al., J. Electrochem. Soc., 2017, vol. 164, p. A2138.
Cao, Q., Zhang, H.P., Wang, G.J., et al., Electrochem. Commun., 2007, vol. 9, p. 1228.
Zhang, H., Xu, Y., Zhao, C., et al., Electrochim. Acta, 2012, vol. 83, p. 341.
Zhu, C., Mu, X., van Aken, P.A., et al., Angew. Chemie Int. Ed., 2014, vol. 53, p. 2152.
Sun, X., Li, J., Shi, C., et al., J. Power Sources, 2012, vol. 220, p. 264.
Liu, X.-M., Huang, Z., Dong, Oh, S. Woon, et al., Compos. Sci. Technol., 2012, vol. 72, p. 121.
Novoselov, K.S., Geim, A.K., Morozov, S.V, et al., Science, 2004, vol. 306, p. 666.
Kucinskis, G., Bajars, G., and Kleperis, J., J. Power Sources, 2013, vol. 240, p. 66.
Lerf, A., He, H., Forster, M., and Klinowski, J., J. Phys. Chem. B, 1998, vol. 102, p. 4477.
He, H., Klinowski, J., Forster, M., and Lerf, A., Chem. Phys. Lett., 1998, vol. 287, p. 53.
Zhou, X., Wang, F., Zhu, Y., and Liu, Z., J. Mater. Chem., 2011, vol. 21, p. 3353.
Su, C., Bu, X., Xu, L., et al., Electrochim. Acta, 2012, vol. 64, p. 190.
Yang, J., Wang, J., Wang, D., et al., J. Power Sources, 2012, vol. 208, p. 340.
Zhang, Y., Wang, W., Li, P., et al., J. Power Sources, 2012, vol. 210, p. 47.
Tang, Y., Huang, F., Bi, H., et al., J. Power Sources, 2012, vol. 203, p. 130.
Xu, H., Chang, J., Sun, J., and Gao, L., Mater. Lett., 2012, vol. 83, p. 27.
Wang, L., Wang, H., Liu, Z., et al., Solid State Ionics, 2010, vol. 181, p. 1685.
Ding, Y., Jiang, Y., Xu, F., et al., Electrochem. Commun., 2010, vol. 12, p. 10.
Wang, Y., Feng, Z.-S., Chen, J.-J., and Zhang, C., Mater. Lett., 2012, vol. 71, p. 54.
Toprakci, O., Toprakci, H.A.K., Ji, L., et al., J. Renew Sustain Energy, 2012, vol. 4, p. 013121.
Su, F.-Y., You, C., He, Y.-B., et al., J. Mater. Chem., 2010, vol. 20, p. 9644.
Wang, H., Yang, Y., Liang, Y., et al., Angew. Chemie Int. Ed. Engl., 2011, vol. 50, p. 7364.
Kim, J.-G., Kim, H.-K., Jegal, J.-P., et al., Proc. Int. Conf. Nanomater Appl. Prop., 2012, vol. p. 3.
Bak, S.-M., Nam, K., Lee, C., et al., J. Mater. Chem., 2011, vol. 21, p. 17309.
Zhang, W., Zeng, Y., Xu, C., et al., Beilstein J. Nanotechnol., 2012, vol. 3, p. 513.
Rui, X., Sim, D., Wong, K., et al., J. Power Sources, 2012, vol. 214, p. 171.
Jiang, Y., Xu, W., Chen, D., et al., Electrochim. Acta, 2012, vol. 85, p. 377.
Liu, H., Yang, G., Zhang, X., et al., J. Mater. Chem., 2012, vol. 22, p. 11039.
Zhu, J., Yang, R., and Wu, K., Ionics (Kiel), 2012, vol. 0, (доступно только онлайн).
Liu, H., Gao, P., Fang, J., and Yang, G., Chem. Commun. (Camb.), 2011, vol. 47, p. 9110.
Du, Y., Tang, Y., Huang, F., and Chang, C., RSC Advantage, 2016, vol. 6, p. 52279.
