

Электрохимия, 2022, T. 58, № 12, стр. 882-900
Теоретический анализ эволюции состава системы при электролизе кислого водного раствора хлорида в зависимости от pH раствора
П. А. Задер a, *, Д. В. Конев a, b, М. А. Воротынцев a, **
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
Москва, Россия
b Институт проблем химической физики РАН
Черноголовка, Россия
* E-mail: paul.zadyor@gmail.com
** E-mail: mivo2010@yandex.com
Поступила в редакцию 01.03.2022
После доработки 20.06.2022
Принята к публикации 01.07.2022
- EDN: DMUIHG
- DOI: 10.31857/S0424857022120088
Аннотация
Проведен анализ соотношения между (квази)равновесным составом водного раствора хлорсодержащих соединений, потенциалом индикаторного электрода (при нулевом проходящем токе) Е, и средней степенью окисления атомов хлора х, т.е. нормированным редокс-зарядом системы Q при различных значениях рН раствора, равных 0, 3, 6 или 10. Предполагается, что во всех состояниях системы остается постоянной суммарная концентрация атомов хлора сtot, равная 0.1 М. Рассмотрение проведено для различных вариантов относительно скоростей редокс-превращений между компонентами системы путем последовательного выведения из рассмотрения равновесий для кислородсодержащих соединений хлора в наиболее положительных степенях окисления. Результаты расчета представлены в виде диаграмм зависимости относительной доли каждого из участвующих в гипотетическом наборе равновесий компонента от редокс-заряда и потенциала индикаторного электрода в такой системе, в отличие от диаграмм Пурбэ, дающих информацию лишь об основных компонентах и равновесиях с их участием. Сопоставление предсказаний эволюции состава системы при изменении редокс-заряда для каждого из рассмотренных вариантов при различных рН раствора показывает их существенную зависимость от этого параметра. В частности, для сильнокислых растворов характерно сосуществование (при определенном потенциале Е или средней степени окисления х) многочисленных компонентов с большими или заметными вкладами в суммарную концентрацию сtot. Напротив, при повышении рН резко уменьшается вклад соединений с промежуточными степенями окисления, а переход между компонентами с наибольшими концентрациями приобретает ступенчатый характер с узким интервалом значений Е или х. Полученные результаты могут быть использованы для первичного анализа состава раствора в ходе восстановительного или окислительного электролиза соединений хлора по данным потенцио- и кулонометрии, а также для выбора возможных маршрутов реакции при моделировании кинетики редокс-медиаторных процессов с участием оксосоединений хлора в качестве окислителей.
ВВЕДЕНИЕ
Электрохимическое окисление хлорид-иона, Cl–, в водных растворах активно исследовалось, прежде всего, в связи с промышленным производством Cl2 и NaOH посредством электролиза раствора NaCl с образованием как молекулярного хлора в растворенном состоянии Cl2 и в газовой фазе над раствором ${\text{Cl}}_{2}^{{{\text{gas}}}}$ [1, 2], так и соединений с промежуточными степенями окисления, главным образом трихлорид-аниона ${\text{Cl}}_{3}^{ - }$ [3]:
(1)
$2{\text{C}}{{{\text{l}}}^{ - }} - {\text{ }}2{{{\text{e}}}^{ - }} \rightleftarrows {\text{C}}{{{\text{l}}}_{2}},\,\,\,\,{\text{C}}{{{\text{l}}}_{2}} \rightleftarrows {\text{Cl}}_{2}^{{{\text{gas}}}},$(2)
${\text{C}}{{{\text{l}}}_{2}} + {\text{C}}{{{\text{l}}}^{ - }} \rightleftarrows {\text{Cl}}_{3}^{ - }.$Для упрощения записи все компоненты системы, находящиеся внутри водной фазы (в частности Cl– и Cl2 в схеме (1) ), а также их концентрации и активности указываются везде без верхнего индекса “aq”, тогда как компоненты в газовой фазе отмечаются верхним индексом “gas”.
Показана также возможность электрохимического окисления хлорида до более высоких степеней окисления, в частности до хлорат-аниона ${\text{ClO}}_{3}^{ - }$ [4–9]. При специальных условиях и при значительном перенапряжении хлорат-анион может быть окислен до перхлората (${\text{ClO}}_{4}^{ - }$) [4, 5].
Из общих принципов электрохимии можно ожидать, что электродный процесс с одновременным переносом нескольких электронов, который при этом сопровождается разрывом и образованием прочных химических связей Cl–O, вряд ли может протекать с заметной скоростью. Поэтому образование соединений хлора с положительными степенями окисления должно быть связано с реакцией диспропорционирования/гидролиза молекулярного хлора, схема которой зависит от рН раствора [4, 5, 10]:
(3)
$\begin{gathered} {\text{C}}{{{\text{l}}}_{2}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}} \rightleftarrows {\text{HClO}} + {\text{C}}{{{\text{l}}}^{ - }} + {{{\text{H}}}^{ + }} \\ \left( {{\text{кислые и нейтральные рН}}} \right), \\ \end{gathered} $(4)
$\begin{gathered} {\text{C}}{{{\text{l}}}_{2}} + 2{\text{О}}{{{\text{Н}}}^{ - }} \rightleftarrows {\text{Cl}}{{{\text{O}}}^{ - }} + {\text{C}}{{{\text{l}}}^{ - }} + {{{\text{Н}}}_{{\text{2}}}}{\text{О }} \\ \left( {{\text{щелочные рН}}} \right). \\ \end{gathered} $В свою очередь соединения со степенью окисления +1 могут вступать в серию стадий диспропорционирования с образованием соединений с более высокими степенями окисления (см. ниже) вплоть до накопления более устойчивых веществ – соединений Cl(+3), диоксида хлора ClO2 и хлоратов, а затем и перхлоратов [4, 5, 9]:
(5)
${\text{HClО}} \to {\text{HCl}}{{{\text{O}}}_{2}}/{\text{ClO}}_{2}^{ - }/{\text{Cl}}{{{\text{O}}}_{2}}/{\text{ClО}}_{3}^{ - } \to {\text{ClO}}_{4}^{ - }.$При написании этих реакционных схем здесь и ниже принято во внимание, что HCl, HClO3 и HClO4 являются сильными кислотами, т.е. практически полностью диссоциируют, а анионы ${\text{Cl}}_{3}^{ - }$ не протонируются – по крайней мере при рН ≥ 0 [4, 5, 11].
Закономерности процесса трансформации соединений с положительными степенями окисления хлора сложным образом зависят как от параметров электрохимического процесса (выбор его режима, величины потенциала или тока), так и от устройства ячейки (в частности, от использованных материала электрода и разделительной мембраны). Как видно из уравнений (2)–(5), сильное влияние должен оказывать состав раствора, в частности его рН и концентрация хлорид-аниона до начала электролиза. В результате пропускание положительного заряда через границу электрод/раствор хлорида сопровождается многоступенчатой эволюцией состава хлорсодержащих соединений, которая определяется как термодинамическими факторами, так и скоростями проходящих процессов, а также указанными параметрами системы. Кроме того, процесс электроокисления часто проходит выше потенциала выделения кислорода для данного рН, что приводит к наложению этого побочного процесса [9, 12, 13].
Поэтому из общих соображений невозможно достоверно предсказывать, какие из ожидаемых процессов при изучаемых экспериментальных условиях будут протекать достаточно быстро (на масштабе времени электролиза), т.е. будут находиться в своем квазиравновесном состоянии, а какие практически не будут проходить вообще, поскольку детальная информация о механизмах проходящих процессов и об их константах скоростей в настоящее время отсутствует.
