

Журнал эволюционной биохимии и физиологии, 2021, T. 57, № 3, стр. 183-201
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ АПОПТОЗА НЕЙТРОФИЛОВ В НОРМЕ И ПРИ ПАТОЛОГИИ
Е. М. Носейкина 1, *, И. А. Щепеткин 1, 2, **, Д. Н. Аточин 1, 3, ***
1 Томский Политехнический университет
Томск, Россия
2 Department of Microbiology and Immunology, Montana State University
Bozeman, MT, USA
3 Cardiovascular Research Center, Cardiology Division, Massachusetts General Hospital
Charlestown, MA, USA
* E-mail: enoseykina@gmail.com
** E-mail: schepetkin@yahoo.com
*** E-mail: atochin@cvrc.mgh.harvard.edu
Поступила в редакцию 27.10.2020
После доработки 10.02.2021
Принята к публикации 23.02.2021
Аннотация
Нейтрофилы являются одними из основных клеток врожденного иммунитета и выполняют ключевую эффекторную и регуляторную функцию при развитии ответной воспалительной реакции организма. Апоптозные формы нейтрофилов имеют важное значение в регуляции интенсивности воспаления и восстановления тканевого гомеостаза. В настоящем обзоре обобщены современные данные о молекулярных механизмах модуляции апоптоза нейтрофилов основными регуляторными факторами воспалительной реакции – цитокинами, интегринами и структурными компонентами бактерий. Также проанализированы нарушения апоптоза нейтрофилов при стрессе, представлены молекулярные маркеры изменения продолжительности жизни нейтрофилов, наблюдаемые при различных заболеваниях и патологических состояниях, рассмотрена информация о фармакологических средствах модуляции апоптоза.
ВВЕДЕНИЕ
В ходе эволюционного развития системы врожденного иммунитета человека сформировался механизм распознавания сигналов потенциального повреждения структурных компонентов тканей, представленный эволюционно консервативными паттерн-распознающими рецепторами (PRR) иммунокомпетентных клеток. Способность этих рецепторов распознавать молекулярные структуры различных патогенов (PAMP), а также сигналы опасности в форме DAMP, высвобождающиеся из разрушенного экстраклеточного матрикса и распадающихся клеток организма, тесно связана с формированием острого воспалительного ответа в очаге инфицирования или повреждения [1–3]. В отличие от других гранулярных лейкоцитов, на нейтрофилах представлено огромное количество разнообразных PRR [2], и именно эти эффекторные клетки одними из первых реагируют на сигнал опасности, мигрируют из циркуляторного русла к очагу повреждения, идентифицируют природу раздражителя и выбирают оптимальные способы его нейтрализации – генерацию активных форм кислорода (ROS), дегрануляцию, фагоцитоз или формирование экстраклеточных ловушек (нетоз) [1]. На каждом этапе развивающейся при этом воспалительной реакции нейтрофилы играют одну из ключевых ролей как эффекторные и регуляторные участники процесса, а момент и форма их гибели имеют критическое значение для полноценного завершения процесса воспаления и восстановления гомеостаза [1, 4].
Апоптоз является основной формой гибели нейтрофилов, а механизм его индукции не является уникальным и сохраняет основные черты, присущие всем клеткам организма [5]. Так, связывание экстраклеточных лигандов FASL, TNF или TRAIL с соответствующими мембранными “рецепторами смерти” (FAS, TNF-R1, TRAIL-R1/2) активирует цитоплазматические “домены смерти” (FADD или FADD/ TRADD) и запускает многоступенчатый каскад внутриклеточных молекулярных взаимодействий, ключевая роль в которых отводится каспазам-8 и -3, протеинкиназам семейства MAPK и регуляторным белкам семейства Bcl-2, одни из которых проявляют проапоптозные свойства (Bad, Bim, Bax, Bid, Bak), а другие обладают защитным эффектом (Mcl-1, A1, Bcl-XL) [6–8] (рис. 1).
Рис. 1.
Молекулярная регуляция апоптоза нейтрофилов, опосредованного “рецепторами смерти” (FAS, TNF-R1, TRAILR), фагоцитозом и стрессом эндоплазматического ретикулума. Пути супрессии апоптоза обозначены красным цветом.
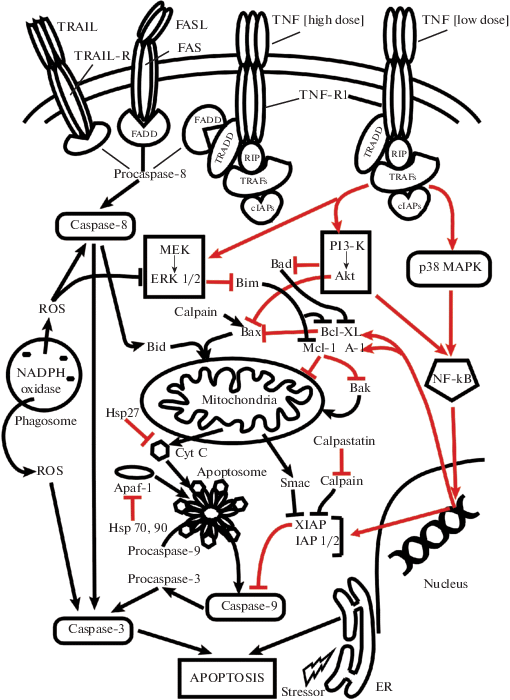
Внутриклеточные триггеры индуцируют пути апоптоза, опосредуемые эндоплазматическим ретикулумом или митохондриями [7–10]. Апоптоз, опосредуемый митохондриями, зависит от доминирования в цитоплазме проапоптозных белков семейства Bcl-2, функционального состояния митохондриальных мембран и активности каспазы-9 [6, 8, 9]. При апоптозе, опосредуемом эндоплазматическим ретикулумом, основная роль отводится так называемой “реакции несвернутых белков”, индуцирующей независимый от каспазного механизма специфический путь программируемой гибели нейтрофилов [10]. Подобно другим фагоцитирующим клеткам, нейтрофилы способны реализовать программу апоптоза, индуцируемого фагоцитозом [7] (рис. 1). Апоптозные формы нейтрофилов теряют функциональную активность, приобретают специфические морфологические характеристики и выводятся из циркуляции фагоцитирующими клетками костного мозга, легких или печени [1, 11].
Стресс представляет собой высококонсервативный биологический ответ на сигнал тревоги или угрозы, связанный с эволюционно древней системой врожденного иммунитета и имеющий решающее значение для выживания организма [12]. Ассоциированные с реализацией программы защиты разнонаправленные изменения продолжительности жизни нейтрофилов отмечаются как при воспалении, так и при стрессе. Нарушения апоптоза нейтрофилов как фактора регуляции их функциональной активности имеют патогенетическое значение при формировании хронических заболеваний различной этиологии.
РЕГУЛЯЦИЯ АПОПТОЗА НЕЙТРОФИЛОВ ПРИ ОСТРЫХ ВОСПАЛИТЕЛЬНЫХ ПРОЦЕССАХ
Продолжительность жизни нейтрофилов в физиологических условиях ограничена 12–18 часами в циркуляторном русле и 1–4 днями в тканях [1, 8, 11, 13, 14]. При развитии воспалительной реакции, характеризующейся активацией нейтрофильного звена, этот показатель может увеличиться в несколько раз, что позволяет иммунной системе с наибольшей эффективностью использовать функциональный потенциал нейтрофилов [1]. После завершения острой фазы воспаления истощенные нейтрофилы подвергаются апоптозу, а поглощение апоптозных телец макрофагами непосредственно в очаге повреждения служит сигналом переключения провоспалительной программы на антивоспалительную с последующим восстановлением тканевого гомеостаза [4, 15]. Продолжительность жизни нейтрофилов в данном случае имеет критическое значение и модулируется комплексом различных сигналов, включая факторы метаболического состояния клетки и экстраклеточного пространства [9, 16, 17].
Показано, что молекулярные механизмы супрессии апоптоза активируются уже в процессе проникновения нейтрофилов в очаг повреждения. Отмечено, что трансэпителиальная миграция этих клеток сопровождается снижением экспрессии прокаспаз -3, -6, -7 и -8 и мРНК прокаспаз-3 и -8 [18], а непосредственный контакт нейтрофилов с эндотелиальным монослоем in vitro тормозит развитие как спонтанного, так и Fas- или TNFR-опосредованного апоптоза [19]. Сигнал подавления апоптоза при трансэндотелиальной миграции в очаг воспаления нейтрофилы получают посредством интегринового взаимодействия, а дополнительными стимулами продления жизни нейтрофилов в острую фазу воспаления являются провоспалительные цитокины, факторы роста, PAMP, DAMP и другие молекулярные регуляторы, каждый из которых четко выполняет свою роль [1, 2, 8]. При сравнении нейтрофилов БАЛ больных тяжелой формой пневмонии с нейтрофилами циркулирующей крови выявлено количественное преобладание антиапоптозных белков Bcl-XL и А1 семейства Bcl-2 в нейтрофилах БАЛ [20]. На этапе разрешения воспаления значительный вклад в модуляцию апоптоза нейтрофилов вносят цитокины, обладающие антивоспалительными свойствами [1, 17].
Интегрины как факторы регуляции апоптоза
Интегрины являются трансмембранными гетеродимерными рецепторами для молекул адгезии и белков экстраклеточного матрикса. Различные α и β субъединицы экспрессируются на поверхности клетки, формируя интегрины с различной специфичностью для лигандов. На цитоплазматической мембране нейтрофилов экспрессированы αLβ2, αMβ2, αVβ3 и α9β1 интегрины [21] с преобладанием αMβ2 или Mac-1 (CD11b/CD18, ITAM антиген), принадлежащего субсемейству β2 интегринов [22, 23]. Активация Mac-1 молекулами межклеточной адгезии ICAM-1, представленными на поверхности эндотелиоцитов капиллярного русла, вносит заметный вклад в повышение жизнеспособности нейтрофилов в процессе их трансэндотелиальной миграции в очаг воспаления [23]. Cвязывание Mac-1 с такими протеинами азурофильных гранул нейтрофилов, как фибриноген, плазминоген и миелопероксидаза, также сопровождается задержкой спонтанного апоптоза [22, 24, 25]. Показано, что костимулирующий сигнал с β2-интегрина необходим для активации транскрипционной активности NF-κB при экспозиции нейтрофилов с воспалительными цитокинами IL-8 и GM-CSF [26]. Механизм передачи сигнала с лигированных интегринов связан с активацией тирозинкиназ из семейства Src, Syk и киназ фокальной адгезии, в частности Pyk2 [27]. В нейтрофилах киназы Syk и Pyk2 модулируют активность киназ Akt и p38 MAPK [28]. Взаимодействие лиганда с Mac-1 вызывает его кластеризацию, активацию Akt и ERK 1/2, а также факторов, блокирующих секвестрацию митохондрий [24, 29]. Кроме того, показано, что растворимый фибриноген способствует транслокации NF-κB в ядро, активируя цитоплазматический ингибитор деградации этого фактора [29]. Выявлено, что для активации фибриногеном или плазминогеном путей выживания, опосредованных ERK1/2 и Akt, необходимо привлечение обеих субъединиц интегрина αMβ2 [24]. При связывании Мас-1 с миелопероксидазой, помимо активации протеинкиназ ERK1/2 и Akt, отмечается накопление в цитоплазме антиапоптозного фактора Mcl-1 семейства Bcl-2, предотвращение дисфункции митохондрий и поддержание эффекторной каспазы-3 в неактивном состоянии [25]. Тем не менее показано, что тиогликолят-индуцированные нейтрофилы, выделенные от мышей, дефицитных по гену CD11b (CD11b –/–), имеют замедленный апоптоз [30], что предполагает существование других интегрин-опосредованных путей регуляции этого процесса. Так, в настоящее время известно, что связывание интегрина α9β1, принадлежащего к субсемейству β1, васкулярными молекулами адгезии VCAM-1 (CD106) приводит к задержке не только спонтанного, но и Fas-индуцированного апоптоза нейтрофилов, и сопровождается стимуляцией PI3-K опосредованного пути выживания и активацией фактора транскрипции NF-κB [21].