Wi, S., Kim, J., Park, K., et al., RSC Advantage, 2016, vol. 6, p. 105081.
Jiang, G., Hu, Z., Xiong, J., et al., J. Alloys Compd., 2018, vol. 767, p. 528.
Yuan, Z., Xue, Y., Sun, L., et al., Ferroelectrics, 2018, vol. 528, p. 1.
Ma, H., Xiang, J., and Xia, X., Mater. Res. Bull., 2018, vol. 101, p. 205.
Rosaiah, P., Zhu, J., Hussain, O.M., et al., J. Electroanal. Chem., 2018, vol. 811, p. 1.
Wang, X., Feng, Z., Huang, J., et al., Carbon N. Y., 2018, vol. 127, p. 149.
Chen, M., Kou, K., Tu, M., et al, Solid State Ionics, 2017, vol. 310, p. 95.
Juarez-Yescas, C., Ramos-Sánchez, G., and González, I., J. Solid State Electrochem., 2018, vol. 22, p. 3225.
Yang, D., Velamakanni, A., Bozoklu, G., et al., Carbon N. Y., 2009, vol. 47, p. 145.
Brunauer, S., Emmett, P.H., and Teller, E., J. Am. Chem. Soc., 1938, vol. 60, p. 309.
Belharouak, I., Johnson, C., and Amine, K., Electrochem. Commun., 2005, vol. 7, p. 983.
Nan, H.Y., Ni, Z.H., Wang, J., et al., J. Raman Spectrosc., 2013, vol. 44, p. 1018.
Mattevi, C., Eda, G., Agnoli, S., et al., Advantage Funct. Mater., 2009, vol. 19, p. 2577.
Erickson, K., Erni, R., Lee, Z., et al., Adv. Mater., 2010, vol. 22, p. 4467.
Krishnan, D., Kim, F., Luo, J., et al., Nano Today, vol. 7, p. 137.
Wu, Z.-S., Zhou, G., Yin, L.-C., et al., Nano Energy, 2012, vol. 1, p. 107.
Gaberscek, M., Dominko, R., and Jamnik, J., Electrochem. Commun., 2007, vol. 9, p. 2778.
Wang, Y., Wang, J., Liao, H., et al., Int. J. Electrochem. Sci., 2013, vol. 8, p. 8730.
Patoux, S., Wurm, C., Morcrette, M., et al., J. Power Sources, 2003, vols. 119–121, p. 278.
Morcrette, M., Wurm, C., and Masquelier, C., Solid State Sci., 2002, vol. 4, p. 239.
Lung-Hao Hu, B., Wu, F.-Y., Lin, C.-T., et al., Nat. Commun., 2013, vol. 4, p. 1687.
Zhao, Q., Zhang, Y., Meng, Y., et al., Nano Energy, 2017, vol. 34, p. 408.
Duan, Y., Zhang, B., Zheng, J., et al., Nano Lett., 2017, vol. 17, p. 6018.
Zhukovskii, Y.F., Balaya, P., Kotomin, E.A., and Maier, J., Phys. Rev. Lett., 2006, vol. 96.
Yu, X.Q., Sun, J.P., Tang, K., et al., PhysChemChemPhys, 2009, vol. 11, p. 9497.
Shin, J.-Y., Samuelis, D., and Maier, J., Adv. Funct. Mater., 2011, vol. 21, p. 3464.
Liu, E., Wang, J., Shi, C., et al., ACS Appl. Mater. Interfaces, 2014, vol. 6, p. 18147.
Kaprans, K., Bajars, G., Kucinskis, G., et al., IOP Conf. Ser. Mater. Sci. Eng., 2015, vol. 77, p. 012042.
Kaprans, K., Mateuss, J., Dorondo, A., et al., Solid State Ionics, 2018, vol. 319, p. 1.
Su, F.-Y., He, Y.-B., Li, B., et al., Nano Energy, 2012, vol. 1, p. 429.
Дополнительные материалы отсутствуют.