В качестве иллюстрации расхождений в выводах относительно механизмов и кинетики можно привести литературные данные относительно процессов:
(6)
$\begin{gathered} {\text{ClO}}_{3}^{ - } + {\text{C}}{{{\text{l}}}^{ - }} + 2{{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows \,\,{\text{Cl}}{{{\text{O}}}_{2}} + \raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} {\text{C}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O }}\left[ {5,14 - 19} \right]{\text{ }}, \\ \end{gathered} $(7)
$\begin{gathered} {\text{ClO}}_{3}^{ - } + {\text{ClO}}_{2}^{ - } + 2{{{\text{H}}}^{ + }} \rightleftarrows \,\,2{\text{Cl}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \\ {\text{или}}\,\,\,\,\,{\text{ClO}}_{3}^{ - } + {\text{HCl}}{{{\text{O}}}_{2}} + {{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows 2{\text{Cl}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O }}\left[ {16,{\text{ }}20 - 28} \right], \\ \end{gathered} $(8a)
$\begin{gathered} {\text{HCl}}{{{\text{O}}}_{2}} + \raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} {\text{C}}{{{\text{l}}}_{2}} \rightleftarrows {\text{Cl}}{{{\text{O}}}_{2}} + {\text{C}}{{{\text{l}}}^{ - }} + {{{\text{H}}}^{ + }}, \\ 2{\text{ClO}}_{2}^{ - } + {\text{HClO}} \rightleftarrows 2{\text{Cl}}{{{\text{O}}}_{2}} + {\text{C}}{{{\text{l}}}^{ - }} + {\text{O}}{{{\text{H}}}^{ - }}; \\ \end{gathered} $(8б)
$\begin{gathered} 2{\text{HCl}}{{{\text{O}}}_{2}} \rightleftarrows {\text{HClO}} + {\text{ClO}}_{3}^{ - } + {{{\text{H}}}^{ + }}, \\ {\text{HCl}}{{{\text{O}}}_{2}} + {\text{C}}{{{\text{l}}}^{ - }} + {{{\text{H}}}^{ + }} \rightleftarrows 2{\text{HClO}}\,\,\,\,\left[ {16} \right]; \\ \end{gathered} $Исследование механизмов реакций [29]:
(8в)
${\text{HCl}}{{{\text{O}}}_{{\text{2}}}} + {{{\text{X}}}^{ - }}\left( {{\text{X}} = {\text{Cl, Br, I}}} \right) + {{{\text{H}}}^{ + }};$(8г)
$\begin{gathered} 2{\text{HCl}}{{{\text{O}}}_{2}} \rightleftarrows {\text{HClО}} + {\text{HCl}}{{{\text{O}}}_{3}}\,\,{\text{и}} \\ {\text{2HClO}} + {\text{HCl}}{{{\text{O}}}_{2}} \rightleftarrows {\text{C}}{{{\text{l}}}_{2}} + {\text{HCl}}{{{\text{O}}}_{3}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}\,\,\,\left[ {30} \right]. \\ \end{gathered} $В то же время имеется надежная информация о равновесных параметрах химических и электрохимических преобразований между всеми стабильными хлорсодержащими соединениями в растворе и в газовой фазе над ним (Cl–, ${\text{Cl}}_{3}^{ - }$, Cl2, ${\text{Cl}}_{2}^{{{\text{gas}}}}$, HClO, ClO–, HClO2, ${\text{ClO}}_{2}^{ - }$, ClO2, ${\text{ClO}}_{2}^{{{\text{gas}}}}$, ${\text{ClO}}_{3}^{ - }$, ${\text{ClO}}_{4}^{ - }$) – об их константах равновесия или их стандартных потенциалах [3, 4, 31, 32], которая приведена в Приложении А работы [33].
Ввиду указанной выше неопределенности относительно скоростей установления этих равновесий в книге Пурбэ [4] было рассмотрено несколько вариантов “равновесных диаграмм” с учетом всех указанных выше соединений хлора; учитывались также соединения HClgas и Cl2Ogas в газовой фазе; напротив, не учитывались ${\text{Cl}}_{3}^{ - }$ и ClO2 внутри раствора. При всех этих расчетах для каждого значения рН раствора и потенциала Е на диаграмме Пурбэ указан растворенный компонент, имеющий наибольшую концентрацию. Кроме того, показаны границы областей, где достаточно велико парциальное давление какого-либо компонента в газовой фазе (прежде всего, Cl2 и HCl).
Расчет диаграмм для указанных соединений хлора в книге Пурбэ [4] был проведен, прежде всего, в рамках стандартного термодинамического подхода, когда все химические и электрохимические реакции между ними считаются находящимися в равновесном состоянии, так что выполняются термодинамические соотношения между концентрациями входящих в них соединений. Принципиальным недостатком такой “термодинамической диаграммы” является ее неприменимость для описания эволюции реального состава даже при сравнительно медленном изменении потенциала электрода, например в ходе электролиза раствора. В частности, как растворы каждого из этих соединений – хлорида или перхлората, так и их смеси в любом соотношении не претерпевают превращений на электроде в широком интервале потенциалов, так что их широко используют как фоновые электролиты. Аналогичную инертность перхлорат демонстрирует и в сочетании с другими хлорсодержащими соединениями.
Поэтому в книге Пурбэ [4] рассматриваются также “квазиравновесные диаграммы” (рис. 2а–2с [4]), в которых из рассмотрения исключены определенные компоненты системы: 1) не учитывается ${\text{ClO}}_{4}^{ - }$; 2) исключены ${\text{ClO}}_{4}^{ - }$ и ${\text{ClO}}_{3}^{ - }$, т.е. рассматриваются HClO2, ${\text{ClO}}_{2}^{ - }$, HClO, ClO–, Cl2 и Cl– в растворе; 3) учитываются только растворенные компоненты HClO, ClO–, Cl2 и Cl–. Для каждого варианта предполагается, что учитываемые компоненты находятся в состоянии термодинамического равновесия между собой при заданных значениях потенциала раствора Е и его рН.
Следует отметить, что все эти диаграммы дают неполную информацию о составе раствора, указывая для каждой точки на плоскости рН, Е только соединение хлора с наибольшей концентрацией (или два таких соединения, если их концентрации равны). В то же время бывает важна и информация о концентрациях неосновных компонентов системы, например, из-за их каталитической роли при прохождении химических процессов, изменяющих состав раствора, в частности соединений хлора с промежуточными степенями окисления от 0 до +4 [16, 18, 23–26, 29]. В качестве примера можно указать реакцию восстановления хлоратов посредством редокс-медиаторного механизма с участием указанных соединений хлора [34] в связи с перспективами ее использования в качестве катодного процесса проточных редокс-батарей с высокой плотностью энергии [35, 36].
Поэтому представляет интерес провести по аналогии с диаграммами, имеющимися в книге [4], такой анализ для процесса электролиза хлорида в рамках нескольких альтернативных гипотез относительно скоростей взаимных преобразований хлорсодержащих соединений:
(а) не происходит образования соединений хлора со степенями окисления выше +1, т.е. электролиз Cl– приводит лишь к образованию молекулярного хлора как в растворенном состоянии Cl2, так и в газовом пространстве над раствором ${\text{Cl}}_{2}^{{{\text{gas}}}}$, аниона трихлорида ${\text{Cl}}_{3}^{ - }$, а также растворенных HClO и ClO– (аналог системы на рис. 2с [4]);
(б) помимо указанных в случае (а) соединений происходит образование соединений хлора в степени окисления +3, т.е. растворенных HClO2 и ${\text{ClO}}_{2}^{ - }$ (аналог системы на рис. 2b [4]);
(в) помимо указанных в случаях (а) и (б) соединений происходит образование соединений хлора в степени окисления +4, т.е. ClO2 в растворенной форме, а также ${\text{ClO}}_{2}^{{{\text{gas}}}}$ в газовой фазе;
(г) процесс электролиза Cl– проходит с образованием как хлорат-иона (${\text{ClO}}_{3}^{ - }$), так и указанных выше для случаев (а)–(в) соединений хлора с более низкими степенями окисления в растворе: ${\text{Cl}}_{3}^{ - }$, Cl2, ${\text{Cl}}_{2}^{{{\text{gas}}}}$, HClO, ClO–, HClO2, ${\text{ClO}}_{2}^{ - }$, ClO2 и ${\text{ClO}}_{2}^{{{\text{gas}}}}$ (аналог системы на рис. 2а [4]);
(д) полный термодинамический анализ, при котором все возможные растворенные и газообразные соединения хлора считаются находящимися в равновесии между собой посредством химических и/или электрохимических процессов. В этом случае помимо компонентов, указанных в предыдущих пунктах, учитывается также возможность образования перхлорат-аниона ${\text{ClO}}_{4}^{ - }$ (аналог системы на рис. 2 [4]).
Не будут приниматься в расчет HClgas и Cl2Оgas в газовой фазе, поскольку их парциальные давления в равновесии с раствором низкие, так что количество атомов хлора в этих компонентах системы практически не влияет на балансовое соотношение, а их давления можно вычислить a posteriori с помощью равновесных соотношений с соответствующими соединениями в растворе.
Отметим, что ни наличие ClO2 в растворенном состоянии, ни вариант (в) в книге [4] вообще не рассматривались, тогда как наш анализ показывает, что в отсутствие равновесий с участием перхлорат- и хлорат-анионов эволюция системы принципиально изменяется из-за присутствия диоксида хлора ClO2.
В работе [33] был проведен анализ эволюций состава системы в зависимости от потенциала для сильнокислого раствора, т.е. при рН 0. Целью настоящей работы является аналогичное исследование при более высоких рН раствора, равных 3, 6 или 10.
Подобный подход ранее был использован для расчета концентраций бромсодержащих соединений при различных рН раствора [37–40] в связи с использованием броматов в качестве окислителя для водородно-броматных [36, 41–44] и цинк-броматных [45] источников тока. Было продемонстрировано, что предсказания для эволюции состава раствора кардинально изменяются в зависимости от выбранного варианта расчета, что и следует ожидать на основе анализа диаграмм Пурбэ в книге [4], см. выше.
МЕТОД РАСЧЕТА ХАРАКТЕРИСТИК СИСТЕМЫ
Расчеты проводятся на основе метода работы [33] по расчету эволюции равновесных характеристик системы, включая состав раствора анолита, т.е. количество каждого из хлорсодержащих соединений Ni, в ходе электролиза раствора хлорида известной концентрации ctot = Ntot/V sol, где Vsol – объем раствора. Ниже дается краткое изложение метода.
Предполагается, что раствор имеет фиксированный рН за счет буфера, не участвующего в редокс-процессах. Не меняются также величины Vsol, Ntot и ctot. Все эти величины рассматриваются как известные параметры системы.
Во время электролиза за счет пропускания зависящего от времени заряда Qelchem(t) происходит постепенное изменение равновесного потенциала раствора Е(t), который может быть также измерен (с помощью индикаторного электрода).