Вовлечение нейтрофилов в β2-интегриновое взаимодействие может не только подавлять, но и активировать апоптоз нейтрофилов. Установлено, что продолжительность жизни клеток в данном случае зависит от функциональной активности интегринового рецептора и костимулирующего действия отдельных цитокинов. Связывание β2 интегриновой субъединицы с лигандами при одновременном действии таких проапоптозных факторов, как FasL, TNF или ультрафиолетовое излучение, может ускорять апоптоз нейтрофилов [31]. Усиление фибронектином TNFR - опосредованного апоптоза нейтрофилов зависит от фосфорилирования протеина Ly-GDI, являющегося регулятором ГТФаз [32]. Возможность связывания Mac-1 с компонентом комплемента iC3b или Fc рецептором обусловливает взаимодействие данного интегрина с опсонизированными бактериями, сопровождающееся их фагоцитозом [33], и, как следствие, индукцией апоптоза [7]. Экспериментально установлено, что антитела к Mac-1 блокируют апоптоз, опосредованный фагоцитозом опсонизированных частиц [30]. Интегрины участвуют в межклеточном взаимодействии нейтрофилов и макрофагов. Это взаимодействие может приводить как к активации апоптоза нейтрофилов, так и к их последующему фагоцитозу макрофагами [34, 35]. Таким образом, посредством интегриновых рецепторов апоптоз нейтрофилов может модулироваться различными лигандами, как экспрессированными на поверхности соседних клеток, так и находящимися в экстраклеточном матриксе или в растворенном состоянии.
Цитокины как факторы регуляции апоптоза
Помимо CR семейства TNF (FAS, TNF-R1 и TRAIL-R1/2), связанных с “доменами смерти” FADD и FADD/TRADD, на цитоплазматической мембране нейтрофилов экспрессируются многочисленные рецепторы, активация которых не затрагивает “домены смерти”, однако оказывает разнонаправленное влияние на различные типы апоптоза на этапе трансдукции нисходящего внутриклеточного сигнала к эффекторной каспазе-3 – ключевой “молекуле смерти” [1, 36].
Многие цитокины и факторы роста, играющие важную роль в формировании воспалительного ответа, могут разнонаправленно изменять продолжительность жизни нейтрофилов. Показано, что IL-1, IL-2, IL-3, IL-6, IL-8, IL-15, IL-18, IL-32γ, TNF, IFNγ, IFNα, IFNβ, GM-CSF, G-CSF, MIF и IGF-1 способны замедлять процесс апоптоза, оказывая провоспалительное действие при развитии острой фазы воспаления [5, 11, 20, 36–45]. Из этих факторов TNF и IL-6 обладают бифункциональными свойствами, т.е. могут при определенных условиях активировать апоптоз этих клеток [6, 8, 16, 41].
TNF является лигандом CR семейства TNF, молекулярный комплекс которого индуцирует как проапоптозный путь, опосредуемый каспазой-8 и связанный исключительно с TNF-R1, так и анти-апоптозный, опосредуемый обоими типами рецепторов TNF-R1 и TNF-R2 [16, 36]. Модуляция выживаемости нейтрофилов посредством TNF осуществляется различными факторами [46], в конечном итоге сдвигающими баланс проапоптозных и антиапоптозных сигналов в ту или иную сторону, однако в большей степени зависит от концентрации TNF в среде инкубации и напряжения кислорода в экстраклеточном пространстве [8, 16].
Действие большинства цитокинов опосредуется CR I и II типов. Одними из наиболее изученных лигандов CR типа I являются факторы роста G-CSF и GM-CSF [1, 11, 36, 47]. Продолжительность жизни нейтрофилов, выделенных из крови добровольцев после курсового введения рекомбинантного G-CSF человека (10 мкг/кг подкожно, ежедневно, однократно, в течение 7 дней), была значительно выше, чем в контрольной группе [48]. Как G-CSF, так и GM-CSF подавляют Fas-опосредованный апоптоз нейтрофилов человека [49, 50], однако G-CSF не оказывает влияния на нейтрофилы мышей [51].
Предполагается, что основной механизм антиапоптозного действия цитокинов на нейтрофилы заключается в изменении соотношения экспрессии белков семейства Bcl-2 в сторону защитных белков. Так, GM-CSF, G-CSF, IL-3, IL-6, IL-15 и IFN-γ снижают экспрессию Bax [5, 52]. GM-CSF и TNF вызывают PI3-K-зависимое фосфорилирование и цитозольную транслокацию Bad [53]. G-CSF и GM-CSF поддерживают экспрессию белков выживания A1 и Mcl-1 [20, 47], но не влияют на уровни экспрессии других белков семейства Bcl-2 (Bcl-XL, Bax, Bcl-w) как в нейтрофилах мыши, так и человека [47, 51]. Следует отметить, что данные об индукции экспрессии Bim, Bax и Bcl-XL факторами роста G-CSF и GM-CSF противоречивы [20, 47, 51], а неоднозначность результатов, по-видимому, связана с такими методическими особенностями проведения исследований in vitro, как доза и продолжительность экспозиции исследуемых факторов, а также со степенью очистки популяции нейтрофилов.
Антиапоптозные эффекты провоспалительных цитокинов на нейтрофилы могут быть опосредованы не только балансом белков семейства Bcl-2, но и функциональной активностью ключевых протеинкиназных каскадов. CR типа I (IL-4R, -6R, -12R, -15R, G-CSFR, GM-CSFR) и CR типа II (IFNAR, IFNBR, IFNGR, IL-10R) конститутивно связаны с протеинкиназой JAK, субстратом для которой является активатор транскрипции протеинов STAT. Связывание лиганда с этим типом рецепторов индуцирует активацию протеинкиназных каскадов по сигнальному пути JAK-STAT, регулирующему многие цитологические процессы, в том числе и апоптоз [36]. Среди протеинов семейства JAK ключевая роль в трансдукции антиапоптозного сигнала принадлежит JAK2 [54], инициирующей сигнальный путь JAK2→STATs. Активация данного пути показана для многих лигандов CR I и II типов, в том числе G-CSF, GM-CSF [54, 55], IL-15 [56], IFNα и IFNγ [36, 57]. Особенности путей активации конкретного типа CR, по-видимому, определяются комбинацией рецептор-специфических вариантов JAK и STAT, обусловленной широким спектром их молекулярного разнообразия. Важную дополнительную регуляторную роль в пострецепторных механизмах передачи сигнала выполняют протеинкиназа PI3-K и ее субстрат Akt, ядерный фактор транскрипции NF-κB, белки семейства Bcl-2, ингибиторы апоптоза семейства IAP и ряд других вспомогательных энзимов [6, 36]. Так, например, при обработке нейтрофилов фактором G-CSF активация сигнального пути G-CSFR → JAK2 → STAT3/5 сопровождается увеличением экспрессии cIAP2 [55]. GM-CSF вызывает задержку апоптоза через сигнальные пути, опосредованные протеинкиназами ERK и PI3-K/Akt [45], но не p38 MAPK [58]. В то же время другой лиганд CR типа I, IL-15, активирует не только киназы JAK2 и ERK, но и p38 MAPK, что также приводит к задержке апоптоза [56].
Среди лигандов CR типа II наибольший интерес представляют IFNα/β и IFNγ. Пролонгирование выживаемости нейтрофилов посредством IFNα и IFNγ связано с увеличением экспрессии cIAP2, но не cIAP1, Mcl-1 и A1 [57]. Антиапоптозное действие IFNβ на нейтрофилы обусловлено активацией протеинкиназ PI3-К, Сδ и транслокацией NF-κB в ядро [59, 60]. Показано также, что IFNγ ингибирует Fas-опосредованный апоптоз нейтрофилов здорового человека [44].
Антиапоптозным потенциалом обладают также некоторые интерлейкины, активирующие другие типы цитокиновых рецепторов. Прежде всего к ним относятся IL-1α /β и IL-18, являющиеся лигандами суперсемейства IL-1R/TLR [36] и принимающие активное участие в регуляции воспалительного процесса. В отличие от CR I и II типов, при активации рецепторного комплекса IL-1R/TLR сигнал инициируется белком-адаптером MyD88 и киназами семейства IRAK с дальнейшей трансдукцией на фактор транскрипции NF-κB и киназы семейства MAPK [36]. Выявлено, что IL-1β оказывает супрессирующее действие как на спонтанный [38], так и на Fas-индуцированный апоптоз [50]. Инкубация очищенной популяции нейтрофилов крови здорового человека с IL-18 также сопровождается супрессией апоптоза, а механизм увеличения продолжительности жизни нейтрофилов связан с увеличением активности фактора выживания A1, но не Mcl-1, и опосредуется PI3-K и ERK, но не p38MAPK, киназными путями [61].
Некоторые протеины, обладающие свойствами воспалительных цитокинов, являются лигандами рецепторов, не относящихся к цитокиновому типу. Один из них представлен гормоном роста IGF-1. Выявлено, что IGF-1 вызывает выраженную задержку как спонтанного, так и FAS-индуцированного апоптоза нейтрофилов здорового человека в условиях in vitro [44], причем механизм действия IGF-1 не затрагивает экспрессию Fas и активность каспазы-8, но связан с PI3-K и ингибирующим, но не блокирующим, эффектом на деполяризацию митохондриальных мембран. При этом эффективность действия IGF-1 сравнима с эффектом GM-CSF и IFN-γ, которые почти на 50% ингибируют Fas-опосредуемый апоптоз [44].
Хорошо изученный цитокин IL-8, функционально принадлежащий к группе хемокинов и являющийся лигандом хеморецептора CXCR семейства GPCR [36], не только усиливает внедрение нейтрофилов в очаг воспаления, но и пролонгирует их выживаемость [6]. В условиях культивирования нейтрофилов человека выявлено, что IL-8 задерживает развитие как спонтанного, так и Fas- или TNF-индуцированного апоптоза [6, 45, 50], а сигнальные пути выживания опосредованы ERK/Akt [45]. Другим, не менее важным цитокином плейотропного действия, обладающим хемокино-подобной активностью и антиапоптозным эффектом, является MIF. Показано, что механизм влияния MIF на апоптоз нейтрофилов человека связан с предотвращением высвобождения из митохондрий цитохрома С и Smac, участвующих в активации эффекторной каспазы-3, а также с поддержанием в неактивном состоянии проапоптозных факторов Bid и Bax [40].
Среди цитокинов, высвобождающихся в процессе развития фазы разрешения воспалительного ответа, интерес представляют IL-4 и IL-10 как наиболее изученные с точки зрения влияния на апоптоз [1, 17]. В эту фазу нейтрофилы вступают уже в активированном состоянии, а продолжительность их жизни увеличивается комплексом модуляторов. Показано, что IL-10 отменяет супрессию спонтанного апоптоза, опосредованную провоспалительными факторами TNF, GM-CSF, G-CSF, IFNγ или ЛПС в условиях in vitro, но при этом не оказывает влияние на выживаемость непримированных нейтрофилов [62, 63]. Изучая молекулярный механизм реверсии задержки апоптоза нейтрофилов, С. Ward и соавт. делают вывод, что трансдукция IL-10-опосредованного сигнала ассоциируется с подавлением активности ERK, однако отличается от путей передачи сигнала к ERK от рецепторных комплексов, активируемых GM-CSF или TNF [64]. Проапоптозный эффект также продемонстрирован при изучении влияния IL-4 на активированные ЛПС и культивируемые в условиях гипоксии нейтрофилы человека [17]. IL-10 и IL-4 являются лигандами IL-10R и IL-4R, и большинство исследователей указывают на важность Jak1/STAT3 взаимодействий в проведении опосредованного данными интерлейкинами сигнала [1, 17, 65].
Модуляция апоптоза компонентами бактерий
Среди нескольких типов PRR одними из наиболее изученных являются трансмембранные TLR, принадлежащие семейству IL-1R/TLR рецепторов и имеющие первостепенное значение в распознавании различных структурных компонентов микроорганизмов с последующей активацией выраженной ответной воспалительной реакции [36, 66]. Посредством связывания с TLR компоненты микроорганизмов могут оказывать влияние на апоптоз нейтрофилов и при отсутствии прямого инфицирования клетки. В нейтрофилах человека представлены все десять известных типов TLR, исключая TLR3 [7, 36], три из которых находятся во внутриклеточных компартментах (TLR-7, -8, -9), а остальные экспрессированы на цитоплазматической мембране [66]. Наиболее изученными PAMP бактериальной природы считаются мембранные ЛПС, ЛП, ПГ и липотейхоевые кислоты, обладающие антиапоптозным потенциалом различной степени выраженности [6, 7, 22, 61, 64, 67, 68]. Экспериментально продемонстрировано, что ЛПС, являющийся компонентом наружной мембраны большинства грамотрицательных бактерий и одновременно лигандом TLR4, вызывает супрессию спонтанного апоптоза нейтрофилов в условиях in vitro и in vivo [6, 22, 61, 64, 68], а выраженность эффекта сравнима с действием таких медиаторов воспаления, как IL-18, G-CSF или GM-CSF [6, 61]. Экспериментально продемонстрировано, что после введения ЛПС в нейтрофилах легких крыс повышаются активности PI3-K, Akt, p38 MAPK и ERK, транскрипционная активность NF-κB, а также усиливается экспрессия фактора выживания Mcl-1 [69].
ЛП и ПГ клеточных мембран бактерий являются основными агонистами TLR2, формирующими сигнал в комплексе с TLR1 или TLR6 [36, 67]. Активация TLR2, как правило, сопровождается супрессией апоптоза различной степени выраженности [22, 68]. Так, например, ЛП грамотрицательной F. tularensis или ПГ грамположительного S. aureus, связываясь с TLR2, способны значительно увеличивать продолжительность жизни нейтрофилов, модулируя пути апоптоза [67, 68]. В настоящее время появились данные, указывающие на взаимодействие бактериальных ПГ не только с рецепторами TLR2 типа, но и с внутриклеточными PRR семейства NOD. В частности, показано, что агонист NOD2 рецептора мурамилдипептид грамотрицательных бактерий активирует классические провоспалительные пути выживания нейтрофилов, опосредованные активацией NF-κB и MAPK [70].
Бактериальная ДНК, высвобождающаяся при лизисе или пролиферации бактерий, имеет все иммунологические свойства PAMP, а короткие последовательности Cp-DNA селективно распознаются TLR9 рецепторами нейтрофилов. Активация TLR9 бактериальной ДНК стимулирует функциональную активность нейтрофилов, что проявляется в индукции экспрессии хемокинов и интегрина Mac-1, регулировании молекул адгезии, усилении фагоцитарной активности и супрессии спонтанного апоптоза [66], что в итоге способствует поддержанию острой фазы ответной воспалительной реакции. Молекулярные механизмы антиапоптозного сигнала агонистов TLR на нейтрофилы опосредованы белком-адаптером MyD88 [66, 71] и ассоциированы с активацией киназных путей PI3-K/Akt и MEK/ERK, запуском NF-κB-опосредованной транскрипции генов провоспалительных цитокинов, увеличением экспрессии факторов выживания A1 и Mcl-1, а также снижением активности эффекторной каспазы-3 [8, 61, 64, 66, 68]. Менее изучен механизм подавления FasL-индуцированного апоптоза лигандами TLR. Известно, что в нейтрофилах мыши цитопротективный эффект, обусловленный взаимодействием с TLR1/2, TLR2/6 или TLR4, связан с ингибированием расщепления каспазы-8 и не затрагивает баланс белков семейства Bcl-2 [71].
Следует также отметить, что в дополнение к антиапоптозному действию структурных компонентов бактерий, токсины некоторых микроорганизмов, например, веротоксин E. coli, Шига токсин и фенол-растворимые модулины, также способны пролонгировать продолжительность жизни нейтрофилов [11], тем самым эффективно увеличивая их количество во время развития острой фазы воспаления.
НАРУШЕНИЯ АПОПТОЗА ПРИ ПАТОЛОГИИ
В основе патогенеза большинства заболеваний лежит воспаление, представляющее собой ответную реакцию иммунологического характера, направленную на локализацию и нейтрализацию повреждающего агента [72]. В большинстве случаев острые воспалительные ответы разрешаются путем удаления тканевого раздражителя и постепенного снижения содержания лейкоцитов в очаге воспаления в результате прекращения их рекрутирования из системы циркуляции, а также вследствие их апоптоза c последующим фагоцитозом и восстановлением гомеостаза в очаге повреждения. Нарушение регуляции апоптоза при развитии воспалительных реакций ассоциируется с нарушением алгоритма воспалительного ответа, способствует хронизации патологического процесса и развитию заболеваний [4, 7, 73].
Так, известно, что при воспалениях инфекционной этиологии увеличение продолжительности жизни активированных нейтрофилов сопровождается избыточным локальным высвобождением комплекса цитотоксических бактерицидных субстанций и регуляторных цитокинов, однако, в случае выраженного воспалительного процесса продукция цитокинов часто приобретает неконтролируемый характер и вносит существенный вклад в формирование системной воспалительной реакции [72]. В то же время несвоевременная акселерация апоптоза при острых инфекционных заболеваниях сопровождается незавершенным фагоцитозом и часто провоцирует развитие вторичного некроза, связанного с чрезмерным локальным повреждающим действием продуктов клеточного распада на ткани [7, 11]. Подобные нарушения облегчают ускользание микроорганизмов от лизиса и благоприятствуют диссеминации в тканях хозяина [7, 67], что способствует поддержанию рецидивирующих воспалительных процессов и развитию хронической патологии. Например, персистирующий в организме репираторный синцитиальный вирус является причиной обостряющихся бронхиолитов, связанных с патогенезом бронхиальной астмы [74].
Некоторые исследователи предполагают, что при воспалительных процессах в механизме ускорения апоптоза нейтрофилов человека важную роль играет увеличение экспрессии Fas. Показано, что у пациентов, часто болеющих острыми респираторными заболеваниями и имеющих клинические признаки интоксикации, отмечается увеличение экспрессии нейтрофилами Fas и повышается количество апоптозных форм нейтрофилов [75].
Разнонаправленные нарушения процесса апоптоза нейтрофилов наблюдаются не только при развитии инфекционных процессов, но и при многих неинфекционных заболеваниях и патологических состояних [5, 15, 40, 76–105 ] (табл. 1). Следует отметить, что некоторые молекулярные регуляторы апоптоза нейтрофилов, такие как CD69, SAA и SHP, у здоровых людей практически не выявляются, однако их количество резко возрастает при некоторых заболеваниях. При этом повышенная экспрессия CD69 и SAA ассоциируется с супрессией апоптоза [91, 96, 97], а SHP, напротив, c его активацией [76, 46 ] (табл. 1). Локальные повреждения тканей, связанные с острым нарушением кровообращения или с травмами, являются причиной развития стерильных воспалений, сопровождающихся высвобождением DAMP и нейтрофильной инфильтрацией. DAMP-опосредованное увеличение продолжительности активного функционирования нейтрофилов при стерильном воспалении может привести к повреждению тканей и органов, выступая дополнительным патогенетическим фактором заболевания [1]. Так, опираясь на экспериментальные данные, некоторые исследователи полагают, что при острой сердечной недостаточности нейтрофилы способны оказывать прямое повреждающее действие на кардиомиоциты в очаге ишемии, расширяя зону инфаркта посредством активации интегриновых взаимодействий [3]. У пациентов с хронической сердечной недостаточностью воспаление миокарда также может быть частично связано со снижением скорости спонтанного апоптоза нейтрофилов [106]. Подобный механизм модуляции апоптоза также является одним из патофизиологических факторов формирования так называемого синдрома полиорганной дисфункции при эндогенной интоксикации различного генеза [62]. Критерием выраженности эндогенной интоксикации может служить количественная оценка содержания апоптозных форм нейтрофилов в периферической крови больного [75]. При заболеваниях, сопровождаемых уремией, обычно наблюдается ускорение апоптоза нейтрофилов [81, 90], однако при состояниях, когда интоксикация организма достигает значительной интенсивности, например, при сепсисе или ожоговой травме, отмечается увеличение продолжительности жизни нейтрофилов [78, 84, 85, 87, 92].
Таблица 1.