Для каждого из вариантов эволюции системы (а)–(д) проводится теоретический анализ соотношения между мгновенными значениями равновесного потенциала раствора Е и суммарного редокс-заряда системы Q, связанного с зарядом электролиза Qelchem:
где Qini = –FNtot (начальное значение редокс-заряда), а также состава системы, т.е. количеств Ni каждого ее компонента i в зависимости от потенциала Е или редокс-заряда системы Q.При анализе в рамках варианта (д) принимается в расчет максимальное число (12) хлорсодержащих соединений со степенями окисления от –1 до +7 (табл. 1), включая 10 веществ внутри раствора и два (Cl2 и ClO2) в газовой фазе. Для каждого из них в таблице указано число атомов хлора ni в этом соединении, а также суммарная степень окисления атомов Cl в этом соединении типа i [33, 38].
Таблица 1.
Список Cl-содержащих компонентов системы Ai, которые могут присутствовать в системе в ходе электроокисления хлорид-аниона при сценарии (д), и величин их параметров ni и xi
| i | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ai | Cl– | ${\text{Cl}}_{3}^{ - }$ | Cl2 | ${\text{Cl}}_{2}^{{{\text{gas}}}}$ | HClO | ClO– | HClO2 | Cl${\text{O}}_{2}^{ - }$ | ClО2 | Cl${\text{O}}_{2}^{{{\text{gas}}}}$ | Cl${\text{O}}_{3}^{ - }$ | Cl${\text{O}}_{4}^{ - }$ |
| ni* | 1 | 3 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| xi** | –1 | –1 | 0 | 0 | +1 | +1 | +3 | +3 | +4 | +4 | +5 | +7 |
По сравнению с набором соединений хлора, который рассматривался в книге [4], включены дополнительно соединения ${\text{Cl}}_{3}^{ - }$ и ClO2 в растворенном состоянии. Вклад первого из них быстро возрастает с увеличением суммарной концентрации атомов хлора в системе ctot и станет существенным при концентрациях ctot порядка 1 М и выше. Второй компонент (ClO2) становится доминирующим вкладом в суммарную концентрацию ctot при высоких степенях окисления в отсутствие хлорат- и перхлорат-анионов и полностью меняет эволюцию системы, а также ее диаграмму Пурбэ, для варианта (в), см. ниже.
Суммарное число молей атомов Cl в системе Ntot и ее суммарный редокс-заряд Q удовлетворяют балансовым условиям:
где суммирование проводится по соединениям табл. 1, сходящим в соответствующий вариант эволюции, Ni – число молей соединения Ai типа i, величины параметров ni и xi даны в таблице, F – постоянная Фарадея.Вместо суммарного редокс-заряда системы Q для графических иллюстраций ниже используется соответствующая удельная характеристика – средняя степень окисления x в этом состоянии системы:
(12)
$x = Q{\text{/}}F{{N}_{{{\text{tot}}}}} = {{\sum {{x}_{i}}{{N}_{i}}} \mathord{\left/ {\vphantom {{\sum {{x}_{i}}{{N}_{i}}} {\sum {{n}_{i}}{{N}_{i}}}}} \right. \kern-0em} {\sum {{n}_{i}}{{N}_{i}}}}.$Согласно уравнению (9), величина х равна xini = –1 в начальном состоянии системы, а ее значения в последующие моменты электролиза х (х > –1) известны из измерений заряда электролиза Qelchem.
Вместо количеств каждого компонента системы Ni удобно ввести их концентрации по формуле (13):
где V sol – объем раствора. Заметим, что определение (13) при i = 4 и i = 10 (для хлора ${\text{Cl}}_{2}^{{{\text{gas}}}}$ и диоксида хлора ${\text{ClO}}_{2}^{{{\text{gas}}}}$ в газовой фазе) является чисто формальным, так как ее объем не равен объему раствора Vsol. Поэтому они связаны с молярными концентрациями этих веществ в газовой фазе $c_{i}^{{{\text{gas}}}}$ [моль/дм3] соотношениями ci = ${{c_{i}^{{{\text{gas}}}}{{V}^{{{\text{gas}}}}}} \mathord{\left/ {\vphantom {{c_{i}^{{{\text{gas}}}}{{V}^{{{\text{gas}}}}}} {{{V}^{{{\text{sol}}}}}}}} \right. \kern-0em} {{{V}^{{{\text{sol}}}}}}}$, т.е. зависят от отношения объемов газовой фазы и раствора.Для концентраций закон сохранения суммарного количества атомов Cl в системе (10) принимает вид:
где величина суммарной концентрации атомов хлора в системе ctot = Ntot/Vsol считается известной. Суммарный редокс-заряд Q и средняя степень окисления атомов хлора х также могут быть выражены через концентрации компонентов ci:(15)
$\begin{gathered} Q = F{{V}^{{{\text{sol}}}}}\sum {{x}_{i}}{{c}_{i}}, \\ x = Q{\text{/}}F{{N}_{{{\text{tot}}}}} = {{\left( {{{c}_{{{\text{tot}}}}}} \right)}^{{ - 1}}}\sum {{x}_{i}}{{c}_{i}}. \\ \end{gathered} $Активности всех Cl-содержащих компонентов системы внутри раствора (1 ≤ i ≤ 3, 5 ≤ i ≤ 9, 11 ≤ i ≤ 12) связаны между собой либо уравнениями Нернста с потенциалом индикаторного электрода, либо уравнениями кислотно-основного равновесия. Используются также термодинамические условия равновесия между молекулярным хлором (i = 4) или диоксидом хлора (i = 10) в растворе и газовой фазе над ним. Эти соотношения, включая величины соответствующих стандартных потенциалов $Е_{i}^{{\text{o}}}$ и констант равновесия Кi приведены в табл. 2 Приложения А работы [33].
При расчетах входящие в них активности компонентов заменялись соответствующими концентрациями, что означает пренебрежение поправками за счет коэффициентов активности fi. Для нейтральных веществ коэффициенты активности близки к 1, тогда как для однозарядных ионов при ионной силе раствора, отвечающей 0.1 М, величины lg fi для всех однозарядных ионов раствора близки к друг другу и составляют около –0.1, что пренебрежимо мало по сравнению с отношениями концентраций различных компонентов раствора, как видно из результатов расчетов состава раствора ниже.
При заданных величинах суммарной концентрации ctot в соотношении (14), а также рН раствора и его равновесного потенциала Е в термодинамических соотношениях между компонентами после перехода от активностей к концентрациям ci для последних получается замкнутая система алгебраических уравнений, число которых равно числу компонентов системы в рассматриваемом варианте ее эволюции (а)–(д). Процедура ее численного решения, а также доказательство единственности находимого решения для каждого набора значений ctot, рН и Е изложены в Приложении А работы [33]. В результате расчетов находятся зависимости состава раствора, а также редокс-заряда системы Q и средней степени окисления атомов хлора х по формулам (15) от величин параметров ctot, рН и Е.
Ниже даются графические иллюстрации (рис. 1 и 3) результатов расчета характеристик системы (потенциала Е и состава раствора) в зависимости от средней степени окисления атомов хлора х, т.е. нормированного редокс-заряда системы Q. Такой расчет проводился для каждого из вариантов (а)–(д) при суммарной концентрации атомов хлора в системе ctot, равной 0.1 М. На рис. 2 даются диаграммы Пурбэ для вариантов эволюции (б) и (в) для того, чтобы подчеркнуть важность учета растворенного диоксида хлора ClO2 в качестве одного из компонентов системы. Кроме того, даются иллюстрации зависимости состава системы от ее равновесного потенциала Е (рис. 4).
Рис. 1.
Зависимость равновесного (индикаторного) электрода системы E (в В, шкала с. в. э.) от ее средней степени окисления х для 5 различных сценариев (а)–(д) (соответствующие обозначения кривых) при рН раствора 0, 3, 6 или 10. Суммарная концентрация атомов хлора ctot = 0.1 M, отношение объемов газовой фазы над раствором и раствора Vgas/Vsol = 0.8.
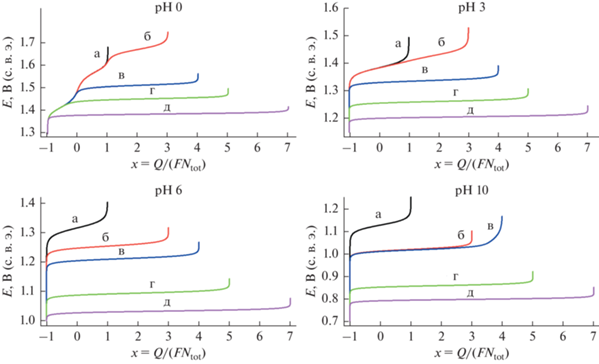
Рис. 2.
Квазиравновесные диаграммы Пурбэ для составов системы, отвечающих варианту (б), аналогичному рис. 2b в секции 20.2 книги [4], и варианту (в) (аналоги в книге [4] отсутствуют). Учитываются только компоненты внутри раствора. Номера около разделительных прямых линий соответствуют номеру соотношения в Приложении А работы [33].

Рис. 3.
Зависимость состава системы, т.е. концентраций атомов хлора в каждом компоненте системы [Cl]i = nici (в логарифмическом масштабе) от средней степени окисления атомов хлора х для вариантов эволюции системы (а)–(д) (варианты указаны над каждым рисунком). pH равен 0. Остальные параметры указаны в подписи к рис. 1.