Нарушения процесса апоптоза нейтрофилов и связанные с ними изменения в системе вне- и внутриклеточной регуляции при некоторых заболеваниях и патологических состояниях
| Патологическое состояние, заболевание | Изменение в системе регуляции апоптоза нейтрофилов* | Ссылки | |
|---|---|---|---|
| Замедление апоптоза | Атопия | Повышение индуцированной GM-CSF экспрессии CD69 | [91] |
| Болезнь Кавасаки | Повышение отношения A1/Bax | [94] | |
| Болезнь Крона и язвенный колит | ↓ Экспрессия прокаспазы-3; ↑ концентрация IL-8 в системе циркуляции и G-CSF в участке воспаления | [80, 88] | |
| Бронхиальная астма в период обострения | ↓ Секреция проапоптозного IL-10 макрофагами и моноцитами | [15] | |
| Ревматоидный артрит | ↑ Экспрессия CD69, A1 и Mcl-1 mРНК; ↑ уровень GM-CSF и SAA в плазме крови | [95–97] | |
| Обширные механические травмы | ↑ Уровень Mcl-1; ↓ уровень Bax; ↑ уровень GM-CSF в сыворотке; ↑ экспрессия Fas/FasL mРНК | [98] | |
| Муковисцидоз | ↑ Уровень кальпастатина, ↓ уровень кальпаина-1; значительно ↑ уровень MIF в плазме крови | [40, 77] | |
| Ожоговая травма | ↑ Экспрессия Bcl-XL, Hsp27, Hsp60, Hsp70; ↓ экспрессия Bax | [87, 92] | |
| Острый панкреатит | ↓ Экспрессия каспаз | [93] | |
| Острый коронарный синдром | ↑ Количество TNF, IFNγ, GM-CSF и IL-1β в сыворотке | [99] | |
| Пароксизмальная гемоглобиноурия | ↑ Экспрессия генов A1 и Mcl-1 | [86] | |
| Перидонтит | ↑ Уровень GM-CSF в участке воспаления | [83] | |
| Сепсис | ↑ Уровень Mcl-1 mRNA, ↑ транскрипционная активность NFκB; ↑ экспрессия Hsp27, Hsp60, Hsp70 и Hsp90; ↓ Активность каспаз-3, -9, -1 | [78, 84, 85, 100] | |
| Хроническая нейтрофильная лейкемия |
↓ Активность кальпаина | [89] | |
| Ускорение апоптоза | Врожденный миелокатексис | ↓ Экспрессия Bcl-XL | [79] |
| Гликогеноз Ib | Нарушение в системе антиоксидантной защиты, усиление ER-стресса, активация Bax, каспаз-3,-9 | [101] | |
| Синдром Костманна (тяжелая врожденная нейтропения) | ↑ Активность SHP-1 и SHP-2; нарушения Г6ФК3 и мутации эластаз, усиливающие ER-стресс; дестабилизация мембранного потенциала митохондрий вследствие дефицита HAX1 и AK2 | [76, 102] | |
| Системная красная волчанка | Появление в системе циркуляции аутоантител к нейтрофилам; нарушение фагоцитарной активности; ↑ экспрессия каспаз -7, -8, -9 mРНК; ↓ экспрессия IAP1/2 и XIAP, а также mРНК к ним; ↑ экспрессия каспазы-3, Fas, и FADD; ↑ концентрация Fas и TRAIL в сыворотке крови | [103–105] | |
| Уремия | ↑ Экспрессия Fas, ↑ концентрация апоптозных факторов в системе циркуляции | [81, 90] | |
| Хирургическая операция с общей анестезией | Дисфункция митохондрий | [82] |
Антимикробная активность нейтрофилов в сочетании с наследственными особенностями функционирования иммунной системы хозяина вносит заметный вклад в развитие широко распространенных заболеваний аутоиммунного генеза. Генетически обусловленное снижение продукции ROS NADPH-оксидазным комплексом нейтрофилов связано с ослаблением их функциональной активности и нарушением апоптоза, что значительно увеличивает риск развития системной красной волчанки, ревматоидного артрита, хронических воспалительных заболеваний кишечника [107–110], а также приводит к серьезным осложнениям инфекционного характера у больных хроническим гранулематозом [111]. У больных системной красной волчанкой обнаружено увеличение количества апоптозных форм нейтрофилов в крови, и этот показатель коррелирует с тяжестью заболевания [112]. При активном ANCA-ассоциированном васкулите примированные нейтрофилы также отличаются ускоренным апоптозом [113]. В то же время ANCA-ассоциированный васкулит в стадии ремиссии, а также ревматоидной артрит и истинная полицетемия характеризуются отложенным апоптозом нейтрофилов, выявленного в условиях эксперимента in vitro [95]. При синдроме Костманна и гликогенозе 1b обнаружены мутации, ассоциированные с нарушением метаболизма нейтрофилов (табл. 1), которые способствуют преждевременной активации внутриклеточных индукторов апоптоза и гибели клеток на этапе созревания, что и является основным этиологическим фактором в патогенезе хронической нейтропении при этих заболеваниях [76, 101, 102]. В настоящее время известно несколько групп врожденных нейтропений, развивающихся вследствие генетически обусловленного преждевременного апоптоза миелоидных предшественников нейтрофилов [14].
Факторы, увеличивающие продолжительность жизни нейтрофилов, появляются в сыворотке крови больных при воспалительных процессах различной этиологии. Такие регуляторы были обнаружены у пациентов с ожоговыми повреждениями, сепсисом, синдромом системного воспалительного ответа, реперфузионным повреждением, болезнью Крона, язвенным колитом, ревматоидным артритом, а также после обширных полостных операций [13, 80, 97, 114–117]. В синовиальной жидкости больных ревматоидным артритом выявляются факторы разнонаправленной регуляции апоптоза, и их количественный анализ может иметь клиническое значение. Так, на ранней стадии ревматоидного артрита (длительность заболевания менее 3 мес) нейтрофилы синовиальной жидкости и периферической крови демонстрируют отложенный апоптоз, что связано с высоким уровнем содержания антиапоптозных цитокинов IL-2, IL-4, IL-15, GM-CSF и G-CSF. В то же время при активном ревматоидном артрите в синовиальной жидкости появляется растворимый FasL, и локальное количество апоптозных форм нейтрофилов увеличивается, несмотря на повышенное содержание G-CSF [13]. По-видимому, это связано с редукцией ответа нейтрофилов больных ревматоидным артритом на воспалительные цитокины GM-CSF, G-CSF и TNF, сопровождающейся увеличением базального уровня фосфорилирования киназ ERK и p38MAPK [118]. Экспериментальным путем выявлено, что развитие индуцированного иммунными комплексами артрита опосредовано активацией Fcγ рецепторов и связанной с ними тирозинкиназы Syk, модулирующей скорость апоптоза нейтрофилов. Низкая продукция воспалительных цитокинов и ускорение апоптоза являются отличительными особенностями дефицитных по Syk нейтрофилов суставной сумки [119]. Аналогичный механизм супрессии апоптоза нейтрофилов обнаружен при связывании иммунорецепторов FcγIIα с анти-IL-8/IL-8 иммунными комплексами, выделенными из легочной отечной жидкости больных с ОРДС [120]. R. Fudala и соавт. продемонстрировали, что в данное взаимодействие вовлечены протеины семейства тирозинкиназы Srk, тирозинкиназа Syk, киназы ERK и PI3-K, а в результате активации путей выживания баланс белков семейства Bcl-2 сдвигается в сторону антиапоптозного Bcl-XL, снижая активность эффекторных каспаз -3 и -9. С учетом полученных данных и сообщений об обнаружении других аутоантител в легочной отечной жидкости больных с ОРДС, R. Fudala и соавт. предполагают, что аутоиммунные механизмы вносят существенный вклад в увеличение продолжительности жизни функционально активных нейтрофилов и имеют важное значение в патогенезе данного синдрома [120].
СТРЕСС КАК ФАКТОР РЕГУЛЯЦИИ АПОПТОЗА
Реакция организма на психоэмоциональный или физиологический стресс сопровождается усилением поступления в систему циркуляции катехоламинов и ГК [121]. Активация симпатической системы при стрессе затрагивает иммунорегуляторные механизмы посредством синтеза и высвобождения ряда провоспалительных медиаторов, в том числе IL-6, IL-1β и TNF [121], известных как модуляторы апоптоза нейтрофилов [5, 8, 36, 38, 52]. В то же время стимуляция парасимпатической системы и поступающие в циркуляцию ГК вносят существенный вклад в угнетение воспаления [121]. Характер и длительность стрессорного воздействия выступают факторами взаиморегуляции активности нейроэндокринной и иммунной систем, что проявляется в неоднозначном влиянии стресса на жизнеспособность нейтрофилов. Так, у лиц, находящихся под влиянием продолжительного экзаменационного стресса или имеющих расстройство адаптации, отмечена активация спонтанного апоптоза нейтрофилов [8, 122]. Однако у здоровых людей, перенесших краткосрочный стресс в форме голодания, усиленной спортивной нагрузки или нарушения сна, выявлена задержка апоптоза [8].
Исследования in vitro показали, что лекарственные средства группы ГК значительно повышают жизнеспособность нейтрофилов здоровых доноров [123]. Агонисты β2-адренорецепторов (сальбутамол, формотерол, сальметерол) потенцируют антиапоптозный эффект ГК (будесонид, флутиказон) в аналогичных экспериментальных условиях, однако собственного действия на спонтанный апоптоз нейтрофилов не оказывают [124]. Среди терапевтических средств группы ГК наиболее изученным по влиянию на процессы апоптоза является дексаметазон. В течение первых 24 ч инкубации дексаметазон снижает спонтанный апоптоз нейтрофилов на 50%, тогда как сочетанная обработка нейтрофилов дексаметазоном и лейкотриеном В4 угнетает их апоптоз на 90% [125]. В то же время антиапоптозный эффект GM-CSF на нейтрофилы незначительно усиливается глюкокортикоидами, в том числе и дексаметазоном [123, 124]. Интересно отметить, что дексаметазон увеличивает экспрессию на нейтрофилах рецепторов для лейкотриена B4 [125], который обладает собственным антиапоптозным потенциалом [124]. Этот препарат также подавляет апоптоз нейтрофилов, индуцированный окислительным стрессом (инкубация клеток в ферментативной системе глюкоза/глюкозоксидаза) [126], что, по-видимому, обусловлено его способностью подавлять NADPH-зависимую продукцию ROS [127]. ГК-опосредованное угнетение апоптоза сопровождается увеличением экспрессии белков-ингибиторов апоптоза семейства IAP и фактора выживания Mcl-1 в нейтрофилах человека, а в экспериментах с использованием нейтрофилов быка обнаружены увеличение экспрессии фактора выживания А1, снижение экспрессии проапоптозного фактора Bak и уменьшение количества Fas-рецепторов на клеточной поверхности [127]. Следует отметить, что при хроническом стрессе отмечается резистентность к воздействию ГК вследствие ингибирования функциональной активности ГК рецепторов [128].
Несмотря на обнаруженный антиапоптозный эффект ГК на нейтрофилы в условиях in vitro, способный поддержать в определенной степени развитие острой фазы воспалительной реакции, синтетические ГК активно применяются в клинической практике в качестве противовоспалительных средств, демонстрируя широкий спектр регуляторного воздействия на интенсивность иммунного ответа. Усиливая фагоцитарную активность макрофагов, направленную на удаление апоптозных форм нейтрофилов, ГК вносят существенный вклад в развертывание фазы разрешения воспаления [129]. Регулируя транскрипционную активность множества генов, ГК индуцируют экспрессию в нейтрофилах двух протеинов, обладающих проапоптозным потенциалом и выраженным антивоспалительным действием – аннексина 1, также известного как липокортин-1 [130], и протеина, кодируемого геном GILZ [127]. Стимулируя апоптоз нейтрофилов, эти пептиды, по-видимому, способны сглаживать прямой антиапоптозный эффект ГК.
Таким образом, стресс посредством связанных с ним гормонов и нейромедиаторов вызывает разнонаправленные нарушения в системе комплексной регуляции продолжительности функционально активной жизни нейтрофилов, оказывая влияние на чувствительность организма к инфекциям и способность к восстановлению гомеостаза в очаге повреждения.
ФАРМАКОЛОГИЧЕСКАЯ МОДУЛЯЦИЯ АПОПТОЗА НЕЙТРОФИЛОВ
Нарушения функциональной активности нейтрофилов, включая нарушения апоптоза, отмечаются при заболеваниях различного генеза (табл. 1), что привлекает внимание исследователей к этим клеткам как к объекту терапевтического воздействия [22, 23, 130]. В качестве потенциальных мишеней фармакологического воздействия могут рассматриваться ключевые индукторы и модуляторы апоптоза нейтрофилов (рис. 1). К настоящему времени на различных моделях воспаления у мышей показано, что некоторые соединения проявляют противовоспалительное действие, сопровождающееся усилением апоптоза нейтрофилов. В частности, такой терапевтический эффект был обнаружен у R-росковитина (ингибитор CDK) на моделях каррагенан-индуцированного плеврита, блеомицин-индуцированного повреждения легких и пневмококкового менингита [132, 133], ролипрама (ингибитор PDE-4) на модели ЛПС-индуцированного плеврита [134] и rTRAIL (рекомбинантный лиганд TRAIL-R) на моделях зимозан-индуцированного перитонита и ЛПС-индуцированного острого повреждения легких [135]. В качестве потенциальных противовоспалительных средств, усиливающих как апоптоз, так и эффероцитоз нейтрофилов, представляют интерес эндогенные медиаторы резолвин E1, аннексин А1 и эпимер липоксина А4, демонстрирующие терапевтичекую эффективность на экспериментальных моделях воспаления [15, 23]. Основными молекулярными мессенджерами, обусловливающими проапоптозный эффект вышеупомянутых соединений, являются белки семейства Bcl-2, регуляторные киназы и эффекторная каспаза-3 (рис. 2).
Рис. 2.
Молекулярные механизмы модуляции апоптоза нейтрофилов потенциальными фармакологическими средствами. Противовоспалительные эффекты ролипрама, R-росковитина и эндогенных медиаторов фазы разрешения воспаления (аннексин A1, резолвин Е1, липоксин A4) опосредуются сдвигом баланса белков семейства Bcl-2 в сторону преобладания проапоптозных факторов, а также блокадой проведения сигнала выживания посредством киназы PI3-K и фактора транскрипции NF-κB. Рекомбинантный лиганд “рецептора смерти” TRAILR (rTRAIL) индуцирует апоптоз по каспазному механизму (см. рис. 1). Эффект саргамостима индуцируется связыванием с рецептором GM-CSF (GM-CSFR) цитокинового типа и аналогичен антиапоптозному эффекту GM-CSF. Экспрессия фактора выживания Mcl-1 усиливается под действием глюкокортикоидов.
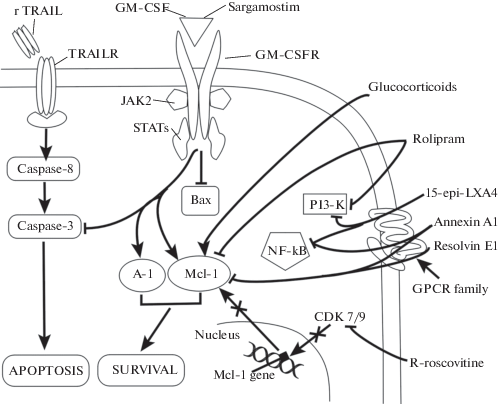
В настоящее время ряд соединений, обладающих плейотропным действием, в том числе и про-апоптозным эффектом в отношении нейтрофилов, находится на различных фазах клинических испытаний. Так, росковитин проходит клинические испытания не только как средство терапии некоторых онкологических заболеваний, но и как потенциальный противовоспалительный препарат для лечения ревматоидного артрита, гломерулонефрита и муковисцедоза, осложненного хронической инфекцией [136]. При других патологиях, напротив, терапевтический эффект фармакологических средств связан с пролонгацией жизнеспособности нейтрофилов. Так, при химиотерапии опухолей часто развивается нейтропения. В этих случаях применение рекомбинантного GM-CSF (саргамостим) приводит к замедлению апоптоза нейтрофилов, вызванного действием противоопухолевых препаратов. Сходный эффект описан и для синтетических кортикостероидов, таких как флутиказон и преднизолон [137] (рис. 2).
Следует также отметить, что апоптозные формы нейтрофилов обладают собственным противовоспалительным потенциалом, возможно, благодаря высвобождению из них α-дефензимов – небольших катионных пептидов, подавляющих продукцию макрофагами провоспалительных цитокинов [138]. При введении апоптозных форм нейтрофилов экспериментальным животным с ЛПС-индуцированным сепсисом отмечались снижение количества циркулирующих провоспалительных цитокинов и предотвращение эндотоксического шока [139], что позволяет рассматривать эти клетки в качестве потенциального средства борьбы с “цитокиновым штормом”.
ДРУГИЕ ФОРМЫ ГИБЕЛИ НЕЙТРОФИЛОВ ПРИ ВОСПАЛЕНИИ
С точки зрения регуляции интенсивности воспалительного ответа, форма гибели нейтрофилов в очаге воспаления является не менее важным иммуномодулирующим фактором, чем степень их функциональной активности. Литические формы гибели клеток (нетоз, пироптоз, некроз и некроптоз) часто сопровождаются усилением интенсивности острой фазы воспаления вследствие высвобождения из гибнущих клеток DAMP, ROS и протеаз [1, 140, 141]. Апоптоз, напротив, представляет собой нелитическую эволюционно консервативную форму гибели клеток, не обладающую воспалительным потенциалом [7, 140]. В отличие от других форм гибели, нетоз характеризуется формированием экстраклеточных антимикробных сетей из покидающих клетку деконденсированных нитей ядерных ДНК, цитруллинированных гистонов и бактерицидных протеаз гранулярного аппарата. Следует отметить, что нетоз не всегда сопровождается лизисом цитоплазматической мембраны, а безъядерная форма нейтрофила сохраняет ряд эффекторных функций, включая фагоцитоз [1, 7, 140]. Пироптоз направлен на элиминацию внутриклеточных патогенов с помощью внутриклеточных антимикробных ловушек, формирование которых опосредовано образованием специфических мембранных пор [140, 142]. Некроптоз нейтрофилов (регулируемый некроз) индуцируется различными сигналами и, по-видимому, представляет собой дополнительный фактор противовирусной защиты [140, 142].
Аутофагия – это крайне консервативный защитный ответ клетки на стресс, в том числе на недостаток питательных веществ [7, 141, 143]. При чрезмерной силе стрессового воздействия аутофагия может обернуться гибелью клетки [141, 144]. Морфологическим маркером аутофагии являются ограниченные двойной мембраной крупные цитоплазматические вакуоли, или аутофагосомы, которые после слияния с лизосомами трансформируются в аутолизосомы, где и происходит литический распад собственных макромолекул и поврежденных органелл [7, 141, 143]. Аутофагия часто сопровождает апоптоз, нетоз и некроз нейтрофилов [143, 145], а при некоторых инфекциях встраивается в систему иммунной защиты от патогена, обеспечивая его элиминацию в аутолизосомах (феномен ксенофагии) [144, 145].
Механизмы литических форм гибели нейтрофилов в патогенезе изучены недостаточно, но, тем не менее, выявлено, что они имеют существенные отличия от апоптоза, при котором протеолиз субстратов обеспечивается преимущественно активацией эффекторных каспаз [6–8, 140] (рис. 1). Кроме некроза, представляющего собой процесс неконтролируемого набухания клетки с последующим разрушением мембран и распадом [141], все иные формы гибели запрограммированы в виде специфических последовательностей молекулярных событий [6, 140, 142], предусматривающих перекрестные внутриклеточные взаимодействия между различными путями проведения терминальных сигналов [136, 142, 143, 146]. Полученные к настоящему времени данные формируют общее представление, что именно программа апоптоза, в отличие от других программ гибели нейтрофилов, встроена в механизм комплексной супрессии острого воспалительного ответа в качестве основного регулятора.
ЗАКЛЮЧЕНИЕ
В ходе эволюционного развития сформировалось несколько механизмов индукции апоптоза, которые поддерживаются разнонаправленными регуляторными путями, вовлеченными в конвергирующее взаимодействие при трансдукции внешнего и внутреннего сигнала к эффекторным каспазам. Например, GM-CSF и TNF способны, с одной стороны, в значительной степени подавлять спонтанный апоптоз нейтрофилов периферической крови человека, а с другой, – ускорять апоптоз, опосредованный митохондриями по зависимому от каспазы-9 механизму [20]. Также выявлено, что индуцированный фагоцитозом апоптоз отменяет увеличение продолжительности жизни нейтрофилов, опосредованное провоспалительными цитокинами [7], а резистентность нейтрофилов к внутренним сигналам апоптоза, обнаруженная у тяжелобольных пациентов, преодолевается активацией Fas рецепторов [98]. Интересно отметить, что нейтрофилы крови пациентов с септическим шоком отличаются отложенным апоптозом, несмотря на заметное превышение экспрессии проапоптозного белка Bim [47]. Многофакторная регуляция продолжительности жизни нейтрофилов позволяет в ряде случаев нивелировать влияние инфекционного агента на апоптоз. Показано, что IL-6 и GM-CSF отменяют ускорение гибели нейтрофилов, индуцированной вирусом гриппа А(N1N1) и вирусом Эпштейна-Барр соответственно [42, 147]. Также показано, что ЛПС, c одной стороны, вызывает увеличение продолжительности жизни нейтрофилов, а с другой, стимулирует синтез рецептора IL-10, индуцирующих ускорение апоптоза в ответ на IL-10 [148]. Несмотря на то что все этапы воспалительного ответа четко отрегулированы, хронизация патологического процесса может ассоциироваться с изменением базальной активности нейтрофилов и сопровождаться нарушением основного алгоритма регуляции апоптоза [73, 118]. Так, обнаружено, что нейтрофилы синовиальной жидкости больных ревматоидным артритом имеют изначально высокий уровень активации NF-κB, и их стимуляция TNF не сопровождается индукцией апоптоза в отличие от нейтрофилов циркуляторного русла [149]. Таким образом, судьбу нейтрофила определяет результирующий сдвиг внутриклеточного баланса про- и антиапопотозных факторов в ту или иную сторону под действием комплекса регуляторных сигналов, модулируемых уровнем базальной активности клетки.
Понимание механизмов мультимодальной регуляции апоптоза как фактора интегральной функциональной активности нейтрофилов имеет важнейшее значение для выработки терапевтической стратегии и выявления ключевых регуляторных молекул в качестве фармакологических мишеней новых лекарственных средств.
Список литературы
Liew P.X., Kubes P. (2019) The Neutrophil’s Role During Health and Disease. Physiol Rev 99 (2): 1223–1248.
Geering B., Stoeckle C., Conus S., Simon H.U. (2013) Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol 34 (8): 398–409.
Frangogiannis N.G. (2014) The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol 63 (3): 185–195.
Schett G., Neurath M.F. (2018) Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun 9 (1): 3261. https://doi.org/10.1038/s41467-018-05800-6
Dibbert B., Weber M., Nikolaizik W.H., Vogt P., Schöni M.H., Blaser K., Simon H.U. (1999) Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci U S A 96 (23): 13330–13335.
Akgul C., Moulding D.A., Edwards S.W. (2001) Molecular control of neutrophil apoptosis. FEBS Lett 487 (3): 318–322.
Kennedy A.D., DeLeo F.R. (2009) Neutrophil apoptosis and the resolution of infection. Immunol Res 43 (1–3): 25–61.
Luo H.R., Loison F. (2008) Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83 (4): 288–295.
Kumar S., Dikshit M. (2019) Metabolic Insight of Neutrophils in Health and Disease. Front Immunol 10:2099. https://doi.org/10.3389/fimmu.2019.02099
Binet F., Chiasson S., Girard D. (2010) Evidence that endoplasmic reticulum (ER) stress and caspase-4 activation occur in human neutrophils. Biochem Biophys Res Commun 391 (1): 18–23.