Что касается рН раствора, рис. 1 сравнивает результаты для значений рН, равных 0, 3, 6 и 10. Так как эволюция системы при рН 0 была рассмотрена в работе [33], рис. 3 и 4 ниже представляют иллюстрации для других значений рН.
ЗАВИСИМОСТИ ПОТЕНЦИАЛА ИНДИКАТОРНОГО ЭЛЕКТРОДА ОТ РЕДОКС-ЗАРЯДА
На рис. 1 показаны зависимости потенциала Е, который может быть измерен индикаторным электродом, от средней степени окисления атомов Cl системы х, т.е. от нормированного суммарного редокс-заряда системы Q = FNtotx, который изменяется за счет заряда электролиза Qelchem. Эти результаты показаны для всех 5 вариантов (а)–(д) ее эволюции при фиксированных значениях суммарной концентрации атомов хлора сtot = 0.1 М и рН раствора (указано около каждого рисунка).
Отметим, что ввиду соотношения (11) началу процесса электролиза (раствор хлорида) отвечает х = –1 и относительно низкие потенциалы электрода Е, тогда как максимальная степень окисления зависит от предположения о пути эволюции системы: х = +1 (HClO и ClO–) для варианта (а), х = +3 (HClO2 и ${\text{ClO}}_{2}^{ - }$) для варианта (б), х = +4 (ClO2) для варианта (в), х = +5 (${\text{ClO}}_{3}^{ - }$) для варианта (г), х = +7 (${\text{ClO}}_{4}^{ - }$) для варианта (д). При приближении редокс-заряда к соответствующему максимальному значению рассчитанный потенциал индикаторного электрода начинает неограниченно возрастать (рис. 1) в соответствии с уравнением Нернста из-за убывания концентраций соединений с более низкими степенями окисления. Поэтому обнаружение такого поведения в ходе эксперимента позволяет сделать вывод в пользу реализации соответствующего сценария эволюции системы. А именно, обнаружение такого резкого возрастания потенциала при приближении величины х к целочисленному значению, меньшему или равному +5, позволило бы сделать вывод о том, что химические и электрохимические стадии, которые могли бы привести к дальнейшему росту степени окисления атомов хлора, не реализуются в условиях эксперимента из-за их замедленного характера.
Специфической особенностью соединений хлора по сравнению с их бромными аналогами [38–40] является относительно низкий стандартный потенциал редокс-пары ${\text{ClO}}_{4}^{ - }$/Cl– (ниже 1.4 В), близкий к потенциалу пары Cl2/Cl– (около 1.4 В). Из-за этого – в соответствии с диаграммой Пурбэ (рис. 1 [4]) – повышение потенциала в случае термодинамической эволюции системы (д) приводило бы к непосредственному переходу хлорида в перхлорат без промежуточного перехода к молекулярному хлору (кривая д на рис. 1). Хорошо известно [4], что такого перехода не наблюдается экспериментально из-за очень высокого энергетического барьера для этого процесса – даже через соединения с промежуточными степенями окисления.
Предсказания термодинамического расчета для соединений брома существенно отличаются. Образование пербромата требует намного более высокого потенциала (около 1.9 В при рН 0) даже в отсутствие перенапряжений и кинетических ограничений. Поэтому для Br-содержащей системы в рамках термодинамического варианта эволюции при повышении потенциала бромид-анион Br– практически полностью переходит в Br2 (возможно, через ${\text{Br}}_{3}^{ - }$ – в зависимости от суммарной концентрации атомов брома) до того, как могут начаться переходы в соединения с положительными степенями окисления – даже в отсутствие кинетических ограничений.
В вариантах эволюции (а)–(г), в которых перхлорат исключен из рассмотрения, закономерности перехода существенно зависят от рН раствора.
Для варианта (г) прямой переход между Cl– и ${\text{ClO}}_{3}^{ - }$ без каких-либо промежуточных компонентов имеет место для рН 3, 6 или 10, о чем свидетельствуют горизонтальные линии в интервале ‒1 < x < +5 на рис. 1. Это поведение существенно отличается от эволюции при рН 0, где окисление Cl– приводит к почти полному переходу в Cl2 – с последующим переходом в ${\text{ClO}}_{3}^{ - }$.
Вариант (в) не был рассмотрен в книге [4]. В этом случае, согласно рис. 1, ClO2 играет ключевую роль. А именно, при промежуточных значениях рН (3 и 6) окисление Cl– ведет непосредственно к состоянию, где доминирующей является концентрация ClО2 (горизонтальная линия при –1 < x < +4 на рис. 1). Отличием эволюции при рН 10 являются сопоставимые концентрации ClO2 и ${\text{ClO}}_{2}^{ - }$ в широком интервале редокс-зарядов системы. Эволюция при рН 0 проходит существенно иначе: переход между предельными состояниями Cl– и ClО2 происходит через промежуточную область редокс-зарядов, где доминирует Cl2.
Графики для вариантов (б) и (а) при кислых и нейтральных рН раствора (0, 3 и 6) для положительных значений х заметно смещены в сторону более положительных потенциалов по сравнению с вариантом (в). Это означает, что включение компонента ClO2 в окислительно-восстановительные равновесия (в отсутствие ${\text{ClO}}_{3}^{ - }$ и ${\text{ClO}}_{4}^{ - }$) полностью изменяет эволюцию. А именно, окисление Cl– (для рН 3 и 6) или Сl2 (для рН 0) должно давать ClO2, а не компоненты со степенями окисления +1 или +3. Компонент ClO– выступает в роли основного компонента системы (в интервале –1 < x < +3) только при рН 10.
В то же время все варианты от (а) до (г) приводят к одинаковым выводам в области отрицательных значений х при рН 0, в частности окисление Cl– около потенциала редокс-пары Cl2/Cl– (примерно 1.4 В при всех рН). При более высоких рН (3 и 6) начало окисления Сl– смещается постепенно в сторону все более положительных потенциалов в серии вариантов (д), (г), (в) и (б), что означает переход к различным компонентам: соответственно, ${\text{ClO}}_{4}^{ - }$, ${\text{ClO}}_{3}^{ - }$, ClO2 и HClO. Для кислых растворов (рН 0 и рН 3) графики для вариантов (б) и (а) разделяются только в окрестности максимальной степени окисления для вариант (а), т.е. при x ≅ 1. Напротив, для нейтральных и щелочных растворов (рН 6 и рН 10) даже окисление Cl– происходит при более положительных, притом зависящих от рН, потенциалах для сценария (а) по сравнению с вариантом (б), что связано с образованием ${\text{ClO}}_{2}^{ - }$ в варианте (б) (так как pKa кислоты HClO2 близко к 2), тогда как в варианте (а) происходит прямой переход в HClO и/или ClO–.
В качестве общего вывода можно также отметить глобальный сдвиг всех графиков к менее положительным потенциалам при возрастании рН от 0 до 10, которое связано с участием протонов в большинстве редокс-трансформаций, приводящих к изменению степеней окисления атомов хлора.
Все эти выводы, основанные на графиках на рис. 1, подтверждаются и расширяются ниже при описании эволюции состава системы.
КВАЗИРАВНОВЕСНЫЕ ДИАГРАММЫ ПУРБЭ
Как известно [4], такие диаграммы показывают области на плоскости потенциал–рН, в которых определенный компонент системы имеет максимальную концентрацию по сравнению с остальными редокс-активными компонентами. В книге [4] построены диаграммы для хлорсодержащих систем как в “термодинамическом” варианте (д), так и для трех квазиравновесных сценариев (а), (б) и (г), где исключаются определенные соединения. Результаты наших расчетов (со слегка отличными значениями некоторых параметров системы) полностью согласуются с этими диаграммами.
Выше было отмечено, что при анализе в книге [4] не учитываются два стабильных хлорсодержащих компонента внутри раствора, которые включены в наше рассмотрение: анион ${\text{Cl}}_{3}^{ - }$ и ClO2. Для сравнительно низкой суммарной концентрации: ctot = 0.1 М наличие трихлорид-аниона не проявляется в диаграммах, хотя очевидно, что для вариантов (а)–(г) он должен играть важную роль при отрицательных степенях окисления (–1 < x < 0), если суммарная концентрация ctot достигнет величины в несколько моль/л. Что касается второго компонента (ClO2), то его учет в варианте (в) (т.е. при отсутствии анионов ${\text{ClO}}_{4}^{ - }$ и ${\text{ClO}}_{3}^{ - }$) приводит к радикальному изменению диаграммы по сравнению с вариантом (б), как видно из сопоставления рис. 2в и 2б.
Согласно диаграмме (б), основная часть плоскости занята областями доминирования компонентов со степенями окисления –1 и +3: Cl– и ${\text{ClO}}_{2}^{ - }$, разделенными соответствующим уравнением Нернста (все разделительными линии на диаграммах рис. 2 даются соотношениями из табл. 2 работы [33]). Это означает, что при повышении потенциала из области доминирования Cl– происходит одноступенчатая эволюция через единственное редокс-превращение (Cl–/${\text{ClO}}_{2}^{ - }$). При достаточно высоких кислотностях раствора (рН < 3.90) имеется несколько областей малого размера, где наибольшими являются концентрации других компонентов: Cl2, НClО и НClО2, так что в этом случае при изменении потенциала может происходить двухступенчатая и даже трехступенчатая эволюция.