Kobayashi S.D., Malachowa N., DeLeo F.R. (2017) Influence of Microbes on Neutrophil Life and Death. Front Cell Infect Microbiol 7: 159. https://doi.org/10.3389/fcimb.2017.00159
Slavich G.M., Irwin M.R. (2014) From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull 140 (3): 774–815.
Cascão R., Rosário H.S., Souto-Carneiro M.M., Fonseca J.E. (2010) Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun Rev 9 (8): 531–535.
Bartels M., Murphy K., Rieter E., Bruin M. (2016) Understanding chronic neutropenia: life is short. Br J Haematol 172 (2): 157–169.
Barnig C., Bezema T., Calder P.C., Charloux A., Frossard N., Garssen J., Haworth O., Dilevskaya K., Levi-Schaffer F., Lonsdorfer E., Wauben M., Kraneveld A.D., Te Velde A.A. (2019) Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons From Asthma and Inflammatory Bowel Disease. Front Immunol 10: 1699. https://doi.org/10.3389/fimmu.2019.01699
Dyugovskaya L., Polyakov A., Ginsberg D., Lavie P., Lavie L. (2011) Molecular pathways of spontaneous and TNF-{alpha}-mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol 45 (1): 154–162.
Harris A.J., Mirchandani A.S., Lynch R.W., Murphy F., Delaney L., Small D., Coelho P., Watts E.R., Sadiku P., Griffith D., Dickinson R.S., Clark E., Willson J.A., Morrison T., Mazzone M., Carmeliet P., Ghesquiere B., O’Kane C., McAuley D., Jenkins S.J., Whyte M.K.B., Walmsley S.R. (2019) IL4Rα Signaling Abrogates Hypoxic Neutrophil Survival and Limits Acute Lung Injury Responses In Vivo. Am J Respir Crit Care Med 200 (2): 235–246.
Le’Negrate G., Rostagno P., Auberger P., Rossi B., Hofman P. (2003) Downregulation of caspases and Fas ligand expression, and increased lifespan of neutrophils after transmigration across intestinal epithelium. Cell Death Differ 10 (2): 153–162.
Tennenberg S.Dl., Finkenauer R., Wang T. (2002) Endothelium down-regulates Fas, TNF, and TRAIL-induced neutrophil apoptosis. Surg Infect (Larchmt) 3 (4): 351–357.
Cowburn A.S., Summers C., Dunmore B.J., Farahi N., Hayhoe R.P., Print C.G., Cook S.J., Chilvers E.R. (2011) Granulocyte/macrophage colony-stimulating factor causes a paradoxical increase in the BH3-only pro-apoptotic protein Bim in human neutrophils. Am J Respir Cell Mol Biol 44 (6): 879–887.
Ross E.A., Douglas M.R., Wong S.H., Ross E.J., Curnow S.J., Nash G.B., Rainger E., Scheel-Toellner D., Lord J.M., Salmon M., Buckley C.D. (2006) Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood 107 (3): 1178–1183.
Filep J.G., El Kebir D. (2009) Neutrophil apoptosis: a target for enhancing the resolution of inflammation. J Cell Biochem 108 (5): 1039–1046.
El Kebir D., Filep J.G. (2013) Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cells. 2 (2): 330–348.
Pluskota E., Soloviev D.A., Szpak D., Weber C., Plow E.F. (2008) Neutrophil apoptosis: selective regulation by different ligands of integrin alphaMbeta2. J Immunol 181 (5): 3609–3619.
El Kebir D., József L., Pan W., Filep J.G. (2008) Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res 103 (4): 352–359.
Kettritz R., Choi M., Rolle S., Wellner M., Luft F.C. (2004) Integrins and cytokines activate nuclear transcription factor-kappaB in human neutrophils J Biol Chem. 279 (4): 2657–2665.
Gu J., Nada S., Okada M., Sekiguchi K. (2003) Csk regulates integrin-mediated signals: involvement of differential activation of ERK and Akt. Biochem Biophys Res Commun 303 (3): 973–977.
Avdi N.J., Nick J.A., Whitlock B.B., Billstrom M.A., Henson P.M., Johnson G.L., Worthen G.S. (2001) Tumor necrosis factor-alpha activation of the c-Jun N-terminal kinase pathway in human neutrophils. Integrin involvement in a pathway leading from cytoplasmic tyrosine kinases apoptosis. J Biol Chem 276 (3): 2189–2199.
Rubel C., Gomez S., Fernandez G.C., Isturiz M.A., Caamano J., Palermo M.S. (2003) Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur J Immunol 33: 1429–1438.
Coxon A., Rieu P., Barkalow F.J., Askari S., Sharpe A.H., von Andrian U.H., Arnaout M.A., Mayadas T.N. (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5 (6), 653–666.
Whitlock B.B., Gardai S., Fadok V., Bratton D., Henson P.M. (2000) Differential roles for alpha (M) beta (2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J Cell Biol. 151 (6): 1305–1320.
Kettritz R., Xu Y.X., Faass B., Klein J.B., Müller E.C., Otto A., Busjahn A., Luft F. C, Haller H. (2000) TNF-alpha-mediated neutrophil apoptosis involves Ly-GDI, a Rho GTPase regulator. J Leukoc Biol 68 (2): 277–283.
Yuki K., Hou L. (2020) Role of β2 Integrins in Neutrophils and Sepsis. Infect Immun. 88 (6): e00031. https://doi.org/10.1128/IAI.00031-20
Meszaros A.J., Reichner J.S., Albina J.E. (2000) Macrophage-induced neutrophil apoptosis. J Immunol 165 (1): 435–441.
Kourtzelis I., Hajishengallis G., Chavakis T. (2020) Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front Immunol 11:553. https://doi.org/10.3389/fimmu.2020.00553
Futosi K., Fodor S., Mócsai A. (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 17 (3): 638–650.
Щепеткин И.А., Чердынцева Н.В., Васильев Н.В. (1994) Регуляция функциональной активности нейтрофилов цитокинами. Иммунология. 15 (1): 4–6. 58. [Schepetkin I.A., Cherdyntseva N.V., Vasil’ev N.V. (1994) Regulation of functional activity of neutrophils by cytokines (review). Immunologiya/Immunology (Russia). 15 (1): 4–6. 58. (In Russ)].
McNamee J.P., Bellier P.V., Kutzner B.C., Wilkins R.C. (2005) Effect of pro-inflammatory cytokines on spontaneous apoptosis in leukocyte sub-sets within a whole blood culture. Cytokine. 31 (2): 161–167.
Pericle F., Liu J.H., Diaz J.I., Blanchard D.K., Wei S., Forni G., Djeu J.Y. (1994) Interleukin-2 prevention of apoptosis in human neutrophils. Eur J Immunol 24 (2): 440–444.
Baumann R., Casaulta C., Simon D., Conus S., Yousefi S., Simon H.U. (2003) Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J 17 (15): 2221–2230.
Biffl W.L., Moore E.E., Moore F.A., Barnett C.C.Jr. (1995) Interleukin-6 suppression of neutrophil apoptosis is neutrophil concentration dependent. J Leukoc Biol 58 (5): 582–584.
Dienz O., Rud J.G., Eaton S.M., Lanthier P.A., Burg E., Drew A., Bunn J., Suratt B.T., Haynes L., Rincon M. (2012) Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol 5 (3): 258–266.
Allaeys I., Gymninova I., Canet-Jourdan C., Poubelle P.E. (2014) IL-32γ delays spontaneous apoptosis of human neutrophils through MCL-1, regulated primarily by the p38 MAPK pathway. PLoS One 9 (10): e109256. https://doi.org/10.1371/journal.pone.0109256
Himpe E., Degaillier C., Coppens A., Kooijman R. (2008) Insulin-like growth factor-1 delays Fas-mediated apoptosis in human neutrophils through the phosphatidylinositol-3 kinase pathway. J Endocrinol 199 (1): 69–80.
Klein J.B., Rane M.J., Scherzer J.A., Coxon P.Y., Kettritz R., Mathiesen J.M., Buridi A., McLeish K.R. (2000) Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol 164 (8): 4286–4291.
Щепеткин И.А., Буданова О.П., Малышев И.Ю., Аточин Д.Н. (2018) Молекулярные механизмы апоптоза нейтрофилов (обзор литературы). Патогенез 16 (4): 5–18. [Schepetkin I.A., Budanova O.P., Malyshev I.Yu., Atochin D.N. (2018) Molecular mechanisms of neutrophil apoptosis (review). Patogenez /Pathogenesis (Russia) 16 (4): 5–18. (in Russ)].
Andina N., Conus S., Schneider E.M., Fey M.F., Simon H.U. Induction of Bim limits cytokine-mediated prolonged survival of neutrophils. Cell Death Differ 16 (9): 1248–1255. 2009.
Leavey P.J., Sellins K.S., Thurman G., Elzi D., Hiester A., Silliman C.C., Zerbe G., Cohen J.J., Ambruso D.R. (1998) In vivo treatment with granulocyte colony-stimulating factor results in divergent effects on neutrophil functions measured in vitro. Blood 92 (11): 4366–4374.
Liles W.C., Kiener P.A., Ledbetter J.A., Aruffo A., Klebanoff S.J. (1996) Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med 184 (2): 429–440.
Iwase M., Takaoka S., Uchida M., Kondo G., Watanabe H., Ohashi M., Nagumo M. (2006) Accelerative effect of a selective cyclooxygenase-2 inhibitor on Fas-mediated apoptosis in human neutrophils. Int Immunopharmacol 6 (3): 334–341.
Villunger A., O’Reilly L.A., Holler N., Adams J., Strasser A. (2000) Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase: Regulators of distinct cell death and survival pathways in granulocytes. J Exp Med 192 (5): 647–658.
Ottonello L., Frumento G., Arduino N., Bertolotto M., Dapino P., Mancini M., Dallegri F. (2002) Differential regulation of spontaneous and immune complex-induced neutrophil apoptosis by proinflammatory cytokines. Role of oxidants, Bax and caspase-3. J Leukoc Biol 72 (1): 125–132.
Cowburn A.S., Cadwallader K.A., Reed B.J., Farahi N., Chilvers E.R. (2002) Role of PI3-kinase-dependent Bad phosphorylation and altered transcription in cytokine-mediated neutrophil survival. Blood 100 (7): 2607–2616.
Sakamoto C., Suzuki K., Hato F., Akahori M., Hasegawa T., Hino M., Kitagawa S. (2003) Antiapoptotic effect of granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and cyclic AMP on human neutrophils: protein synthesis-dependent and protein synthesis-independent mechanisms and the role of the Janus kinase-STAT pathway. Int J Hematol 77 (1): 60–70.
Hasegawa T., Suzuki K., Sakamoto C., Ohta K., Nishiki S., Hino M., Tatsumi N., Kitagawa S. (2003) Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood 101 (3): 1164–1171.
Pelletier M., Ratthé C., Girard D. (2002) Mechanisms involved in interleukin-15-induced suppression of human neutrophil apoptosis: role of the anti-apoptotic Mcl-1 protein and several kinases including Janus kinase-2, p 38 mitogen-activated protein kinase and extracellular signal-regulated kinases-1/2. FEBS Lett 532 (1–2): 164–170.
Sakamoto E., Hato F., Kato T., Sakamoto C., Akahori M., Hino M., Kitagawa S. (2005) Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol 78 (1): 301–309.
Aoshiba K., Yasui S., Hayashi M., Tamaoki J., Nagai A. (1999) Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J Immunol 162 (3): 1692–1700.
Scheel-Toellner D., Wang K., Henriquez N.V., Webb P.R., Craddock R., Pilling D., Akbar A.N., Salmon M., Lord J.M. (2002) Cytokine-mediated inhibition of apoptosis in non-transformed T cells and neutrophils can be dissociated from protein kinase B activation. Eur J Immunol 32 (2): 486–493.
Wang K., Scheel-Toellner D., Wong S.H., Craddock R., Caamano J., Akbar A.N., Salmon M., Lord J.M. (2003) Inhibition of neutrophil apoptosis by type 1 IFN depends on cross-talk between phosphoinositol 3-kinase, protein kinase C-delta, and NF-kappa B signaling pathways. J Immunol 171 (2): 1035–1041.
Hirata J., Kotani J., Aoyama M., Kashiwamura S., Ueda H., Kuroda Y., Usami M., Okamura H., Marukawa S. (2008) A role for IL-18 in human neutrophil apoptosis. Shock 30 (6): 628–633.
Keel M., Ungethüm U., Steckholzer U., Niederer E., Hartung T., Trentz O., Ertel W. (1997) Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood 90 (9): 3356–3363.
Cox G. (1996) IL-10 enhances resolution of pulmonary inflammation in vivo by promoting apoptosis of neutrophils. Am J Physiol 271 (4 Pt 1): L566–L571.
Ward C., Murray J., Clugston A., Dransfield I., Haslett C., Rossi A.G. (2005) Interleukin-10 inhibits lipopolysaccharide-induced survival and extracellular signal-regulated kinase activation in human neutrophils. Eur J Immunol 35 (9): 2728–2737.
Bazzoni F., Tamassia N., Rossato M., Cassatella M.A. (2010) Understanding the molecular mechanisms of the multifaceted IL-10-mediated anti-inflammatory response: lessons from neutrophils. Eur J Immunol 40 (9): 2360–2368.
El Kebir D., József L., Filep J.G. (2008) Neutrophil recognition of bacterial DNA and Toll-like receptor 9-dependent and -independent regulation of neutrophil function. Arch Immunol Ther Exp (Warsz) 56 (1): 41–53.
Kinkead L.C., Whitmore L.C., McCracken J.M., Fletcher J.R., Ketelsen B.B., Kaufman J.W., Jones B.D., Weiss D.S., Barker J.H., Allen L.H. (2018) Bacterial lipoproteins and other factors released by Francisella tularensis modulate human neutrophil lifespan: Effects of a TLR1 SNP on apoptosis inhibition. Cell Microbiol 20 (2): e12795. https://doi.org/10.1111/cmi.12795
François S., El Benna J., Dang P.M., Pedruzzi E., Gougerot-Pocidalo M.A., Elbim C. (2005) Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol 174 (6): 3633–3642.
Sunil V.R., Connor A.J., Lavnikova N., Gardner C.R., Laskin J.D., Laskin D.L. (2002) Acute endotoxemia prolongs the survival of rat lung neutrophils in response to 12-O-tetradecanoyl-phorbol 13-acetate. J Cell Physiol 190 (3): 382–389.
Jeong Y.J., Kang M.J., Lee S.J., Kim C.H., Kim J.C., Kim T.H., Kim D.J., Kim D., Núñez G., Park J.H. (2014) Nod2 and Rip2 contribute to innate immune responses in mouse neutrophils. Immunology 143 (2): 269–276.
O'Donnell J.A., Kennedy C.L., Pellegrini M., Nowell C.J., Zhang J.G., O’Reilly L.A., Cengia L., Dias S., Masters S.L., Hartland E.L., Roberts A.W., Gerlic M., Croker B.A. (2015) Fas regulates neutrophil lifespan during viral and bacterial infection. J Leukoc Biol 97 (2): 321–326.
Черешнев В.А., Гусев Е.Ю. (2012) Иммунологические и патофизиологические механизмы системного воспаления. Медицинская иммунология 14 (1–2): 9–20. [Chereshnev V.A., Gusev E.Yu. (2012) Immunological and Pathophysiological mechanisms of Systemic Inflammation. Meditsinskaya Immunologiya/Med Immunol (Russia) 14 (1–2): 9–20. (In Russ)]
Wright H.L., Moots R.J., Edwards S.W. (2014) The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol 10 (10): 593–601.
Wang S.Z., Forsyth K.D. (2000) The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 5 (1): 1–10.
Понякина И.Д. (2003) Активация апоптоза нейтрофилов периферической крови как показатель аутоинтоксикации организма. Клин лаб диагн 7: 19–21. [Poniakina I.D. (2003) Activation of apoptosis of the peripheral blood neutrophils as an indicator of the body autointoxication. Klin Lab Diagn/Clin Lab Diagn (Russia) (7): 19–21. (in Russ)].
Tidow N., Kasper B., Welte K. (1999) SH2-containing protein tyrosine phosphatases SHP-1 and SHP-2 are dramatically increased at the protein level in neutrophils from patients with severe congenital neutropenia (Kostmann’s syndrome). Exp Hematol 27 (6): 1038–1045.
Altznauer F., Conus S., Cavalli A., Folkers G., Simon H.U. (2004) Calpain-1 regulates Bax and subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J Biol Chem 279 (7): 5947–5957.
Abraham E. (2003) Nuclear factor-kappaB and its role in sepsis-associated organ failure J Infect Dis 187 Suppl 2: S364–369.
Aprikyan A.A., Liles W.C., Park J.R., Jonas M., Chi E.Y., Dale D.C. (2000) Myelokathexis, a congenital disorder of severe neutropenia characterized by accelerated apoptosis and defective expression of bcl-x in neutrophil precursors. Blood 95 (1): 320–327.
Brannigan A.E., O’Connell P.R., Hurley H., O’Neill A., Brady H.R., Fitzpatrick J.M., Watson RW (2000) Neutrophil apoptosis is delayed in patients with inflammatory bowel disease. Shock 13 (5): 361–366.
Cohen G., Rudnicki M., Hörl W.H. (2001) Uremic toxins modulate the spontaneous apoptotic cell death and essential functions of neutrophils. Kidney Int Suppl 78: S48–52.
Delogu G., Moretti S., Famularo G., Antonucci A., Signore L., Marcellini S., Lo Bosco L., De Simone C. (2001) Circulating neutrophils exhibit enhanced apoptosis associated with mitochondrial dysfunctions after surgery under general anaesthesia. Acta Anaesthesiol Scand 45 (1): 87–94.
Gamonal J., Sanz M., O’Connor A., Acevedo A., Suarez I., Sanz A., Martínez B., Silva A. (2003) Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 30 (7): 616–623.
Härter L., Mica L., Stocker R., Trentz O., Keel M. (2003) Mcl-1 correlates with reduced apoptosis in neutrophils from patients with sepsis. J Am Coll Surg 197 (6): 964–973.
Hashiguchi N., Ogura H., Tanaka H., Koh T., Nakamori Y., Noborio M., Shiozaki T., Nishino M., Kuwagata Y., Shimazu T., Sugimoto H. (2001) Enhanced expression of heat shock proteins in activated polymorphonuclear leukocytes in patients with sepsis. J Trauma 51 (6): 1104–1109.
Heeney M.M., Ormsbee S.M., Moody M.A., Howard T.A., DeCastro C.M., Ware R.E. (2003) Increased expression of anti-apoptosis genes in peripheral blood cells from patients with paroxysmal nocturnal hemoglobinuria. Mol Genet Metab 78 (4): 291–294.
Hu Z., Sayeed M.M. (2004) Suppression of mitochondria-dependent neutrophil apoptosis with thermal injury. Am J Physiol Cell Physiol 286 (1): C170–178.
Ina K., Kusugami K., Hosokawa T., Imada A., Shimizu T., Yamaguchi T., Ohsuga M., Kyokane K., Sakai T., Nishio Y., Yokoyama Y., Ando T. (1999) Increased mucosal production of granulocyte colony-stimulating factor is related to a delay in neutrophil apoptosis in Inflammatory Bowel disease. J Gastroenterol Hepatol 14 (1): 46–53.
Kobayashi S., Yamashita K., Takeoka T., Ohtsuki T., Suzuki Y., Takahashi R., Yamamoto K., Kaufmann S.H., Uchiyama T., Sasada M., Takahashi A. (2002) Calpain-mediated X-linked inhibitor of apoptosis degradation in neutrophil apoptosis and its impairment in chronic neutrophilic leukemia. J Biol Chem 277 (37): 33968–33977.
Majewska E., Baj Z., Sulowska Z., Rysz J., Luciak M. (2003) Effects of uraemia and haemodialysis on neutrophil apoptosis and expression of apoptosis-related proteins. Nephrol Dial Transplant 18 (12): 2582–2588.
Nopp A., Stridh H., Grönneberg R., Lundahl J. (2002) Lower apoptosis rate and higher CD69 expression in neutrophils from atopic individuals. Inflamm Res 51 (11): 532–540.
Ogura H., Hashiguchi N., Tanaka H., Koh T., Noborio M., Nakamori Y., Nishino M., Kuwagata Y., Shimazu T., Sugimoto H. (2002) Long-term enhanced expression of heat shock proteins and decelerated apoptosis in polymorphonuclear leukocytes from major burn patients. J Burn Care Rehabil 23 (2): 103–109.
O'Neill S., O’Neill A.J., Conroy E., Brady H.R., Fitzpatrick J.M., Watson R.W. (2000) Altered caspase expression results in delayed neutrophil apoptosis in acute pancreatitis. J Leukoc Biol 68 (1): 15–20.
Tsujimoto H., Takeshita S., Nakatani K., Kawamura Y., Tokutomi T., Sekine I. (2001) Delayed apoptosis of circulating neutrophils in Kawasaki disease. Clin Exp Immunol 126 (2): 355–364.
Abdgawad M., Pettersson Å., Gunnarsson L., Bengtsson A.A., Geborek P., Nilsson L., Segelmark M., Hellmark T. (2012) Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PloS one 7 (3): e32439. https://doi.org/10.1371/journal.pone.0032439
Atzeni F., Del Papa N., Sarzi-Puttini P., Bertolazzi F., Minonzio F., Capsoni F. (2004) CD69 expression on neutrophils from patients with rheumatoid arthritis. Clin Exp Rheumatol 22 (3): 331–334.
Christenson K., Björkman L., Tängemo C., Bylund J. (2008) Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J Leukoc Biol 83 (1): 139–148.
Paunel-Görgülü A., Zörnig M., Lögters T., Altrichter J., Rabenhorst U., Cinatl J., Windolf J., Scholz M. (2009) Mcl-1-mediated impairment of the intrinsic apoptosis pathway in circulating neutrophils from critically ill patients can be overcome by Fas stimulation. J Immunol 183 (10): 6198–6206.
Garlichs C.D., Eskafi S., Cicha I., Schmeisser A., Walzog B., Raaz D., Stumpf C., Yilmaz A., Bremer J., Ludwig J., Daniel W.G. (2004) Delay of neutrophil apoptosis in acute coronary syndromes. J Leukoc Biol 75 (5): 828–835.