Учет диоксида хлора ClO2 среди хлорсодержащих компонентов раствора для варианта эволюции (в) радикально изменяет диаграмму. Основная часть плоскости занята компонентами Cl– и ClО2, причем в интервале рН от 2 до 10 между ними происходит прямой переход на линии, отвечающей уравнению Нернста для этой редокс-пары. Хлорит-анион ${\text{ClO}}_{2}^{ - }$ остался доминирующим только в промежуточной области при рН > 10, а молекулярный хлор – в узкой области при рН < 2, тогда как другие соединения с промежуточными степенями окисления HClO, ClO– и HClO2 вообще не представлены на диаграмме (в).
Как всегда, диаграммы Пурбэ построены в предположении, что соответствующие редокс-преобразования происходят достаточно быстро (возможно, через промежуточные соединения), чтобы равновесные соотношения между концентрациями успевали устанавливаться. Доступные экспериментальные данные относительно электрохимического превращения соединений Cl(+3) в ClO2 [25, 27, 28] свидетельствуют о достаточно высоких скоростях этого процесса, по крайней мере при высоких кислотностях раствора.
Диаграмма на рис. 2б согласуется с диаграммой на рис. 2b раздела 20.2 книги [4], однако можно ожидать, что ввиду достаточно высокой скорости переходов между соединениями Cl(+3) в ClO2 исключение диоксида вряд ли может быть обосновано.
Более детальная информация об эволюции системы дается результатами расчетов концентраций всех компонентов, которые приведены ниже в следующих разделах.
ЗАВИСИМОСТИ СОСТАВА СИСТЕМЫ ОТ РЕДОКС-ЗАРЯДА
В этом разделе рассматривается изменение состава раствора и газовой фазы над ним в зависимости от средней степени окисления атомов Cl х, т.е. от нормализованного по формуле (15) суммарного редокс-заряда системы Q для различных значений рН раствора (0, 3, 6 или 10).
Результаты расчета при каждом значении рН представлены в виде 5 рисунков, которые отвечают вариантам эволюции (а)–(д) и имеют один и тот же масштаб по осям Y (десятичные логарифмы концентраций компонентов системы) и оси Х. При этом интервал изменения переменой х для каждого из этих рисунков выбран в соответствии с интервалом изменения этой величины для соответствующего варианта эволюции системы, которые видны на рис. 1.
Вместо концентрации каждого соединения ci на рисунках дается концентрация атомов хлора в нем, т.е. величины [Cl]i = nici, где числа niданы в табл. 1. Преимущество этого выбора состоит в том, что именно такая величина отражает относительный вклад данного соединения в суммарную концентрацию атомов Cl системы ctot ввиду соотношения (14), т.е. при каждом значении х сумма концентраций на любом рисунке равна ctot.
рН 0
Результаты при рН 0 в виде зависимостей величин [Cl]i= nici от х представлены в работе [33] в линейном масштабе вдоль обеих координатных осей. Ниже на рис. 3 они показаны в модифицированном виде – в полулогарифмических координатах, т.е. для зависимостей lg nici от х, что позволяет показать поведение как для компонентов с наибольшими концентрациями, так и для компонентов с очень малыми концентрациями.
Для компонентов системы с наибольшими концентрациями (Cl–, Cl2, ${\text{Cl}}_{{\text{2}}}^{{{\text{gas}}}}$ и ${\text{Cl}}_{3}^{ - }$) в интервале –1 < x < 0 предсказания для вариантов (а), (б) и (в) практически идентичны, тогда как дополнительные соединения (НClО2 и ${\text{ClO}}_{2}^{ - }$ для варианта (б) или ClО2 и ${\text{ClO}}_{2}^{{{\text{gas}}}}$ для варианта (в)) представляют относительно малые добавки, хотя и быстро растущие при приближении величины х к 0. Для варианта (б) эти добавки остаются относительно малыми и в следующем интервале х до +1, тогда как ClО2 в растворе и в газовой фазе становятся сопоставимыми для варианта (в) с основными компонентами (Cl2 внутри обеих фаз), тогда как вклад НClО для этого варианта остается относительно малым, в то время как он становился доминирующим для вариантов (а) и (б).
В последующем интервале (x > 1) состав раствора резко различается для вариантов (б) и (в), как уже указано выше, доминирующие компоненты НClО и НClО2 для варианта (б) сменяются компонентами Cl2 и ClО2 в обеих фазах для варианта (в). Существенно изменяются и величины концентраций “малых компонентов”: в варианте (в) на много десятичных порядков возрастают (по сравнению с вариантом (б)) концентрации Cl– и ${\text{Cl}}_{3}^{ - }$; напротив, концентрации компонентов Cl(+3) уменьшаются примерно на 6–7 десятичных порядков. Так как в сильнокислых растворах на электроде происходит быстрое установление равновесия между компонентами редокс-пары ClО2/НClО2, то можно сделать вывод о том, что в этом случае расчет состава системы для варианта (б) приводит к заведомо неверным результатам, поскольку ClО2 образуется автоматически, причем в больших количествах, что резко изменяет все остальные концентрации.
В то же время резкое возрастание компонента с наивысшей степенью окисления в вариантах (г) (${\text{ClO}}_{3}^{ - }$) и (д) (${\text{ClO}}_{4}^{ - }$) уже в области x < 0 приводит к подавлению вкладов компонентов с промежуточными степенями окисления +3 и +4 (а также +5 для варианта (д)), тогда как компоненты с низкими степенями окисления (от –1 до +1 включительно) дают заметные вклады для этих вариантов во всем интервале значений х.
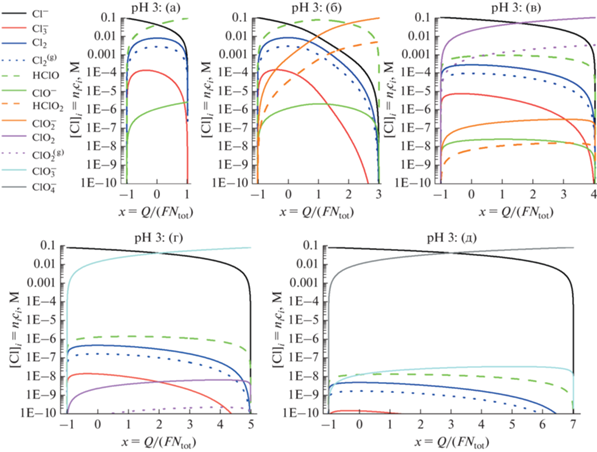
рН 3
На рис. 4 показаны соответствующие результаты для рН раствора, равного 3, что позволяет сравнить предсказания для сильно- и слабокислых растворов. Бросается в глаза резкое изменение общего характера эволюции для вариантов (а)–(г): если при рН 0 он имел ступенчатый вид – с ростом степени окисления между х = –1 и х = 0 происходил переход Cl– в Cl2 (в обеих фазах) и ${\text{Cl}}_{3}^{ - }$, который затем сменялся соответствующим соединением с положительной степенью окисления – то при рН 3 в большинстве вариантов (кроме (б)) имеет место прямой переход между Cl– и соединением с наивысшей положительной степенью окисления для этого варианта.
Это глобальное изменение связано, прежде всего, с величиной константы равновесия процесса гидролиза Cl2:
(16)
$\begin{gathered} {\text{C}}{{{\text{l}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} = {\text{C}}{{{\text{l}}}^{ - }} + {\text{HClO}} + {{{\text{H}}}^{ + }}, \\ {{К}_{{{\text{hydr}}}}} = \left\{ {{\text{HClO}}} \right\}\left\{ {{\text{C}}{{{\text{l}}}^{ - }}} \right\}/\left\{ {{\text{C}}{{{\text{l}}}_{2}}} \right\} = {{10}^{{{\text{pH}} - 3.33}}} \\ \end{gathered} $В результате даже для вариантов (а) и (б) концентрация Cl2 при рН 3 оказывается в несколько раз меньше меньшего из сомножителей в формуле (16), т.е. концентраций Cl– и НClО, которые становятся наивысшими для этих вариантов при –1 < x < 1 – с последующим переходом между НClО и ${\text{ClO}}_{2}^{ - }$ в окрестности х = 2 для варианта (б). Это изменение основных концентраций заметно деформирует кривые для “малых компонентов”.
Еще сильнее меняется – по этой же причине – картина для вариантов (в) и (г), где вместо сложной эволюции в промежуточным основным соединением (Cl2) имеет место прямой переход между начальным и конечным компонентами, тогда как концентрации “малых компонентов” намного ниже – на 2–3 десятичных порядка в варианте (в) и на 5–6 порядков в варианте (г). Еще сильнее разница для варианта (д).
рН 6
Обнаруженная при переходе от рН 0 к рН 3 тенденция исчезновения промежуточных состояний на диаграмме Пурбэ и, соответственно, ступенчатых переходов основной части атомов хлора между соединениями с наинизшей (Cl–) и наивысшей (для данного варианта) степенями окисления приводит к полному отсутствию таковых при рН 6 (рис. 5). Более того, заметные вклады “малых компонентов” наблюдаются только для ClО– в варианте (а), для НClО, ClО– и НClО2 в варианте (б) и для ${\text{ClO}}_{2}^{{{\text{gas}}}}$, ${\text{ClO}}_{2}^{ - }$ и НClО в варианте (в), тогда как концентрации Cl2 и ${\text{Cl}}_{3}^{ - }$ еще намного ниже. Вклады “малых компонентов” вообще очень малы для вариантов (г) и (д) из-за энергетической выгодности начального и конечного состояний системы.

рН 10
Тенденция к уменьшению вкладов “малых компонентов” продолжается и при переходе от рН 6 к рН 10 (рис. 6). Единственным исключением является вариант (в), где в переход между начальным (Cl–) и конечным (ClО2) компонентами системы вмешивается ${\text{ClO}}_{2}^{ - }$, который становится наибольшим вкладом в интервале х примерно от 1.5 до 3.5, тогда концентрации двух других компонентов заметно ниже – впрочем, не более, чем в несколько раз. Такое поведение обусловлено близостью величин потенциалов редокс-переходов между любой парой из этих трех компонентов – около 1.0 В с. в. э. – в соответствии с рис. 1 для рН 10. Заметные вклады к ним дают ${\text{ClO}}_{2}^{{{\text{gas}}}}$ и НClО, тогда как концентрации остальных компонентов очень малы.

ЗАВИСИМОСТИ СОСТАВА СИСТЕМЫ ОТ РАВНОВЕСНОГО ПОТЕНЦИАЛА ЭЛЕКТРОДА
При каждом значении рН раствора и любом варианте эволюции (а)–(д) существует однозначное соответствие между равновесным потенциалом системы Е, который может быть измерен индикаторным электродом, и средней степенью окисления атомов хлора системы х, т.е. ее нормированным суммарным редокс-зарядом Q в формуле (15). Поэтому все иллюстрации для зависимости состава системы от х могут быть преобразованы в зависимости состава от потенциала Е, которые даются ниже (рис. 7–10).
Рис. 7.
Зависимость состава системы, т.е. концентраций атомов хлора в каждом компоненте системы [Cl]i = nici (в логарифмическом масштабе) от равновесного потенциала индикаторного электрода Е для вариантов эволюции системы (а)–(д) (варианты указаны над каждым рисунком). pH равен 0. Остальные параметры указаны в подписи к рис. 1.




Можно отметить следующую общую закономерность: как видно из рис. 1 и всех рисунков предыдущего раздела переход от варианта (а) к варианту (д) через промежуточные варианты (б), (в) и (г) при постоянном рН раствора сопровождается монотонным расширением интервала изменения х от 2 (интервал от –1 до +1) до 8 (интервал от –1 до +7). Как видно из рисунков ниже, противоположная тенденция имеет место для зависимости состава от потенциала: тот же переход между вариантами приводит к монотонному сужению интервала потенциалов.
Еще одной закономерностью рисунков данного раздела является прямолинейность графиков для всех “малых компонентов” i в этих полулогарифмических координатах. Такая форма является следствием линейных зависимостей логарифмов концентраций от потенциала в соотношениях Нернста для редокс-перехода между этим “малым” (i) и доминирующим компонентами, тогда как концентрация последнего в этом интервале потенциалов близка к константе ввиду условия (14) для суммы концентраций всех компонентов. В результате наклон графика, деленный на нернстовский множитель RT/F, определяется частным от деления числа переносимых электронов n на стехиометрический коэффициент при “малом компоненте” i в схеме этой редокс-реакции – со знаком плюс или минус в зависимости от того, является ли “малый компонент” в этой схеме реакции окислителем или восстановителем. Так как смена доминирующего компонента системы происходит, как правило, в узком интервале потенциалов, при этом изменяется редокс-реакция, которая определяет концентрацию того же “малого компонента” i, и, соответственно, в этом же интервале потенциалов изменяется и наклон графика.
Так как все графики, отвечающие определенным значениям рН и определенному варианту эволюции, получаются непосредственным пересчетом величины х на величину Е, все качественные выводы предыдущего раздела остаются справедливыми и для рисунков ниже.
ЗАКЛЮЧЕНИЕ
Проведенный выше анализ показал, что эволюция как равновесного потенциала индикаторного электрода Е (рис. 1), так и состава системы (рис. 3–6) в зависимости от средней степени окисления атомов хлора в системе х существенно различается для 5 вариантов ее описания (а)–(д), а также в зависимости от рН раствора. При сопоставлении с экспериментальными данными по изменению как потенциала Е, так и состава раствора посредством окислительного или восстановительного электролиза следует учитывать:
– измерение потенциала должно проводиться индикаторным электродом, т.е. без пропускания через него тока и, по возможности, без изменения им локального состава раствора у поверхности электрода;
– для регистрации эволюции суммарного редокс-заряда системы необходимо минимизировать долю заряда электролиза на посторонние процессы, не связанные с изменением степени окисления атомов хлора. К ним относятся, прежде всего, реакция выделения кислорода и редокс-процессы из-за деградации материала электрода. Поэтому следует использовать соответствующие электродные материалы, в частности допированный бором алмаз (BDD) или смешанные оксиды (DSA) [46, 47].
Так как из-за длительного процесса при очень высоких анодных потенциалах трудно полностью избежать этих проблем, то важно дополнять эти электрохимические сведения дополнительными данными, прежде всего спектроскопическими, в частности in situ спектрами в УФ-видимом диапазоне.
Можно ожидать, что из-за высокой устойчивости анионов Cl${\text{O}}_{4}^{ - }$ и Cl${\text{O}}_{3}^{ - }$ далеко за пределами их термодинамической стабильности варианты (д) и (г) с их участием в редокс-равновесиях вряд ли реализуются в реальных условиях. Напротив, сравнительно быстрое редокс-равновесие между соединениями Cl(+3) и ClО2, особенно в сильнокислых растворах, представляет свидетельство в пользу варианта (в) с участием ClО2 по сравнению с вариантом (б), где это соединение игнорируется. В этом контексте вывод о кардинальном изменении эволюций для этих двух вариантов (см. в частности, диаграммы для них на рис. 2) приобретает особую важность. В то же время остается открытым вопрос об условиях, когда переходы между этими соединениями Cl(+3) и ClО2, с одной стороны, и компонентами со степенями окисления хлора от –1 до +1, с другой стороны, являются достаточно быстрыми, чтобы обеспечить их равновесное распределение в соответствии с предсказаниями варианта (в).
Сопоставление предсказаний для каждого из рассмотренных вариантов эволюции при различных рН раствора показывает их существенную зависимость от этого глобального параметра системы. В частности, для сильнокислых растворов характерно сосуществование (при определенном потенциале Е или средней степени окисления х) многочисленных компонентов с большими или заметными вкладами в суммарную концентрацию сtot, а также ступенчатый характер изменения компонента с наибольшей концентрацией. Эта тенденция сменяется с ростом рН раствора: число компонентов с достаточно большими вкладами в суммарную концентрацию существенно уменьшается, а переход между доминированием соединений с наиболее низкой (Cl–) и наиболее высокой (для данного варианта эволюции) степенями окисления происходит в узком интервале Е или х и без перехода в промежуточные соединениями.
Непосредственное применение проведенных в работе расчетов относится прежде всего к окислительному электролизу хлорида щелочного металла при различных рН раствора. При этом можно исключить “термодинамический вариант (д), согласно которому уже при относительно низких потенциалах должен происходить прямой переход Cl– в перхлорат-анион Cl${\text{O}}_{4}^{ - }$, поскольку его отсутствие даже на продолжительных интервалах времени хорошо известно.
Что касается остальных вариантов эволюции состава (а)–(г), то в сильнокислых растворах (рН 0) первым этапом окисления должен быть переход Cl– в Cl2/${\text{Cl}}_{3}^{ - }$, и только при дальнейшем значительном повышении потенциала следует переход в соответствующее кислородсодержащее соединение хлора, причем на этом этапе варианты (а)–(б), (в) и (г) начинают резко различаться. Это расхождение происходит, по расчетам, в интервале потенциалов 1.45–1.5 В, однако из-за омических эффектов при пропускании достаточно сильного тока в ходе электролиза приходится накладывать еще более высокие потенциалы, что приводит к побочным реакциям выделения кислорода из воды и/или деградации материала электрода. Поэтому для проведения таких экспериментов предпочтительно использовать наиболее подходящие электродные материалы – прежде всего допированный бором алмаз.
Что касается первого этапа процесса окисления при рН 0 (до Cl2/${\text{Cl}}_{3}^{ - }$), он представляет аналог превращения Br– в Br2/${\text{Br}}_{3}^{ - }$, который успешно изучался с использованием комбинации электрохимических и спектроскопических методов [37]. Соответствующий переход для случая хлора представляет заметно бóльшие трудности как из-за намного более высокого потенциала перехода (около 1.4 В по сравнению примерно с 1.1 В для брома), так и из-за более медленной кинетики этого процесса, особенно на каталитически неактивных электродных материалах.
Предсказанные эволюции для вариантов (а)–(г) при более высоких рН резко отличаются от случая рН 0. Это различие обусловлено смещением равновесия реакции гидролиза Cl2 при этих условиях в сторону продуктов гидролиза, так что Cl2 и ${\text{Cl}}_{3}^{ - }$ образуются в ходе эволюции лишь в относительно небольших количествах, а окисление Cl– приводит непосредственно к кислородсодержащему компоненту, специфическому для каждого из вариантов эволюции; единственными исключениями являются эволюции для варианта (б) при рН 3, а также для варианта (в) при рН 10. С ростом рН потенциалы переходов заметно снижаются, однако следует учитывать, что аналогичное снижение потенциала имеет место и для кислородной реакции.
Из-за указанных выше проблем с надежным измерением как заряда, истраченного на изменение степени окисления атомов хлора в этих соединениях, так и индикаторного потенциала становится особенно важным сочетать электрохимические измерения с данными других методов, в частности спектроскопии в УФ-видимом диапазоне. В то время как наиболее стабильные соединения: Cl–, ${\text{ClO}}_{3}^{ - }$ и ${\text{ClO}}_{4}^{ - }$ имеют очень низкие экстинкции, остальные соединения: Cl2, ${\text{Cl}}_{3}^{ - }$, HClO, ClO–, HClO2, ${\text{ClO}}_{2}^{ - }$ и ClO2 обладают характерными полосами поглощения [10, 48–50], причем максимальные экстинкции трихлорид-аниона и диоксида хлора довольно велики, так что основной проблемой для измерения всех их концентраций является перекрывание этих полос поглощения – с трудностью разделения вкладов каждого вещества в суммарное поглощение.
Восстановительный электролиз кислородсодержащих соединений имеет то преимущество, что омические эффекты, приводящие к сдвигу приложенных потенциалов по сравнению с равновесными, в данном случае понижают их величины, таким образом снижая вклады в ток побочных процессов. Ввиду высокой устойчивости ${\text{ClO}}_{4}^{ - }$ и ${\text{ClO}}_{3}^{ - }$ анионов далеко за пределами их термодинамической стабильности подобные эксперименты представляют интерес для соединений со степенями окисления +4 и +3, поскольку высокая обратимость процесса гидролиза Cl2 c образованием Cl– и соединения Cl(+1) (HClO или/и ClO– в зависимости от рН) надежно установлена [10]. Согласно [23–26], возможны взаимные превращения ClO2 и ${\text{ClO}}_{2}^{ - }$ или/и HClO2 (в зависимости от рН) на электроде, что является аргументом в пользу варианта (в) по сравнению с вариантом (б). Необходимы дальнейшие эксперименты в области достаточно высоких значений потенциала индикаторного электрода, когда различаются предсказания расчетов для вариантов (а)–(в).
Список литературы
Chen, R., Trieu, V., Schley, B., Natter, H., J. Kintrup, J., Bulan, A., Weber, R., and Hempelmann, R., Anodic Electrocatalytic Coatings for Electrolytic Chlorine Production: A Review, Z. Phys. Chem., 2013, vol. 227, p. 651. https://doi.org/10.1524/zpch.2013.0338
Crook, J. and Mousavi, A., The chlor-alkali process: A review of history and pollution, Environmental Forensics, 2016, vol. 17, no. 3, p. 21. https://doi.org/10.1080/15275922.2016.1177755
Kelsall, G.H., Welham, N.J., and Diaz, M.A., Thermodynamics of Cl–H2O, Br–H2O, I–H2O, Au–Cl–H2O, Au–Br–H2O and Au–I–H2O systems at 298 K, J. Electroanal. Chem., 1993, vol. 361, nos. 1–2, p. 13. https://doi.org/10.1016/0022-0728(93)87034-S
Pourbaix, M., Atlas of Electrochemical Equilibria in Aqueous Solutions, Houston: National Association of Corrosion Engineers, 1974. Section 20.2 (Chlorine), p. 590–603.
Vogt, H., Balej, J., Bennett, J.E., Wintzer, P., Sheikh, S.A., Gallone, P., Vasudevan, S., and Pelin, K., Chlorine Oxides and Chlorine Oxygen Acids, in Ullmann’s Encyclopedia of Industrial Chemistry, Ullmann, F., Ed, Berlin: Wiley Online Library, 2010, p. 622. https://doi.org/10.1002/14356007.a06_483.pub2
Viswanathan, K. and Tilak, B.V., Chemical, electrochemical, and technological aspects of sodium chlorate manufacture, J. Electrochem. Soc., 1984, vol. 131, no. 7, p. 1551. https://doi.org/10.1149/1.2115908
Wulff, J. and Cornell, A., Cathodic current efficiency in the chlorate process, J. Appl. Electrochem., 2007, vol. 37, p. 181. https://doi.org/10.1007/s10800-006-9263-3
Vacca, A., Mascia, M., Palmas, S., Mais, L., and Rizzardini, S., On the formation of bromate and chlorate ions during electrolysis with boron doped diamond anode for seawater treatment, J. Chem. Technol. & Biotechnol., 2013, vol. 88, no. 12, p. 2244. https://doi.org/10.1002/jctb.4095
Karlsson, R.K.B. and Cornell, A., Selectivity between Oxygen and Chlorine Evolution in the Chlor-Alkali and Chlorate Processes, Chem. Rev., 2016, vol. 116, no. 5, p. 2982. https://doi.org/10.1021/acs.chemrev.5b00389
Wang, T.X. and Margerum, D.W., Kinetics of Reversible Chlorine Hydrolysis: Temperature Dependence and General-Acid/Base-Assisted Mechanisms, Inorg. Chem., 1994, vol. 33, no. 6, p. 1050. https://doi.org/10.1021/ic00084a014
Alves, W.A., Téllez, C.A., Sala, S.O., Santos, P.S., and Faria, R.B., Dissociation and rate of proton transfer of HXO3 (X = Cl, Br) in aqueous solution determined by Raman spectroscopy, J. Raman Spectroscopy, 2001, vol. 32, p. 1032. https://doi.org/10.1002/jrs.794
Tarasevich, M.R., Sadkowsky, A., and Yeager, E., Oxygen Electrochemistry, In Comprehensive Treatise of Electrochemistry, Horsman, P., Conway, B.E., and Yeager, E., Eds, Boston: Springer, 1983, p. 301.
Reier, T., Oezaslan, M., and Strasser, P., Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials, ACS Catal., 2012, vol. 2, p. 1765. https://doi.org/10.1021/cs3003098
Skrabal, A. and Schreiner, H., Die Reduktionsgeschwindigkeit der Chlorsäure und Bromsäure, Monatshefte für Chemie und verwandte Teile anderer Wissenschaften, 1934, Bd. 65, Nr. 1, S. 213. https://doi.org/10.1007/bf01522061
Bray, W.C., An oxide of iodine, I2O2, an intermediate compound, J. Amer. Chem. Soc., 1930, vol. 52, p. 3580. https://doi.org/10.1021/ja01372a017
Taube, H. and Dodgen, H., Applications of Radioactive Chlorine to the Study of the Mechanisms of Reactions Involving Changes in the Oxidation State of Chlorine, J. Amer. Chem. Soc., 1949, vol. 71, no. 10, p. 3330. https://doi.org/10.1021/ja01178a016
Lenzi, F. and Rapson, W.H., Effets ioniques spécifiques sur le taux de formation du ClO2 par la réaction chlorure–chlorate, Canad. J. Chem., 1968, vol. 46, no. 6, p. 979. https://doi.org/10.1139/v68-160
Schmitz, G., Kinetics and mechanism of the iodate–iodide reaction and other related reactions, Phys. Chem. Chem. Phys., 1999, vol. 1, no. 8, p. 1909. https://doi.org/10.1039/a809291e
Sant’Anna, R.T.P., Santos, C.M.P., Silva, G.P., Ferreira, R.J.R., Oliveira, A.P., Côrtes, C.E.S., and Faria, R.B., Kinetics and mechanism of chlorate-chloride reaction, J. Brazil. Chem. Soc., 2012, vol. 23, no. 8, p. 1543. https://doi.org/10.1590/S0103-50532012005000017
Pillai, K.C., Kwon, T.O., Park, B.B., and Moon, I.S., Studies on process parameters for chlorine dioxide production using IrO2 anode in an un-divided electrochemical cell, J. Hazardous Mater., 2009, vol. 164, no. 2–3, p. 812. https://doi.org/10.1016/j.jhazmat.2008.08.090
Pillai, K.C., Kwon, T.O., Park, B.B., and Moon, I.S., Using RuO2 anode for chlorine dioxide production in an un-divided electrochemical cell, Water Sci. & Technol., 2010, vol. 61, no. 8, p. 2151. https://doi.org/10.2166/wst.2010.131
Tian, M., Li, Y.Y., Sun, H.C., Yang, L.J., and Li, Z.L., Preparation of Chlorine Dioxide by Electrocatalytic Reduction of Sodium Chlorate, Advanced Mater. Res., 2013, vols. 781–784, p. 342. https://doi.org/10.4028/www.scientific.net/amr.781-784.342
Raspi, G. and Pergola, F., Voltammetric behaviour of chlorites and chlorine dioxide on a platinized-platinum microelectrode with periodical renewal of the diffusion layer and its analytical applications, J. Electroanal. Chem. and Interfac. Electrochem., 1969, vol. 20, no. 3, p. 419. https://doi.org/10.1016/s0022-0728(69)80171-3
Pergola, F., Guidelli, R., and Raspi, G., Potentiostatic study of heterogeneous chemical reactions. ClO2–ClO2–Cl– system on platinized platinum, J. Amer. Chem. Soc., 1970, vol. 92, no. 9, p. 2645. https://doi.org/10.1021/ja00712a010
Lipsztajn, M., Electrolytic protection of chlorine dioxide, Patent 4,767,510 (USA), 1988.
Sinkaset, N., Nishimura, A.M., Pihl, J.A., and Trogler, W.C., Slow Heterogeneous Charge Transfer Kinetics for the ${\text{ClO}}_{2}^{ - }$/ClO2 Redox Couple at Platinum, Gold, and Carbon Electrodes. Evidence for Nonadiabatic Electron Transfer, J. Phys. Chem. A, 1999, vol. 103, no. 49, p. 10461. https://doi.org/10.1021/jp992693f10.1021/jp992693f
omez-Gonzalez, A., Ibanez, J.G., Vasquez-Medrano, R., Zavala-Araiza, D., and Paramo-Garcia, U., Electrochemical Paired Convergent Production of ClO, ECS Trans., 2009, vol. 20, no. 1, p. 91. 10.1149/1.326837
Gomez-Gonzalez, A., Ibanez, J.G., Vasquez-Medrano, R.C., Paramo-Garcia, U., and Zavala-Araiza, D., Cathodic Production of ClO2 from NaClO3, J. Electrochem. Soc., 2009, vol. 156, no. 7, p. E113. https://doi.org/10.1149/1.3121588
Schmitz, G. and Rooze, H., Mécanisme des réactions du chlorite et du dioxyde de chlore. 5. Cinétique de la réaction chlorite–bromure, Canad. J. Chem., 1987, vol. 65, no. 3, p. 497. https://doi.org/10.1139/v87-086
Ni, Y. and Yin, G., Disproportionation of Chlorous Acid at a Strong Acidity, Industrial & Engineering Chem. Res., 1998, vol. 37, no. 6, p. 2367. https://doi.org/10.1021/ie970608p
Mussini, T. and Longhi, P., The Halogens. Bromine, in Standard potentials in aqueous solutions, Bard, A.J., Parsons, R., and Jordan J., Eds., New York: Marcel Dekker Inc., 1985, p. 78. https://books.google.ru/ books?id=fuJV1H18KtEC&pg=PA67&hl=ru&source= gbs_toc_r&cad=4#v=onepage&q&f=false.
Lide, D.R., CRC handbook of chemistry and physics, vol. 84, New York: CRC press, 2004.
Задер, П.А., Конев, Д.В., Гун, Дж., Лев, О., Воротынцев, М.А. Теоретический анализ изменения состава системы при электролизе кислого водного раствора хлорида. Электрохимия. 2022. Т. 58. С. 614. [Zader, P.A., Konev, D.V., Gun, J., Lev, O., and Vorotyntsev, M.A., Theoretical analysis of system’s composition changes in the course of electrolysis of acidic aqueous chloride solution, Russ. J. Electrochem., 2022, vol. 58, № 10.]
Конев, Д.В., Гончарова, О.А., Толмачев, Ю.В., Воротынцев, М.А. О роли диоксида хлора в процессе электровосстановления хлоратов при низких рН. Электрохимия. 2022. Т. 58. в печати. [Konev, D.V., Goncharova, O.A., Tolmachev, Yu.V., and Vorotyntsev, M.A., On the role of chlorine dioxide in chlorate electroreduction process at low pH, Russ. J. Electrochem., 2022, vol. 58, in press.]
Tolmachev, Y.V., Pyatkivskiy, A., Ryzhov, V.V., Konev, D.V., and Vorotyntsev, M.A., Energy cycle based on a high specific energy aqueous flow battery and its potential use for fully electric vehicles and for direct solar-to-chemical energy conversion, J. Solid State Electrochem., 2015, vol. 19, p. 2711. https://doi.org/10.1007/s10008-015-2805-z
Vorotyntsev, M.A., Antipov, A.E., and Konev, D.V., Bromate anion reduction: novel autocatalytic (EC") mechanism of electrochemical processes. Its implication for redox flow batteries of high energy and power densities, Pure Appl. Chem., 2017, vol. 89, no. 10, p. 1429. https://doi.org/10.1515/pac-2017-0306
Петров, М.М., Локтионов, П.А., Конев, Д.В., Антипов, А.E., Астафьев, Е.А., Воротынцев М.А. Эволюция состава анолита при окислительном электролизе сернокислого раствора бромида натрия. Электрохимия. 2019. Т. 55. С. 95. [Petrov, M.M., Loktionov, P.A., Konev, D.V., Antipov, A.E., Astafiev, E.A., and Vorotyntsev, M.A., Evolution of Anolyte Composition in the Oxidative Electrolysis of Sodium Bromide in a Sulfuric Acid Medium, Russ. J. Electrochem., 2018, vol. 54. p. 1223.]
Petrov, M.M., Konev, D.V., Kuznetsov, V.V., Antipov, A.E., Glazkov, A.T., and Vorotyntsev, M.A., Electrochemically driven evolution of Br-containing aqueous solution composition, J. Electroanal. Chem., 2019, vol. 836, p. 125. https://doi.org/10.1016/j.jelechem.2019.01.070
Петров, М.М., Конев, Д.В., Антипов, А.E., Карташова, Н.В., Кузнецов, В.В., Воротынцев, М.А. Теоретический анализ изменения состава раствора при анодном электролизе бромида. Электрохимия. 2019. Т. 55. С. 1307. [Petrov, M.M., Konev, D.V., Antipov, A.E., Kartashova, N.V., Kuznetsov, V.V., and Vorotyntsev, M.A., Theoretical Analysis of Changes in the Solution Composition during Anodic Electrolysis of Bromide, Russ. J. Electrochem., 2019, vol. 55, p. 1058.]
Петров, М.М., Конев, Д.В., Антипов, А.E., Карташова, Н.В., Кузнецов, В.В., Воротынцев, М.А. Теоретический анализ изменения состава системы при окислительном электролизе раствора бромида: зависимость от рН. Электрохимия. 2020. Т. 56. С. 978. [Petrov, M.M., Konev, D.V., Antipov, A.E., Kartashova, N.V., Kuznetsov, V.V., and Vorotyntsev, M.A., Theoretical Analysis of Changes in the System’s Composition in the Course of Oxidative Electrolysis of Bromide Solution: pH Dependence, Russ. J. Electrochem., 2020, vol. 56, p. 883.]
Vorotyntsev, M.A., Konev, D.V., and Tolmachev, Y.V., Electroreduction of halogen oxoanions via autocatalytic redox mediation by halide anions: novel EC" mechanism. Theory for stationary 1D regime, Electrochim. Acta, 2015, vol. 173, p. 779. https://doi.org/10.1016/j.electacta.2015.05.099
Modestov, A.D., Konev, D.V., Antipov, A.E., Petrov, M.M., Pichugov, R.D., and Vorotyntsev, M.A., Bromate electroreduction from sulfuric acid solution at rotating disk electrode: experimental study, Electrochim. Acta, 2018, vol. 259, p. 655. https://doi.org/10.1016/j.electacta.2017.10.199
Modestov, A.D., Konev, D.V., Tripachev, O.V., Antipov, A.E., Tolmachev, Y.V., and Vorotyntsev, M.A., A Hydrogen–Bromate Flow Battery for Air-Deficient Environments, Energy Technol., 2018, vol. 6, p. 242. https://doi.org/10.1002/ente.201700447
Modestov, A.D., Konev, D.V., Antipov, A.E., and Vorotyntsev, M.A., Hydrogen-bromate flow battery: can one reach both high bromate utilization and specific power? J. Solid State Electrochem., 2019, vol. 23, p. 3075. https://doi.org/10.1007/s10008-019-04371-w
Modestov, A.D., Andreev, V.N., Antipov, A.E., and Petrov, M.M., Novel Aqueous Zinc–Halogenate Flow Batteries as an Offspring of Zinc–Air Fuel Cells for Use in Oxygen-Deficient Environment, Energy Technol., 2021, vol. 9, no. 9, p. 1. https://doi.org/10.1002/ente.202100233
Bergman, M.E.H., Iourtchouk, T., and Rollin, J., The occurrence of bromate and perbromate on BDD anodes during electrolysis of aqueous systems containing bromide: first systematic experimental studies, J. Appl. Electrochem., 2011, vol. 41, no. 9, p. 1109. https://doi.org/10.1007/s10800-011-0329-5
Sáez, C., Sánchez-Carretero, A., Cañizares, P., and Rodrigo, M.A., Electrochemical synthesis of perbromate using conductive-diamond anodes, J. Appl. Electrochem., 2010, vol. 40, no. 10, p. 1715. https://doi.org/10.1007/s10800-010-0108-8
Buser, W. und Hanisch, H., Spektrophotometrische Untersuchung saurer Chloritlosungen, Helvetica Chimica Acta, 1952, vol. 35, no. 310, p. 2547. https://doi.org/10.1002/hlca.19520350740
Chen, T., Spectrophotometric Determination of Microquantities of Chlorate, Chlorite, Hypochlorite, and Chloride in Perchlorate, Anal. Chem., 1967, vol. 39. no. 7, p. 804. https://doi.org/10.1021/ac60251a023
Wang, T.X., Kelley, M.D., Cooper, J.N., Beckwith, R.C., and Margerum, D.W., Equilibrium, Kinetic, and UV-Spectral Characteristics of Aqueous Bromine Chloride, Bromine, and Chlorine Species, Inorg. Chem., 1994, vol. 33, no. 25, p. 5872. https://doi.org/10.1021/ic00103a040
Дополнительные материалы отсутствуют.
Инструменты
Электрохимия