Taneja R., Parodo J., Jia S.H., Kapus A., Rotstein O.D., Marshall J.C. (2004) Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med 32 (7): 1460–1469.
Kim S.Y., Jun H.S., Mead P.A., Mansfield B.C., Chou J.Y. (2008) Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood 111 (12): 5704–5711.
Klein C. (2011) Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol 29: 399–413.
Tsai C.Y., Li K.J., Hsieh S.C., Liao H.T., Yu C.L. (2019) What’s wrong with neutrophils in lupus? Clin Exp Rheumatol 37 (4): 684–693.
Midgley A., McLaren Z., Moots R.J., Edwards S.W., Beresford M.W. (2009) The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum 60(8):2390–2401.
Midgley A., Mayer K., Edwards S.W., Beresford M.W. (2011) Differential expression of factors involved in the intrinsic and extrinsic apoptotic pathways in juvenile systemic lupus erythematosus. Lupus 20 (1): 71–79.
Rumalla V.K., Calvano S.E., Spotnitz A.J., Krause T.J., Hilkert R.J., Lin E., Lowry S.F. (2002) Alterations in immunocyte tumor necrosis factor receptor and apoptosis in patients with congestive heart failure. Ann Surg 236 (2): 254–260.
Olsson L.M., Lindqvist A.K., Källberg H., Padyukov L., Burkhardt H., Alfredsson L., Klareskog L., Holmdahl R. (2007) A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther 9 (5): R98. https://doi.org/10.1186/ar2299
Olsson L.M., Johansson Å.C., Gullstrand B., Jönsen A., Saevarsdottir S., Rönnblom L., Leonard D., Wetterö J., Sjöwall C., Svenungsson E., Gunnarsson I., Bengtsson A.A., Holmdahl R. (2017) A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis 76 (9): 1607–1613.
Zhao J., Ma J., Deng Y., Kelly J.A., Kim K., Bang S.Y., Lee H.S., Li Q.Z., Wakeland E.K., Qiu R., Liu M., Guo J., Li Z., Tan W., Rasmussen A., Lessard C.J., Sivils K.L., Hahn B.H., Grossman J.M., Kamen D.L., Gilkeson G.S., Bae S.C., Gaffney P.M., Shen N., Tsao B.P. (2017) A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet 49 (3): 433–437.
Dhillon S.S., Fattouh R., Elkadri A., Xu W., Murchie R., Walters T., Guo C., Mack D., Huynh H.Q., Baksh S., Silverberg M.S., Griffiths A.M., Snapper S.B., Brumell J.H., Muise A.M. (2014) Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 147 (3): 680–689.
Nauseef W.M., Clark R.A. (2019) Intersecting Stories of the Phagocyte NADPH Oxidase and Chronic Granulomatous Disease. Methods Mol Biol 1982: 3–16.
Courtney P.A., Crockard A.D., Williamson K., Irvine A.E., Kennedy R.J., Bell A.L. (1999) Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis 58 (5): 309–314.
Harper L., Cockwell P., Adu D., Savage C.O. (2001) Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int 59 (5): 1729–1738.
Chen M.F., Chen J.C., Chiu D.F., Ng C.J., Shyr M.H., Chen H.M. (2001) Prostacyclin analogue (OP-2507) induces delayed ex vivo neutrophil apoptosis and attenuates reperfusion-induced hepatic microcirculatory derangement in rats. Shock 16 (6): 473–478.
Chitnis D., Dickerson C., Munster A.M., Winchurch R.A. (1996) Inhibition of apoptosis in polymorphonuclear neutrophils from burn patients. J Leukoc Biol 59 (6): 835–839.
Ertel W., Keel M., Infanger M., Ungethüm U., Steckholzer U., Trentz O. (1998) Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein-tyrosine phosphorylation. J. Trauma 44 (5): 767–775.
Jimenez M.F., Watson R.W., Parodo J., Evans D., Foster D., Steinberg M., Rotstein O.D., Marshall J.C. (1997) Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg 132 (12): 1263–1269.
Inaba M., Takahashi T., Kumeda Y., Kato T., Hato F., Yutani Y., Goto H., Nishizawa Y., Kitagawa S. Increased basal phosphorylation of mitogen-activated protein kinases and reduced responsiveness to inflammatory cytokines in neutrophils from patients with rheumatoid arthritis. Clin Exp Rheumatol 26 (1): 52–60. 2008
Elliott E.R., Van Ziffle J.A., Scapini P., Sullivan B.M., Locksley R.M., Lowell (2011) CA Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol 187 (8): 4319–4330.
Fudala R., Krupa A., Matthay M.A., Allen T.C., Kurdowska A.K. (2007) Anti-IL-8 autoantibody:IL-8 immune complexes suppress spontaneous apoptosis of neutrophils. Am J Physiol Lung Cell Mol Physiol 293 (2): L364–L374.
Raison C.L., Lowry C.A., Rook G.A. (2010) Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry 67 (12): 1211–1224.
Черных Е.И., Языков К.Г., Семке В.Я. (2002) Апоптоз лейкоцитов периферической крови, индуцированный действием гипертермии и преднизолона, у лиц с расстройством адаптации. Бюл. экспер. биологии и медицины 134 (12): 617–619. [Chernykh E.I., Yazykov K.G., Semke V.Y. (2002) Apoptosis in peripheral blood leukocytes induced by hyperthermia and prednisolone in patients with dysadaptation. Bull Exp Biol Med 134 (6): 531–533.]
Zhang X., Moilanen E., Kankaanranta H. (2001) Beclomethasone, budesonide and fluticasone propionate inhibit human neutrophil apoptosis. Eur J Pharmacol 431 (3): 365–371.
Perttunen H., Moilanen E., Zhang X., Barnes P.J., Kankaanranta H. (2008) Beta2-agonists potentiate corticosteroid-induced neutrophil survival. COPD 5 (3): 163–169.
Stankova J., Turcotte S., Harris J., Rola-Pleszczynski M. (2002) Modulation of leukotriene B4 receptor-1 expression by dexamethasone: potential mechanism for enhanced neutrophil survival J Immunol 168 (7): 3570–3576.
Ruiz L.M., Bedoya G., Salazar J., García de O.D., Patiño P.J. (2002) Dexamethasone inhibits apoptosis of human neutrophils induced by reactive oxygen species. Inflammation 26 (5): 215–222.
Ronchetti S., Ricci E., Migliorati G., Gentili M., Riccardi C. (2018) How Glucocorticoids Affect the Neutrophil Life. Int J Mol Sci 19 (12): 4090. https://doi.org/10.3390/ijms19124090
Pace T.W., Hu F., Miller A.H. (2007) Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21 (1): 9–19.
Heasman S.J., Giles K.M., Ward C., Rossi A.G., Haslett C., Dransfield I. (2003) Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol 178 (1): 29–36.
Sugimoto M.A., Vago J.P., Teixeira M.M., Sousa L.P. (2016) Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res 2016: 8239258. https://doi.org/10.1155/2016/8239258
Chellappan D.K., Yee L.W., Xuan K.Y., Kunalan K., Rou L.C., Jean L.S., Ying L.Y., Wie L.X., Chellian J., Mehta M., Satija S., Singh S.K., Gulati M., Dureja H., Da Silva M.W., Tambuwala M.M., Gupta G., Paudel K.R., Wadhwa R., Hansbro P.M., Dua K. (2020) Targeting neutrophils using novel drug delivery systems in chronic respiratory diseases. Drug Dev Res 81 (4): 419–436.
Rossi A.G., Sawatzky D.A., Walker A., Ward C., Sheldrake T.A., Riley N.A., Caldicott A., Martinez-Losa M., Walker T.R., Duffin R., Gray M., Crescenzi E., Martin M.C., Brady H.J., Savill J.S., Dransfield I., Haslett C. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 12 (9): 1056–1064. 2006
Koedel U., Frankenberg T., Kirschnek S., Obermaier B., Häcker H., Paul R., Häcker G. (2009) Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog 5 (5): e1000461. https://doi.org/10.1371/journal.ppat.1000461
Sousa L.P., Lopes F., Silva D.M., Tavares L.P., Vieira A.T., Rezende B.M., Carmo A.F., Russo R.C., Garcia C.C., Bonjardim C.A., Alessandri A.L., Rossi A.G., Pinho V., Teixeira M.M. (2010) PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-kappaB-independent manner. J Leukoc Biol 87 (5): 895–904.
McGrath E.E., Marriott H.M., Lawrie A., Francis S.E., Sabroe I., Renshaw S.A., Dockrell D.H., Whyte M.K. (2011) TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol 90 (5): 855–865.
Meijer L., Nelson D.J., Riazanski V., Gabdoulkhakova A.G., Hery-Arnaud G., Le Berre R., Loaëc N., Oumata N., Galons H., Nowak E., Gueganton L., Dorothée G., Prochazkova M., Hall B., Kulkarni A.B., Gray R.D., Rossi A.G., Witko-Sarsat V., Norez C., Becq F., Ravel D., Mottier D., Rault G. (2016) Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J Innate Immun 8 (4): 330–349.
Brostjan C., Oehler R. (2020) The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov 6: 26. https://doi.org/10.1038/s41420-020-0255-6
Miles K., Clarke D.J., Lu W., Sibinska Z., Beaumont P.E., Davidson D.J., Barr T.A., Campopiano D.J., Gray M. (2009) Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol 183 (3): 2122–2132.
Ren Y., Xie Y., Jiang G., Fan J., Yeung J., Li W., Tam P.K., Savill J. (2008) Apoptotic cells protect mice against lipopolysaccharide-induced shock. J Immunol 180 (7): 4978–4985.
Lawrence S.M., Corriden R., Nizet V. (2020) How Neutrophils Meet Their End. Trends Immunol 41 (6): 531–544.
Kroemer G., Levine B. (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9 (12): 1004–1010.
Jorgensen I., Rayamajhi M., Miao E.A. (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17 (3): 151–164.
Germic N., Frangez Z., Yousefi S., Simon H.U. (2019) Regulation of the innate immune system by autophagy: neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ 26 (4): 703–714.
Chargui A., Cesaro A., Mimouna S., Fareh M., Brest P., Naquet P., Darfeuille-Michaud A., Hébuterne X., Mograbi B., Vouret-Craviari V., Hofman P. (2012) Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One 7 (12): e51727. https://doi.org/10.1371/journal.pone.0051727
Ullah I., Ritchie N.D., Evans T.J. (2017) The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun 23 (5): 413–423.
Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., Brunner T., Simon H.U. (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8 (10): 1124–1132.
Larochelle B., Flamand L., Gourde P., Beauchamp D., Gosselin J. (1998) Epstein-Barr virus infects and induces apoptosis in human neutrophils. Blood 92 (1): 291–299.
Crepaldi L., Gasperini S., Lapinet J.A., Calzetti F., Pinardi C., Liu Y., Zurawski S., de Waal Malefyt R., Moore K.W., Cassatella M.A. (2001) Up-regulation of IL-10R1 expression is required to render human neutrophils fully responsive to IL-10. J Immunol 167 (4): 2312–2322.
Hotta K., Niwa M., Hara A., Ohno T., Wang X., Matsuno H., Kozawa O., Ito H., Kato K., Otsuka T., Matsui N., Uematsu T. (2001) The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-kappa B activation. Eur J Pharmacol 433 (1): 17–27.
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии