

Журнал эволюционной биохимии и физиологии, 2022, T. 58, № 5, стр. 349-364
РЕГУЛЯЦИЯ КОНЦЕНТРАЦИЙ ИОНОВ КАЛИЯ И ХЛОРИД-ИОНОВ В НЕРВНОЙ ТКАНИ КАК МЕТОД ПРОТИВОСУДОРОЖНОЙ ТЕРАПИИ
Е. Ю. Проскурина 1, 2, *, А. В. Зайцев 2
1 Национальный медицинский исследовательский центр им. В.А. Алмазова Минздрава России
Санкт-Петербург, Россия
2 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: elena.yu.proskurina@gmail.com
Поступила в редакцию 30.08.2021
После доработки 05.08.2022
Принята к публикации 09.08.2022
- EDN: DANOFV
- DOI: 10.31857/S0044452922050096
Аннотация
При некоторых патологических состояниях, например, фармакорезистентной эпилепсии, эпилептическом статусе или определенных формах генетических аномалий спайковая активность ГАМКергических интернейронов может усиливать процессы возбуждения в нервной ткани и провоцировать генерацию иктального разряда. В результате противосудорожные средства, действующие на ГАМКергическую систему, могут оказаться неэффективными или даже усиливать судорожную активность. Этот парадоксальный эффект работы тормозной системы обусловлен нарушением ионного баланса в нервной ткани. В данном обзоре рассмотрены механизмы инициации иктального разряда в нейронных сетях из-за нарушения баланса хлорид-ионов и ионов калия, а также возможные методы воздействия на регуляцию ионных концентраций. Для подавления эпилептической активности, вызванной дисбалансом ионов, эффективным может оказаться как усиление (или ослабление) активности определенных транспортеров ионов и ионных насосов в нейронах, так и их дополнительная экспрессия с помощью генной терапии. В поддержании необходимых концентраций ионов калия и хлора в нервной ткани важное значение имеют NaK-помпа, NKCC1- и KCC2-котранспортеры, которые ранее неоднократно рассматривались в качестве фармакологических мишеней для противоэпилептического воздействия. Препятствием для работы в этом направлении является отсутствие достаточно селективных фармакологических инструментов для воздействия на них, а также методов доставки препаратов к эпилептическому очагу. Более перспективным направлением представляется использование методов генной терапии, таких как гиперэкспрессия транспортера KCC2 в эпилептическом очаге. Другим возможным направлением может стать применение оптогенетических инструментов: специально сконструированных светочувствительных ионных помп или каналов. В этом случае энергия фотонов может быть использована для создания требуемых градиентов хлорид-ионов и ионов калия, однако и у этих методов пока есть существенные ограничения, которые затрудняют их быстрое введение в практику.
ВВЕДЕНИЕ
Эпилепсия – распространенное неврологическое заболевание, основным проявлением которого являются повторяющиеся судорожные приступы. Примерно у 30% больных эпилептические приступы плохо поддаются фармакологическому контролю [1]. Это подтверждает необходимость поиска новых методов лечения, воздействующих на альтернативные мишени и патофизиологические механизмы генерации судорожной активности. В настоящее время для лечения эпилепсии используют препараты, ключевой механизм действия которых заключается в модуляции работы потенциал-зависимых натриевых и кальциевых каналов, либо эти лекарства обеспечивают потенцирование тормозных ГАМК-рецепторов или ингибирование возбуждающих глутаматных рецепторов [2].
Среди множества патологических изменений, наблюдаемых в эпилептическом очаге, в том числе при фармакорезистентной эпилепсии, следует отдельно выделить нарушение ионного баланса, приводящее к нарушению функций тормозной ГАМКергической системы и гипервозбудимости нейронов. К наиболее выраженному эпилептогенному эффекту приводят накопление внутриклеточных хлорид-ионов и повышение внеклеточной концентрации катионов калия. Подробный анализ ионной динамики в здоровой и эпилептической ткани можно найти в работе Raimondo и соавт. [3]. Дисбаланс хлорид-ионов в эпилептической ткани детально обсуждался в обзорах [4, 5]. Эпилептогенный эффект ионов калия описан de Curtis и соавт. [6]. Модельные исследования также указывают на критическую роль ионной динамики в ткани мозга для генерации как интериктальных, так и иктальных разрядов [7, 8].
Таким образом, снизить гипервозбудимость нейронов и восстановить функции ГАМКергической системы в эпилептическом очаге возможно, если направленно изменить ионный баланс в данной области мозга. Цель данного обзора – проанализировать и описать наиболее перспективные способы воздействия на ионный баланс в эпилептическом очаге, что может стать основой для разработки новых подходов лечения фармакорезистентной эпилепсии. Мы последовательно рассмотрим, какие изменения ионной динамики наблюдаются при эпилептической активности, к каким последствиям они приводят, а затем опишем возможные способы воздействия на ионную динамику.
ДИСБАЛАНС ИОНОВ КАЛИЯ ПРИ ЭПИЛЕПТИЧЕСКОЙ АКТИВНОСТИ
Потенциал покоя нейрона определяется в первую очередь градиентом ионов калия внутри и вне клетки. В норме концентрация ионов калия во внеклеточной среде мозга составляет 3 мМ, а в нейронах млекопитающих 140 мM. Согласно уравнению Нернста, потенциал реверсии ионов калия ${{E}_{{{\text{rev}}}}}$ зависит от концентрации этих ионов на внутренней стороне мембраны ${{[{{{\text{K}}}^{ + }}]}_{i}}$ и наружней ${{[{{{\text{K}}}^{ + }}]}_{i}}$ по следующей формуле (Ур. 1):
(1)
${{E}_{{{\text{rev}}}}} = \frac{{RT}}{{zF}}\ln \left( {\frac{{{{{[{{{\text{K}}}^{ + }}]}}_{o}}}}{{{{{[{{{\text{K}}}^{ + }}]}}_{i}}}}} \right),$Движущая сила электрохимического градиента ионов калия $\Delta V$ представляет собой разницу между мембранным потенциалом $V(t)$ и потенциалом реверсии ионов калия ${{E}_{{{\text{rev}}}}}$ (Ур. 2):
(2)
$\Delta V = V(t)--\frac{{RT}}{{zF}}\ln \left( {\frac{{{{{[{{{\text{K}}}^{ + }}]}}_{o}}}}{{{{{[{{{\text{K}}}^{ + }}]}}_{i}}}}} \right)~.$Согласно уравнению 2, повышение концентрации ионов калия во внеклеточной среде в два раза вызовет деполяризацию мембраны на 19 мВ.
Экспериментальную регистрацию внеклеточной концентрации ионов калия обычно производят с помощью калий-чувствительного электрода [9]. Метод изготовления калий-чувствительных электродов для нейрофизиологических экспериментов подробно описан в работах [10, 11].
Впервые повышение концентрации ионов калия во время генерации эпилептиформных разрядов in vitro было описано еще в 70-е годы прошлого столетия [12–14]. В дальнейшем это явление было независимо подтверждено в различных лабораториях. Пример записи регистрации внеклеточной концентрации ионов калия c помощью монополярного микроэлектрода одновременно с регистрацией спайковой активности или постсинаптических токов нейронов методом патч-кламп в срезах гиппокампа и энторинальной коры крысы в 4‑аминопиридиновой модели эпилептиформной активности, проведенной в нашей лаборатории, показан на рис. 1 (приведено из [11]).
Рис. 1.
Одновременная регистрация внеклеточной концентрации ионов калия с помощью калий-чувствительного электрода (зеленая кривая) и постсинаптических токов репрезентативного нейрона (черная кривая) в энторинальной коре крысы Вистар в 4-аминопиридиновой модели эпилептиформной активности in vitro (a). Потенциал для записи постсинаптических токов удерживался на –27 мВ. Иктальный разряд, отмеченный синей полосой, представлен в увеличенном масштабе на панели b. Воспроизведено из [11].
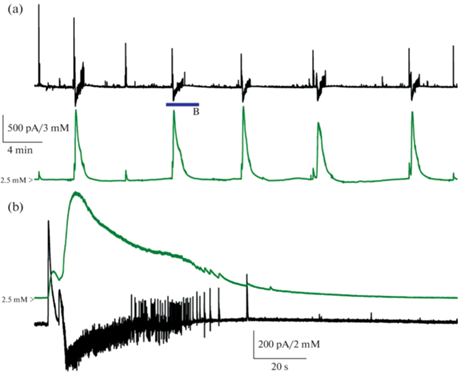
Внеклеточная концентрация ионов калия в каждый момент времени вычисляется по формуле (Ур. 3):
(3)
${{[{{{\text{K}}}^{ + }}]}_{o}}(t) = {{[{{{\text{K}}}^{ + }}]}_{{o,~{\text{baseline}}}}}~{{e}^{{SV(t)}}},$При отсутствии спайковой активности в нейронной сети внеклеточная концентрация ионов калия не изменяется и соответствует значению в омывающем срез растворе (в приведенном примере 2.5 мМ). Во время эпилептических разрядов внеклеточная концентрация ионов калия повышается в разы, достигая в тоническую фазу иктального разряда около 10 мМ и деполяризуя мембрану, соответственно делая нейроны более возбудимыми.
ИЗБЫТОЧНАЯ ВНУТРИКЛЕТОЧНАЯ КОНЦЕНТРАЦИЯ ХЛОРИД-ИОНОВ ПРИ ЭПИЛЕПТИЧЕСКОЙ АКТИВНОСТИ
Функцию торможения в головном мозге выполняет ГАМКергическая система. Медиатор ГАМК оказывает быстрый и сильный гиперполяризующий эффект на постсинаптический нейрон при связывании с ГАМКА-рецептором, являющимся ионотропным (хлорным) каналом. Как правило, при активации этих рецепторов хлорид-ионы по электрохимическому градиенту перемещаются внутрь клетки. Однако ток хлорид-ионов в клетку наблюдается только в том случае, когда потенциал реверсии, определяемый преимущественно равновесным хлорным потенциалом, ниже мембранного потенциала покоя клетки [15]. В противном случае ГАМК не будет оказывать торможения и может даже деполяризовать постсинаптический нейрон.
Так как потенциал реверсии в значительной степени определяется изменением внутриклеточной концентрации хлорид-ионов, то необходимо было разработать доступный способ регистрации. Нетоксичный метод измерения внутриклеточной концентрации хлорид-ионов был разработан в 2000 г. Kuner и Augustine [16] на основе физического явления “флуоресцентный резонансный перенос энергии”. Суть этого явления заключается в том, что между двумя хромофорами происходит безызлучательный перенос энергии от донора, находящегося в возбужденном состоянии, на акцептор через диполь-дипольное взаимодействие. Характерными чертами данного процесса являются тушение флуоресценции донора и возникновение более длинноволновой флуоресценции акцептора. Изучая межбелковые взаимодействия, эти исследователи заметили, что эффективность флуоресцентного резонансного переноса энергии между циановым и желтым флуоресцентными белками зависела от хлорид-ионов. Поэтому авторы предложили оптический индикатор хлорид-ионов в виде соединения желтого и цианового флуоресцентных белков Clomeleon и успешно апробировали его на первичной культуре нейронов гиппокампа [16].
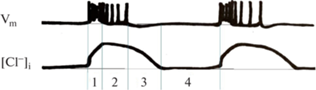
Впервые измерили концентрацию хлорид-ионов во время генерации иктального разряда Glykys и соавт в 2014 г., после экспрессии белка Clomeleon в органотипической культуре гиппокампа, совместив двухфотонную микроскопию и внеклеточные отведения [17]. Если в состоянии покоя концентрация хлорид-ионов составила 25.4 ± 7.4 мМ, то во время генерации иктального разряда она повышалась до 40.8 ± 13.8 мM. Высокая внутриклеточная концентрация хлорид-ионов поддерживается на протяжении генерации эпилептиформной активности и восстанавливается до базового уровня только при продолжительном отсутствии синхронной популяционной активности (рис. 2).
Рис. 2.
Схематическое изображение динамики внутриклеточной концентрации хлорид-ионов [Cl–]i, во время иктальных разрядов в модели эпилептиформной активности in vitro. Во время иктального разряда наблюдается спайковая активность репрезентативного нейрона коры, которая представлена записью мембранного потенциала Vm. Цифры на рисунке: 1 – тоническая фаза иктального разряда, 2 – клоническая фаза иктального разряда, 3 – гиперполяризация, 4 – состояние покоя (по материалам [17]).
Разработаны альтернативные способы оценки концентрации хлорид-ионов с помощью других сенсоров. Например, довольно широко используется ClopHensorN, чувствительный, кроме [Cl–]i, также и к pH [18–22]. Burman и соавт. измерили динамику внутриклеточной концентрации хлорид-ионов во время генерации эпилептиформных разрядов в срезах органотипической культуры гиппокампа [22]. Было показано, что во время эпилептиформной активности внутриклеточная концентрация хлорид-ионов возрастает, соответственно во время генерации эпилептических разрядов потенциал реверсии повышается на 30–40 мВ, а по окончании эпилептиформной активности возвращается к базовому уровню.
ЭПИЛЕПТОГЕННЫЕ ЭФФЕКТЫ ДИСБАЛАНСА ХЛОРИД-ИОНОВ И КАТИОНОВ КАЛИЯ
В норме в зрелых нейронах хлорид-ионы переносятся в межклеточное пространство за счет K+-Cl–котранспортера (KCC2-котранспортера): электрохимический градиент ионов калия способствует выведению хлорид-иона вместе с ионом калия [23]. Таким образом, КСС2-котранспортер препятствует накоплению хлорид-ионов внутри нейронов в результате работы тормозной системы и повышению потенциала реверсии ГАМКА-рецепторов.
Функциональная активность КСС2-котранспортера регулируется рядом механизмов и при высокой активации нейронных сетей обычно резко снижается. Последние данные свидетельствуют о том, что скорость ионного транспорта, а также стабильность и перемещение KCC2 по клеточной поверхности и плазмалемме быстро и обратимо модулируются (де)фосфорилированием некоторых сериновых, треониновых и тирозиновых остатков в С-концевой части этого белка [24].
Этот механизм выведения хлорид-ионов из клеток довольно уязвим при различных повреждениях в нервной системе, что ведет к повышению возбудимости нейронных сетей и эпилептизации ткани мозга. Kaila и соавт. [25] в своем обзоре предлагают следующий механизм развития таких нарушений. В ходе повторяющихся судорожных припадков происходит, с одной стороны, прогрессирующая интернализация постсинаптических ГАМКА-рецепторов [26], а с другой – быстрое снижение экспрессии KCC2 [27, 28], что совместно ведет к длительному накоплению хлорид-ионов в нейронах и последующему ослаблению торможения. Конкретный молекулярный механизм снижения экспрессии KCC2 до конца не выяснен, но наиболее известными на сегодняшний день патологическими ингибиторами функции KCC2 являются избыток глутамата [29] и высокие уровни нейротрофического фактора мозга (BDNF) [30].
Кроме приобретенных нарушений хлорного баланса, могут встречаться и наследственные нарушения. Следует отметить, что при некоторых формах наследственной эпилепсии обнаружены мутации гена SLC12A5, кодирующего KCC2-котранспортер. Например, у австралийской семьи с наследственными фебрильными судорогами и во франко-канадской когорте с тяжелой генетической генерализованной эпилепсией (GGE) обнаружены гетерозиготные миссенс-полиморфизмы в SLC12A5, вызывающих снижение KCC2-зависимой способности к выведению хлорид-ионов [31]. У пациентов с тяжелым, рано проявляющимся заболеванием менделевского типа, названным “эпилепсией младенчества с мигрирующими фокальными припадками” (EIMFS), обнаружена рецессивная мутация в SLC12A5, ведущая к потере функции [32]. Также показано, что полиморфизм гена, кодирующего KCC2 по rs2297201, может быть прогностическим генетическим маркером фебрильных судорог [33], более подробные сведения о мутациях гена KCC2-котранспортера, приводящих к эпилепсии, можно найти в обзорах [31, 34].
Проэпилептогенное действие высокой концентрации внеклеточного калия подтверждается многочисленными экспериментальными данными. Например, повышение концентрации хлорида калия в омывающем растворе с 2.5 до 10 мМ приводит к генерации эпилептиформной активности в переживающем срезе гиппокампа ювенильной мыши [35]. Во время пре- или интериктальных разрядов, обеспеченных в основном синхронной активностью интернейронов, происходит повышение внеклеточной концентрации ионов калия [11, 36], которое, в свою очередь, может спровоцировать генерацию иктального разряда. Основными источниками внеклеточного калия выступают различные типы калиевых каналов и уже описанный выше KCC2-котранспортер.
Так, во время генерации потенциалов действия ионы калия попадают в межклеточное пространство через потенциал-чувствительные калиевые каналы [37], а KCC2-котранспортер обеспечивает совместный выход ионов калия и хлорид-ионов в межклеточное пространство. В результате изменяется потенциал реверсии калиевого тока у всех нейронов, включая пирамидные клетки и соответственно мембранный потенциал становится более деполяризованным у всех нейронов. Это приводит к гипервозбудимости ткани мозга.
Эффективное удаление избыточного K+ из внеклеточного пространства во время активности нейронов имеет первостепенное значение. В этом процессе ключевую роль играют астроциты. Нормализацию внеклеточной концентрации ионов калия обеспечивают NaK-помпа и калиевые каналы внутреннего выпрямления (KIR), а также быстрая пространственная буферизация, обеспеченная щелевыми контактами между астроцитами [38]. Нарушения в работе любого из этих механизмов могут вести к развитию эпилепсии. Есть данные о том, что мутации астроцитарных калиевых каналов внутреннего выпрямления KIR4.1 обнаружены у пациентов, страдающих эпилепсией [39–41]. У больных эпилепсией описаны также мутации субъединиц NaK-помпы, совместимые с жизнью [42]. В случае ювенильной миоклонической эпилепсии описаны мутации гена коннексина 36, обеспечивающего щелевой контакт между астроцитами [43].
МЕХАНИЗМЫ ГЕНЕРАЦИИ ЭПИЛЕПТИЧЕСКОЙ АКТИВНОСТИ, ОБУСЛОВЛЕННЫЕ ГАМКЕРГИЧЕСКИМИ ИНТЕРНЕЙРОНАМИ
Нарушение ионного баланса может изменить функции ГАМКергических интернейронов. В норме ГАМКергическая система в головном мозге выполняет функцию торможения, поэтому оказывает выраженное противоэпилептическое действие. Однако накоплено много экспериментальных и клинических свидетельств ее проэпилептического действия при некоторых патологических состояниях [4, 36, 44–49]. Механизмы генерации эпилептической активности за счет работы ГАМКергической системы подробно обсуждаются в ряде ранее опубликованных обзоров [4, 5], поэтому здесь мы только кратко опишем наиболее часто встречающиеся механизмы.
Иктальный разряд может быть спровоцирован краткой синхронной активацией, а затем выключением тормозных интернейронов в условиях высокой эпилептической готовности. В этом случае сразу после выключения торможения наблюдается синхронизованная генерация потенциалов действия пирамидными нейронами (post-inhibitory rebound synchronization, PIRS). Такой эффект описан в оптогенетических экспериментах с кратковременной 30-миллисекундной фотостимуляцией ГАМКергических интернейронов верхних слоев соматосенсорной коры ювенильной мыши (линия VGAT-ChR2) в 4-аминопиридиновой модели in vitro. Фотостимуляция вызывает одновременную спайковую активность ГАМКергических интернейронов, а выключение света – синхронное прекращение их активности. Сразу после выключения фотостимуляции в срезе мозга генерировался иктальный (или интериктальный) разряд [47]. Используя перфорированный патч и мультиэлектродные отведения, исследователи показали, что во время фотостимуляции интернейронов наблюдается гиперполяризация пирамидных нейронов, а после выключения фотостимуляции возникает деполяризация пирамидных нейронов, и развивается эпилептическое событие. Таким образом, при одновременном выключении многих интернейронов происходит синхронная активация пирамидных нейронов, которая может реализоваться в иктальный или интериктальный разряд. Схожую картину наблюдали в ткани мозга человека, полученной из эпилептического очага, после локальной аппликации ГАМК (100 мМ), а также in vivo на анестезированной взрослой мыши VGAT-ChR2 при фотостимуляции интернейронов [47]. Аналогичный результат был продемонстрирован in vitro при стимуляции только парвальбумин-положительных быстроразряжающихся интернейронов [50].
Другой механизм может быть задействован при более длительной синхронизованной спайковой активности интернейронов (и опционально пирамидных нейронов). Такая активность нейронной сети повышает внеклеточную концентрацию ионов калия настолько, что приближает мембранный потенциал нейронов к порогу возникновения потенциала действия. В результате может активироваться достаточное число для генерации иктального разряда пирамидных нейронов. В пользу этого механизма свидетельствует ряд экспериментальных данных: во время генерации ГАМК-глутаматных интериктальных разрядов регистрируется существенный скачок внеклеточной концентрации ионов калия [36, 51]; в 4-аминопиридиновой модели эпилептиформной активности в срезах энторинальной коры иктальному разряду предшествуют ГАМК-глутаматные преиктальные разряды, зачастую внеклеточная концентрация ионов калия не успевает вернуться к базовому уровню в промежутке между преиктальными разрядами, поэтому обеспечивает больший скачок концентрации [11, 52]. Таким образом может быть достигнуто пороговое значение концентрации, достаточное для генерации иктального разряда.
Одновременно может происходить накопление хлорид-ионов в пирамидном нейроне за счет продолжительной активации ГАМКА-рецепторов, что повышает потенциал реверсии ГАМК. Поэтому гиперполяризующее действие ГАМК может меняться на деполяризующее. Мы предполагаем, что в развитии эпилептогенного эффекта, обеспечиваемого ГАМКергическими интернейронами, важную роль может играть локальное изменение концентрации хлорид-ионов, а также некоторые специализированные тормозные синапсы вблизи начального сегмента аксона постсинаптического пирамидного нейрона. В пользу такого предположения свидетельствует ряд наших собственных и полученных в других лабораториях экспериментальных данных, а также модельные расчеты. Так, в 4-аминопиридиновой модели эпилептиформной активности в срезах гиппокампа и энторинальной коры ювенильной крысы, во время генерации преиктальных разрядов, мы неоднократно регистрировали низкопороговые потенциалы действия в пирамидных нейронах [52], что может быть обусловлено тем, что они вызваны интернейронами.
Известно, что один из подтипов парвальбумин-содержащих интернейронов, клетки-канделябры, формируют особые аксо-аксональные синапсы, обеспечивая выброс ГАМК в области начального сегмента аксона пирамидных нейронов [53–55]. Было показано, что аппликация агониста ГАМКА-рецепторов мусцимола в этой области аксона уменьшает вероятность генерации потенциала действия нейроном при низкой внутриклеточной концентрации хлорид-ионов и увеличивает эту вероятность при высокой концентрации [56]. Получены экспериментальные свидетельства того, что количество KCC2-котранспортера на аксоне нейрона значительно меньше, чем на дендритах и соме [57, 58], тогда как в этой области довольно высока экспрессия NKCC1-котранспортера. Эти особенности локальной экспрессии котранспортеров обеспечивают накопление хлорид-ионов в начальном сегменте аксона [59]. Поскольку объем аксона мал, то продолжительная ГАМКергическая активация приводит к локальному накоплению хлорид-ионов, обеспечивая деполяризующее действие ГАМК. В этом случае активность ГАМКергических клеток-канделябров может приводить к синхронизованному возбуждению пирамидных нейронов и инициации иктального разряда. Однако эта гипотеза требует дальнейших экспериментальных исследований.
Кроме клеток-канделябров важную роль в этих процессах может играть и другой подтип парвальбумин-содержащих интернейронов – корзинчатые интернейроны, образующие синаптические контакты в основном на теле и проксимальных дендритах пирамидных нейронов [60, 61]. Оптогенетическая деполяризация парвальбуминовых интернейронов в пилокарпиновой модели фокальной эпилепсии in vivo меняет эффект с анти- на проэпилептический в течение нескольких секунд [4]. Magloire и соавт. также отмечают, что проэпилептического действия нет при фотостимуляции SOM-интернейронов, синапсы которых расположены преимущественно на дендритах пирамидных нейронов, а также показывают, что проэпилептическое действие фотостимуляции парвальбуминовых интернейронов отсутствует при гиперэкспрессии KCC2-котранспортера [4].
ФАРМАКОЛОГИЧЕСКИЕ ВОЗМОЖНОСТИ РЕГУЛЯЦИИ ИОННОГО БАЛАНСА В ЦНС
Если нарушение ионного баланса ведет к эпилептизации нервной ткани, то фармакологическое воздействие с целью восстановления ионного баланса может стать одним из перспективных способов нормализации функций мозга.
Фармакологическая регуляция баланса хлорид-ионов
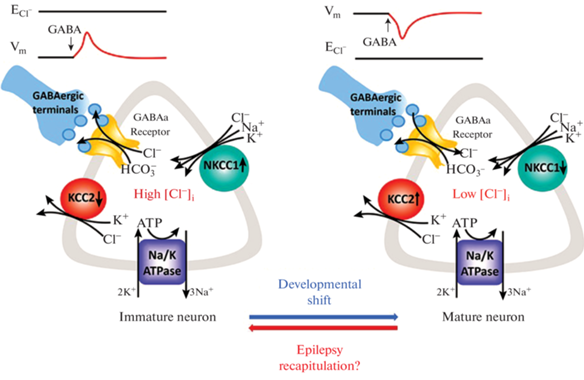
Важнейшую роль в естественной регуляции внутриклеточной концентрации хлорид-ионов осуществляют KCC2- и NKCC1-котранспортеры, действие которых прямо противоположно. KCC2-котранспортер выводит избытки хлорид-ионов из нейрона, тогда как NKCC1, наоборот, способствует их накоплению. В раннем онтогенезе преобладает NKCC1, но по мере созревания нейронов усиливается вклад KCC2 (рис. 3). Эти изменения связывают с ослаблением функций STE20/SPS1-связанной пролин-аланин-обогащенной протеинкиназы (SPAK), которая фосфорилирует как KCC2, так и NKCC1, но оказывает противоположное воздействие на их функции: снижает активность KCC2 и повышает активность NKCC1 [62]. При развитии эпилепсии активность NKCC1 может стать преобладающей [63]. Поэтому можно ожидать противоэпилептический эффект при ингибировании NKCC1 и проэпилептический при инактивации KCC2 [64], тогда как выраженный противоэпилептический эффект можно ожидать только при усилении функций КСС2.
Рис. 3.
Механизмы регуляции концентрации Cl–, лежащие в основе действия ГАМК в незрелых (слева) и зрелых нейронах ЦНС (справа). NKCC1 является основным транспортером в незрелых нейронах ЦНС, который опосредует поступление Cl– в клетку. KCC2 является основным K-Cl котранспортером в зрелых нейронах ЦНС и обеспечивает выведение Cl– из клетки. При некоторых патофизиологических состояниях, таких как эпилепсия, нейроны испытывают “рекапитуляцию” и дедифференцировку на более ранние стадии развития. Остальные пояснения приведены в тексте. Схема воспроизведена из [63].
В настоящее время набор фармакологических агентов, воздействующих на катион-хлорные котранспортеры, сильно ограничен и представлен в основном диуретиками (фуросемид и буметанид) и их производными. Для ингибирования NKCC1 обычно применяют буметанид. Буметанид имеет в ~500 раз большее сродство к NKCC1 (Ki ~0.1 мкМ), чем к KCC2 (Ki ~25–50 мкМ), и поэтому низкие концентрации буметанида (2–10 мкМ) могут быть легко использованы для ингибирования NKCC1 in vitro без существенного влияния на KCC2 [65]. Однако при более высоких концентрациях буметанид будет блокировать оба транспортера. Фуросемид еще менее специфичен и воздействует не только на катион-хлорные котранспортеры, но и Na+-независимый Cl–/${\text{HCO}}_{3}^{ - }$-обменник (AE3), некоторые подтипы ГАМК-рецепторов, а также карбоновую ангидразу [66].
Несмотря на то что в ряде работ отмечались противоэпилептические свойства этих диуретиков (см. обзор [67]), применение их в качестве противоэпилептических препаратов затрудняется плохой проходимостью через гематоэнцефалический барьер и большим числом побочных воздействий, поскольку NKCC1-котранспортер экспрессируется во многих клетках организма.
Недавно были разработаны специфичные ингибиторы KCC2: VU0463271 [68] и VU0240551 [69]. Мы изучили действие ингибитора VU0463271 на эпилептиформную активность в срезах гиппокампа и энторинальной коры крысы в возрасте 3 и 8 нед. Несмотря на то что к возрасту 3 нед у крыс уже должен установиться максимальный уровень экспрессии KCC2, соответствующий уровню у взрослых животных, ингибирование KCC2 действовало по-разному: если в возрасте 3 нед мы не обнаружили существенных изменений формы эпиактивности, то в возрасте 8 нед ингибирование KCC2 привело к замещению иктальной активности на эпилептический статус [52]. Hamidi и Avoli получили схожий результат в 4-аминопиридиновой модели в срезах энторинальной и периринальной коры, применив VU0240551 (10 µМ) [70]. Таким образом, экспериментальные данные подтверждают важную роль KCC2-котранспортера в регуляции эпилептической активности.
Перспективной стратегией усиления функций KCC2, что потенциально будет иметь противоэпилептическое действие, может быть направленное фосфорилирование KCC2 прямо или косвенно через вышележащие регуляторные киназы по некоторым сайтам [24]. Например, выявлено PKC-зависимое усиление функции KCC2 благодаря фосфорилированию по S940 в С-концевом хвосте KCC2 [71]. К такому эффекту может приводить активация метаботропных глутаматных рецепторов (mGluR) группы I. В экспериментах с грамицидин-перфорированным патч-клампом было показано, что одновременная тоническая активация mGluR1 и mGluR5 увеличивает силу тормозных синапсов через активацию PKC-зависимого пути в пирамидных нейронах области СА3 гиппокампа [72]. Однако агонисты mGluR группы I являются проконвульсантами и ведут к эпилептогенезу, так как одновременно модулируют возбудимость нейронов и активность NMDA-рецепторов [73], поэтому терапевтический потенциал такого подхода на настоящий момент неочевиден.
Фармакологическая регуляция баланса катионов калия
Выведение ионов калия из межклеточного пространства на молекулярном уровне в основном обеспечивают NaK-помпа и KIR-каналы. Если NaK-помпа отвечает за выведение избытков K+ из внеклеточного пространства, то активация ее работы должна оказать противоэпилептический эффект и, наоборот, ингибирование помпы должно обладать проэпилептическим действием.
Так как активность NaK-помпы повышается при повышении концентрации Na+ внутри клетки, то в качестве модулятора NaK-помпы в работах in vitro иногда применяют натриевый ионофор монензин [74, 75]. Однако мы не нашли работ, в которых монензин использовался бы в качестве экспериментального противоэпилептического средства. Наши собственные неопубликованные данные экспериментов показали, что монензин обладает очень узким терапевтическим окном. В 4-аминопиридиновой модели эпилептиформной активности в срезах гиппокампа и энторинальной коры мыши в некоторых случаях применение монензина (2–3 мкМ) препятствовало генерации иктальных разрядов, однако увеличение концентрации монензина до 5 мкМ приводило к гибели нейронов.
В отличие от активирующего действия на NaK-помпу эффект ее ингибирования изучен лучше и хорошо согласуется с модельными расчетами [8]. Было показано, что внутрижелудочковая инъекция оуабаина вызывает эпилептический приступ у крыс [76]. В моделях in vitro ослабление работы NaK-помпы с помощью оуабаина тоже имеет выраженный проэпилептический эффект [52].
За выведение ионов калия из внеклеточного пространства отвечают также астроциты. Астроциты осуществляют пространственную калиевую буферизацию (spatial potassium buffering), они поглощают внеклеточный K+, секретируемый нейронами, и высвобождают K+ в регионах с более низким уровнем K+ (например, микрокапиллярах), соединяясь в синцитий через щелевые контакты [77]. Поступление K+ в астроциты в основном опосредуется Kir-каналами, содержащими 4 субъединицы Kir4.1 (гиппокамп) или комбинацию Kir4.1/Kir5.1 субъединиц (неокортекс) [78–80].
Электрофизиологические исследования показали, что Kir-токи значительно снижены в образцах гиппокампа пациентов с рефрактерной височной эпилепсией [81], что может быть обусловлено снижением уровня экспрессии Kir4.1-каналов [82]. Нарушение функций Kir4.1-каналов выявлено при ряде наследственных форм неврологических расстройств, сопровождающихся эпилептическими судорогами, например, восточный синдром (EAST: эпилепсия, атаксия, сенсоневральная глухота и тубулопатия) и синдром SeSAME (судороги, сенсоневральная глухота, атаксия, умственная отсталость и электролитный дисбаланс) [83, 84]. Хотя исследования на моделях эпилепсии в некоторых случаях показывают, наоборот, усиление экспрессии: например, в литий-пилокарпиновой модели было найдено увеличение экспрессии Kir4.1 в коре головного мозга, стриатуме и гипоталамусе, это явление может носить компенсаторный характер [85]. Исследования же с использованием животных с условным нокаутом (conditional knockout), нацеленным на астроцитарный Kir4.1, выявили четкую связь между дисфункцией канала Kir4.1 и предрасположенностью к судорогам [86, 87].
Достаточно обоснованно поэтому можно предположить, что усиление Kir-токов благодаря применению положительных модуляторов или увеличению экспрессии Kir4.1-каналов в астроцитах будет обладать выраженным противоэпилептическим эффектом. Однако долгое время не было известно никаких специфических модуляторов Kir4.1-каналов. Сейчас идентифицировано только несколько антагонистов Kir4.1, таких как VU0134992 (IC50 = 0.97 мкМ) [88], пентамидин (IC50 = 97 нМ) [89] и некоторые другие, обзор которых можно найти у Ohno и соавт. [90]. Также выявлено, что многие препараты, применяемые в клинике, могут уменьшать Kir-токи при использовании их в терапевтических дозах. Например, такой побочный эффект описан у антидепрессантов [91], противоопухолевых препаратов на основе комплексов двухвалентной платины [92, 93].
К сожалению, мы не смогли найти данных о позитивных модуляторах работы Kir4.1-каналов. Зато целый ряд широко применяемых противоэпилептических препаратов (вальпроат, фенитоин и фенобарбитал) могут усиливать экспрессию астроцитарного Kir4.1 в лимбической области, что было показано в экспериментах на крысах [94], возможно, что этот эффект является одним из компонентов их противоэпилептического действия.
Таким образом, на сегодняшний день практически нет доступных фармакологических инструментов, которые позволили бы эффективно воздействовать на ионный баланс в нервной ткани. Кроме того, рассматриваемые нами транспортеры и каналы (или близкие аналоги) присутствуют не только в ЦНС, но и в других системах организма, обеспечивая целый ряд жизненно важных функций, поэтому можно ожидать широкий спектр побочных эффектов фармакологического воздействия на NaK-помпу, катион-хлорные котранспортеры и KIR-каналы.
ГЕННАЯ ТЕРАПИЯ, НАПРАВЛЕННАЯ НА РЕГУЛЯЦИЮ ИОННОГО БАЛАНСА
Экспериментальные генно-терапевтические подходы для лечения фармакорезистентной эпилепсии
Генно-терапевтические подходы подразделяют на четыре типа: (1) замена гена при моногенных заболеваниях, (2) добавление гена при сложных патологиях, (3) изменение уровня экспрессии генов и таргетинг РНК и (4) редактирование генома [95]. Применительно к эпилепсии первый подход практически не разрабатывается, так как идиопатические формы эпилепсии, вызванные мутацией какого-то одного гена и вследствие этого нарушенной функцией канала или рецептора, встречаются относительно редко. Кроме того, доставка генетического материала одновременно в обширные области мозга методически сложна. Третий и четвертый подходы также пока не реализуются для экспериментального лечения эпилепсии. Наиболее перспективным считается второй подход, который может быть использован для лечения фармакорезистентных фокальных форм эпилепсии. В этом варианте необходимые гены доставляются в клетку-мишень с целью локального снижения активности нейронных сетей в эпилептическом очаге.
Для доставки генов наиболее часто используют вирусные конструкты. Среди различных вирусов, испытанных для генной терапии, наиболее перспективными кандидатами для применения в ЦНС являются конструкты на основе аденоассоциированных вирусов (AAV), лентивирусов и вирусов герпеса [96]. AAV-конструкты сейчас считаются наиболее безопасными: при сборке конструкта можно почти полностью удалить ДНК вируса, сам вирусный конструкт относительно нетоксичен для клеток и обладает низкой иммуногенностью. Однако значимым ограничением для широкого применения AAV-конструктов является их относительно небольшой размер [97]. Так как емкость AAV-конструктов не превышает 4.5–5 тысяч пар оснований, а размер кодирующей последовательности многих генов таргетных белков сопоставим по размерам или даже больше, то далеко не все гены могут быть доставлены в ЦНС таким способом. Поэтому в экспериментальных работах нередко применяют конструкты на основе лентивирусов или вирусов герпеса, имеющих большую емкость, но и больший риск побочных эффектов [96].
Сейчас основные способы генной терапии фармакорезистентной эпилепсии направлены на увеличение экспрессии ингибирующих пептидов, таких как галанин [98] и NPY [99, 100], или подавление возбудимости нейронов путем гиперэкспрессии в них калиевых каналов [101–103]. Среди близких подходов, направленных на снижение гиперактивации нейронных сетей, следует отметить хемо- и оптогенетический методы, обзор которых можно найти в статье Walker и Kullmann [104].
Генетические методы регуляции баланса хлорид-ионов в нейронах
Как было описано выше, накопление хлорид-ионов в нейронах при эпилептической активности ведет к нарушению функций ГАМКергической системы и самоподдержанию эпилептической активности. Усиление функций KCC2-транспортера может предотвратить нарушение функций торможения. Экспериментальных работ, в которых генная терапия была бы нацелена именно на модуляцию ионного баланса хлорид-ионов опубликовано относительно мало. Мы смогли найти всего несколько работ, в которых был изучен эффект гиперэкспрессии KCC2-транспортера на эпилептическую активность [105, 106].
Гиперэкспрессию KCC2-транспортера, доставленного с помощью лентивирусного конструкта под промотером CaMKII, осуществляли в коре трансгенной мыши PV-ChR2. У этой линии мышей каналородопсин-2 экспрессируется в парвальбумин-позитивных интернейронах и световая стимуляция вызывает их активацию [105]. Magloire и соавт. показали, что при пилокарпин-индуцированном эпилептическом статусе у PV-ChR2 мышей с нормальным уровнем KCC2-транспортера фотостимуляция коры вместо противосудорожного эффекта обладает выраженным проиктальным действием, если начинается через 2 и более секунд с момента развития иктального разряда в коре. Это наблюдение полностью согласуется с представлениями о накоплении Cl– в нейронах в ходе эпилептического приступа и переходу от тормозного к возбуждающему действию ГАМК. Гиперэкспрессия KCC2-котранспортера в пирамидных нейронах коры препятствовала развитию проиктального действия парвальбуминовых интернейронов и сохраняла тормозное действие ГАМК. Однако гиперэкспрессия KCC2 не повлияла сама по себе на протекание эпилептических припадков [105].
Аналогичный вывод был сделан в другой экспериментальной работе, в которой у линии мышей с доксициклин-индуцибельной гиперэкспрессией KCC2-транспортера моделировали каинатные судороги. Гиперэкспрессия KCC2 также не повлияла на тяжесть эпилептических припадков, однако увеличила эффективность позитивного модулятора ГАМК-рецепторов диазепама в остановке судорожных припадков, регистрируемых на ЭЭГ [106]. Таким образом, одновременное воздействие на KCC2 и ГАМК-рецепторы может стать эффективной противосудорожной терапевтической стратегией.
Сейчас доступно несколько оптогенетических инструментов для направленного транспорта Cl– в нейрон или из нейрона, например: галородопсин (хлорная помпа), который под воздействием одного кванта света 560 нм проводит один хлорид-ион в клетку [107]; хлорный канал, который под воздействием света 490 нм открывает пору для тока хлорид-ионов по электрохимическому градиенту [108, 109]; Cl-Out-комплекс, в котором протонная помпа обеспечивает движущую силу для тока хлорид-ионов из клетки по хлорному каналу [110].
Аналогично работе ГАМКергической системы, в условиях патологической эпилептической активности хлорные помпа и канал могут оказать проэпилептическое действие из-за накопления хлорид-ионов в нейронах. Поэтому оптогенетическая активация светочувствительных хлорных каналов может вызывать как подавление, так и усиление нейронной активности [111, 112].
Применение комплекса Cl-out может оказаться полезным инструментом для нормализации внутриклеточной концентрации хлорид-ионов в эпилептической ткани с нарушенным градиентом хлорид-ионов. Alfonsa и соавт. показали, что активация Cl-out-комплекса оказывает противоэпилептическое действие в том случае, когда причиной эпилептической активности является высокая концентрация хлорид-ионов в нейронах [110]. Эпилептическую активность в срезах гиппокампа мыши авторы вызывали с помощью блокады KCC2. Регистрацию сетевой активности производили с помощью внеклеточных отведений в пирамидном слое CA1, для мониторинга возбудимости нейронов проводилась электрическая стимуляция. Далее проводили фотостимуляцию серией одновременно светом 488 и 561 нм для активации хлорного каналородопсина и протонной помпы соответственно. Alfonsa и соавт. продемонстрировали, что активация Cl-out-комплекса уменьшает возбудимость и в целом оказывает противоэпилептическое действие [110].
Генетические методы регуляции концентрации катионов калия во внеклеточной среде
Разработано довольно много эффективных вариантов генной терапии эпилепсии, усиливающих функции различных типов нейронных калиевых каналов [101–103]. Хотя дополнительная активация калиевых каналов может влиять на повышение концентрации катионов калия во внеклеточной среде, этот эффект не является преобладающим. Поэтому в этой части обзора мы рассмотрим только подходы, направленные на нормализацию внеклеточной концентрации ионов калия. Эффективной стратегией для этого может стать усиление Kir-токов путем увеличения экспрессии Kir4.1-каналов в астроцитах.
Предпосылкой для этого подхода является снижение Kir-токов в образцах гиппокампа пациентов с рефрактерной височной эпилепсией [81]. Мы не смогли найти литературных данных об экспериментальной проверке данного метода для лечения эпилепсии, однако увеличение экспрессии Kir4.1-каналов в астроцитах стриатума оказалось эффективным при лечении болезни Хантингтона в модели на трансгенных линиях мышей R6/2 и Q175 HD [113]. Авторы показало, что дополнительная экспрессия каналов Kir4.1 с помощью вирусной доставки в астроциты стриатума восстановила Kir-токи в этих клетках, в результате чего снизилась концентрация внеклеточного K+, уменьшилась деполяризация мембраны и улучшились другие функциональные характеристики нейронов стриатума. Такое лечение позволило несколько ослабить двигательные нарушение и улучшить выживаемость R6/2 мышей [113].
Следует иметь в виду, что у этого подхода могут оказаться ограничения. Например, избыточное усиление Kir-токов может приводить к негативным последствиям. Выявлено, что мутации гена KCNJ10, приводящие к усилению функций Kir4.1-каналов (gain-of-function), могут способствовать формированию неврологических синдромов, объединяющих расстройства аутистического спектра и эпилепсию [114]. Можно предположить, что усиление Kir-токов в этом случае не допускает естественной деполяризации нейронов и инактивации натриевых каналов из-за накопления внеклеточного калия, вызывающих деполяризационный блок и прерывающих спайковую активность. Однако существующие модели иктальной активности прямо не подтверждают такой вариант развития [8]. Вероятно, что эпилептическая активность в этом случае может быть вызвана функциональными нарушениями в нейрон-глиальных взаимодействиях. Уменьшение концентрации внеклеточного K+ ведет к гиперполяризации астроцитов, что ослабляет глиотрансмиссию, которая обычно ингибирует выброс глутамата пирамидными нейронами [115].
ЗАКЛЮЧЕНИЕ
В эпилептической ткани длительная избыточная нейронная активность приводит к нарушению функций ГАМКергической системы: вместо торможения она может оказывать проэпилептическое действие. В основе этих нарушений лежит дисбаланс концентраций хлорид-ионов и/или катионов калия во вне и внутриклеточной среде. Модельные и экспериментальные работы показывают, что восстановление баланса этих ионов может оказаться наиболее эффективным подходом для лечения таких форм эпилепсии. В большинстве случаев для этого необходимо восстановить или усилить ослабленную функцию каких-либо ионных каналов, транспортеров или насосов. К сожалению, имеющийся арсенал фармакологических инструментов крайне мал и недостаточно эффективен.
Мы полагаем, что для данных целей методы генной терапии, позволяющие усилить функции каких-либо белков путем их дополнительной экспрессии в целевых клетках, могут оказаться наиболее перспективными. Сейчас область генной терапии эпилепсии интенсивно развивается, однако инструменты, направленные на поддержание ионного баланса в эпилептической ткани, пока исследуются относительно мало. Среди доступных вариантов терапии особое внимание привлекают методы гиперэкспрессии KCC2-транспортера и Kir4.1-каналов, позволяющие восстановить концентрации хлорид-ионов и катионов калия. Однако требуется дополнительная экспериментальная проверка эффективности и безопасности данных инструментов в различных моделях эпилепсии и судорожных состояний.
Список литературы
Fattorusso A, Matricardi S, Mencaroni E, Dell’Isola GB, Di Cara G, Striano P, Verrotti A (2021) The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front Neurol 12: 1030. https://doi.org/10.3389/FNEUR.2021.674483
Löscher W, Klitgaard H, Twyman RE, Schmidt D (2013) New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov 12: 757–776. https://doi.org/10.1038/nrd4126
Raimondo JV, Burman RJ, Katz AA, Akerman CJ (2015) Ion dynamics during seizures. Front Cell Neurosci 9: 1–14. https://doi.org/10.3389/fncel.2015.00419
Magloire V, Mercier MS, Kullmann DM, Pavlov I (2019) GABAergic Interneurons in Seizures: Investigating Causality With Optogenetics. Neuroscientist 25: 344–358. https://doi.org/10.1177/1073858418805002
Khazipov R (2016) GABAergic Synchronization in Epilepsy. Cold Spring Harb Perspect Med 6: a022764. https://doi.org/10.1101/CSHPERSPECT.A022764
de Curtis M, Uva L, Gnatkovsky V, Librizzi L (2018) Potassium dynamics and seizures: Why is potassium ictogenic? Epilepsy Res 143: 50–59. https://doi.org/10.1016/J.EPLEPSYRES.2018.04.005
Chizhov AV, Amakhin DV, Zaitsev AV (2017) Computational model of interictal discharges triggered by interneurons. PLoS One 12: e0185752. https://doi.org/10.1371/journal.pone.0185752
Chizhov AV, Zefirov AV, Amakhin DV, Smirnova EY, Zai-tsev AV (2018) Minimal model of interictal and ictal discharges “Epileptor-2.” PLOS Comput Biol 14: e1006186. https://doi.org/10.1371/journal.pcbi.1006186
Ammann D, Chao P, Simon W (1987) Valinomycin-based K+ selective microelectrodes with low electrical membrane resistance. Neurosci Lett 74: 221–226. https://doi.org/10.1016/0304-3940(87)90153-4
Codadu NK, Parrish RR, Trevelyan AJ (2019) Region-specific differences and areal interactions underlying transitions in epileptiform activity. J Physiol 597: 2079–2096. https://doi.org/10.1113/JP277267
Chizhov AV, Amakhin DV, Smirnova EY, Zaitsev AV (2022) Ictal wavefront propagation in slices and simulations with conductance-based refractory density model. PLOS Comput Biol 18: e1009782. https://doi.org/10.1371/journal.pcbi.1009782
Moody WJ, Futamachi KJ, Prince DA (1974) Extracellular potassium activity during epileptogenesis. Exp Neurol 42: 248–263. https://doi.org/10.1016/0014-4886(74)90023-5
Prince DA, Lux HD, Neher E (1973) Measurement of extracellular potassium activity in cat cortex. Brain Res 50: 489–495. https://doi.org/10.1016/0006-8993(73)90758-0
Heinemann U, Lux HD, Gutnick MJ (1977) Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res 27–27: 237–243. https://doi.org/10.1007/BF00235500
Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA (1999) The Neuron-specific K-Cl Cotransporter, KCC2. J Biol Chem 274: 12656–12664. https://doi.org/10.1074/jbc.274.18.12656
Kuner T, Augustine GJ (2000) A Genetically Encoded Ratiometric Indicator for Chloride. Neuron 27: 447–459. https://doi.org/10.1016/S0896-6273(00)00056-8
Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ (2014) Local impermeant anions establish the neuronal chloride concentration. Science 343: 670–675.https://doi.org/10.1126/science.1245423
Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F (2010) Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods 7: 516–518.https://doi.org/10.1038/nmeth.1471
Raimondo J (2013) A genetically-encoded chloride and pH sensor for dissociating ion dynamics in the nervous system. Front Cell Neurosci 7: 202.https://doi.org/10.3389/fncel.2013.00202
Sulis Sato S, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J, Luin S, Arosio D, Beltram F, Cancedda L, Kaila K, Ratto GM (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci U S A114: E8770–E8779https://doi.org/10.1073/pnas.1702861114
Ponomareva D, Petukhova E, Bregestovski P (2021) Simultaneous Monitoring of pH and Chloride (Cl–) in Brain Slices of Transgenic Mice. Int J Mol Sci 22: 13601. https://doi.org/10.3390/ijms222413601
Burman RJ, Selfe JS, Lee JH, van den Berg M, Calin A, Codadu NK, Wright R, Newey SE, Parrish RR, Katz AA, Wilmshurst JM, Akerman CJ, Trevelyan AJ, Raimondo JV (2019) Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain 142: 3482–3501. https://doi.org/10.1093/brain/awz283
Rahmati N, Hoebeek FE, Peter S, De Zeeuw CI (2018) Chloride Homeostasis in Neurons With Special Emphasis on the Olivocerebellar System: Differential Roles for Transporters and Channels. Front Cell Neurosci 12: 101. https://doi.org/10.3389/fncel.2018.00101
Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ (2013) Modulation of neuronal activity by phosphorylation of the K–Cl cotransporter KCC2. Trends Neurosci 36: 726–737. https://doi.org/10.1016/j.tins.2013.08.006
Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M (2014) GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol 26: 34–41. https://doi.org/10.1016/j.conb.2013.11.004
Wasterlain CG, Liu H, Naylor DE, Thompson KW, Suchomelova L, Niquet J, Mazarati AM, Baldwin RA (2009) Molecular basis of self-sustaining seizures and pharmacoresistance during status epilepticus: The receptor trafficking hypothesis revisited. Epilepsia 50: 16–18. https://doi.org/10.1111/j.1528-1167.2009.02375.x
Puskarjov M, Ahmad F, Kaila K, Blaesse P (2012) Activity-Dependent Cleavage of the K-Cl Cotransporter KCC2 Mediated by Calcium-Activated Protease Calpain. J Neurosci 32: 11356–11364. https://doi.org/10.1523/JNEUROSCI.6265-11.2012
Lee HHC, Jurd R, Moss SJ (2010) Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci 45: 173–179. https://doi.org/10.1016/j.mcn.2010.06.008
Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor–mediated currents. Nat Neurosci 14: 736–743. https://doi.org/10.1038/nn.2806
Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation down-regulates the K+–Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol 159: 747–752. https://doi.org/10.1083/jcb.200209011
Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A (2016) The KCC2 Cotransporter and Human Epilepsy. Neuroscience 22: 555–562. https://doi.org/10.1177/1073858416645087
Stödberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, Farabella I, Meyer E, Kawahara A, Vassallo G, Stivaros SM, Bjursell MK, Stranneheim H, Tigerschiöld S, Persson B, Bangash I, Das K, Hughes D, Lesko N, Lundeberg J, Scott RC, Poduri A, Scheffer IE, Smith H, Gissen P, Schorge S, Reith MEA, Topf M, Kullmann DM, Harvey RJ, Wedell A, Kurian MA (2015) Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun 6: 8038https://doi.org/10.1038/ncomms9038
Dimitrijevic S, Jekic B, Cvjeticanin S, Tucovic A, Filipovic T, Novaković I, Ivić B, Nikolic D (2022) KCC2 rs2297201 Gene Polymorphism Might be a Predictive Genetic Marker of Febrile Seizures. ASN Neuro 14: 175909142210932https://doi.org/10.1177/17590914221093257
Duy PQ, David WB, Kahle KT (2019) Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front Cell Neurosci 13: 515. https: //doi.org/https://doi.org/10.3389/FNCEL.2019.00515/BIBTEX
Filatov G, Krishnan GP, Rulkov NF, Bazhenov M (2011) Dynamics of epileptiform activity in mouse hippocampal slices. J Biol Phys 37: 347–360.https://doi.org/10.1007/S10867-011-9216-X/FIGURES/5
Librizzi L, Losi G, Marcon I, Sessolo M, Scalmani P, Carmignoto G, de Curtis M (2017) Interneuronal Network Activity at the Onset of Seizure-Like Events in Entorhinal Cortex Slices. J Neurosci 37: 10398–10407. https://doi.org/10.1523/JNEUROSCI.3906-16.2017
González OC, Krishnan GP, Timofeev I, Bazhenov M (2019) Ionic and synaptic mechanisms of seizure generation and epileptogenesis. Neurobiol Dis 130: 104485. https://doi.org/10.1016/J.NBD.2019.104485
Bellot-Saez A, Stevenson R, Kékesi O, Samokhina E, Ben-Abu Y, Morley JW, Buskila Y (2021) Neuromodulation of Astrocytic K+ Clearance. Int J Mol Sci 22: 2520. https://doi.org/10.3390/ijms22052520
Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, Zamboni D, Schwebel CL, Press DM, Kratzer SO, Zhao H, Berrettini WH, Buono RJ (2004) Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm Genome 2004 154 15: 239–251. https://doi.org/10.1007/S00335-003-2270-3
Olsen ML, Sontheimer H (2008) Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107: 589–601. https://doi.org/10.1111/J.1471-4159.2008.05615.X
Köhling R, Wolfart J (2016) Potassium Channels in Epilepsy. Cold Spring Harb Perspect Med 6: a022871. https://doi.org/10.1101/cshperspect.a022871
Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT (2008) A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci 273: 123–126. https://doi.org/10.1016/J.JNS.2008.06.006
Hempelmann A, Heils A, Sander T (2006) Confirmatory evidence for an association of the connexin-36 gene with juvenile myoclonic epilepsy. Epilepsy Res 71: 223–228. https://doi.org/10.1016/j.eplepsyres.2006.06.021
D’Antuono M, Louvel J, Köhling R, Mattia D, Bernasconi A, Olivier A, Turak B, Devaux A, Pumain R, Avoli M (2004) GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain 127: 1626–1640. https://doi.org/10.1093/BRAIN/AWH181
Mattia D, Olivier A, Avoli M (1995) Seizure-like discharges recorded in human dysplastic neocortex maintained in vitro. Neurology 45: 1391–1395. https://doi.org/10.1212/WNL.45.7.1391
Avoli M, De Curtis M, Gnatkovsky V, Gotman J, Köhling R, Lévesque M, Manseau F, Shiri Z, Williams S (2016) Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J Neurophysiol 115: 3229–3237. https://doi.org/10.1152/JN.01128.2015/ASSET/IMAGES/LARGE/Z9K0071636960005.JPEG
Chang M, Dian JA, Dufour S, Wang L, Moradi Chameh H, Ramani M, Zhang L, Carlen PL, Womelsdorf T, Valiante TA (2018) Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol Dis 109: 102–116. https://doi.org/10.1016/J.NBD.2017.10.007
Elahian B, Lado NE, Mankin E, Vangala S, Misra A, Moxon K, Fried I, Sharan A, Yeasin M, Staba R, Bragin A, Avoli M, Sperling MR, Engel J, Weiss SA (2018) Low-voltage fast seizures in humans begin with increased interneuron firing. Ann Neurol 84: 588–600. https://doi.org/10.1002/ANA.25325
Miri ML, Vinck M, Pant R, Cardin JA (2018) Altered hippocampal interneuron activity precedes ictal onset. Elife 7: e40750. https://doi.org/10.7554/eLife.40750
Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, Carmignoto G (2015) Parvalbumin-Positive Inhibitory Interneurons Oppose Propagation But Favor Generation of Focal Epileptiform Activity. J Neurosci 35: 9544–9557. https://doi.org/10.1523/JNEUROSCI.5117-14.2015
Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S (2015) Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol 113: 1616–1630. https://doi.org/10.1152/jn.00841.2014
Smirnova EY, Sinyak DS, Chizhov AV, Zaitsev AV (2021) Age-Dependent Generation of Epileptiform Activity in the 4-Aminopyridine Model with Slices of the Rat Entorhinal Cortex. J Evol Biochem Physiol 57: 230–240. https://doi.org/10.1134/S0022093021020058
Somogyi P, Freund TF, Hodgson AJ, Somogyi J, Beroukas D, Chubb IW (1985) Identified axo-axonic cells are immunoreactive for GABA in the hippocampus visual cortex of the cat. Brain Res 332: 143–149. https://doi.org/10.1016/0006-8993(85)90397-X
Zaitsev AV, Povysheva NV, Gonzalez-Burgos G, Rotaru D, Fish KN, Krimer LS, Lewis DA (2009) Interneuron Diversity in Layers 2–3 of Monkey Prefrontal Cortex. Cereb Cortex 19: 1597–1615. https://doi.org/10.1093/cercor/bhn198
Schneider-Mizell CM, Bodor AL, Collman F, Brittain D, Bleckert A, Dorkenwald S, Turner NL, Macrina T, Lee K, Lu R, Wu J, Zhuang J, Nandi A, Hu B, Buchanan J, Takeno MM, Torres R, Mahalingam G, Bumbarger DJ, Li Y, Chartrand T, Kemnitz N, Silversmith WM, Ih D, Zung J, Zlateski A, Tartavull I, Popovych S, Wong W, Castro M, Jordan CS, Froudarakis E, Becker L, Suckow S, Reimer J, Tolias AS, Anastassiou CA, Seung HS, Reid RC, Costa NM da (2021) Structure and function of axo-axonic inhibition. Elife 10: e73783. https://doi.org/10.7554/eLife.73783
Ruiz A, Fabian-Fine R, Scott R, Walker MC, Rusakov DA, Kullmann DM (2003) GABAA Receptors at Hippocampal Mossy Fibers. Neuron 39: 961–973. https://doi.org/10.1016/S0896-6273(03)00559-2
Báldi R, Varga C, Tamás G (2010) Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci 32: 1319–1325. https://doi.org/10.1111/j.1460-9568.2010.07361.x
Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G (2006) Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311: 233–235. https://doi.org/10.1126/science.1121325
Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K (2008) GABAergic Depolarization of the Axon Initial Segment in Cortical Principal Neurons Is Caused by the Na-K-2Cl Cotransporter NKCC1. J Neurosci 28: 4635–4639. https://doi.org/10.1523/JNEUROSCI.0908-08.2008
Dudok B, Klein PM, Soltesz I (2022) Toward Understanding the Diverse Roles of Perisomatic Interneurons in Epilepsy. Epilepsy Curr 22: 54–60. https://doi.org/10.1177/15357597211053687
Jiang X, Lachance M, Rossignol E (2016) Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res 226: 81–126. https://doi.org/10.1016/bs.pbr.2016.04.012
Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ (2017) Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci 40: 555–571. https://doi.org/10.1016/j.tins.2017.06.008
Liu R, Wang J, Liang S, Zhang G, Yang X (2019) Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol 10: 1407. https://doi.org/10.3389/fneur.2019.01407
Buchin A, Chizhov A, Huberfeld G, Miles R, Gutkin BS (2016) Reduced efficacy of the KCC2 cotransporter promotes epileptic oscillations in a subiculum network model. J Neurosci 36: 11619–11633. https://doi.org/10.1523/JNEUROSCI.4228-15.2016
Payne JA, Rivera C, Voipio J, Kaila K (2003) Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26: 199–206. https://doi.org/10.1016/S0166-2236(03)00068-7
Uwera J, Nedergaard S, Andreasen M (2015) A novel mechanism for the anticonvulsant effect of furosemide in rat hippocampus in vitro. Brain Res 1625: 1–8https://doi.org/10.1016/j.brainres.2015.08.014
Löscher W, Puskarjov M, Kaila K (2013) Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69: 62–74. https://doi.org/10.1016/j.neuropharm.2012.05.045
Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW (2012) Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22: 4532–4535. https://doi.org/10.1016/j.bmcl.2012.05.126
Deisz RA, Wierschke S, Schneider UC, Dehnicke C (2014) Effects of VU0240551, a novel KCC2 antagonist, and DIDS on chloride homeostasis of neocortical neurons from rats and humans. Neuroscience 277: 831–841. https://doi.org/10.1016/j.neuroscience.2014.07.037
Hamidi S, Avoli M (2015) KCC2 function modulates in vitro ictogenesis. Neurobiol Dis 79: 51–58. https://doi.org/10.1016/j.nbd.2015.04.006
Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ (2007) Direct Protein Kinase C-dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J Biol Chem 282: 29777–29784. https://doi.org/10.1074/jbc.M705053200
Banke TG, Gegelashvili G (2008) Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J Physiol 586: 4925–4934. https://doi.org/10.1113/JPHYSIOL.2008.157024
Ure J, Baudry M, Perassolo M (2006) Metabotropic glutamate receptors and epilepsy. J Neurol Sci 247: 1–9. https://doi.org/10.1016/j.jns.2006.03.018
Kueh D, Barnett WH, Cymbalyuk GS, Calabrese RL (2016) Na(+)/K(+) pump interacts with the h-current to control bursting activity in central pattern generator neurons of leeches. Elife 5: e19322. https: //doi.org/https://doi.org/10.7554/eLife.19322
Picton LD, Nascimento F, Broadhead MJ, Sillar KT, Miles GB (2017) Sodium Pumps Mediate Activity-Dependent Changes in Mammalian Motor Networks. J Neurosci 37: 906–921. https://doi.org/10.1523/JNEUROSCI.2005-16.2016
Donaldson J, Minnich JL, Barbeau A (1972) Ouabain-induced Seizures in Rats: Regional and Subcellular Localization of 3 H-Ouabain Associated with Na+–K + -ATPase in Brain. Can J Biochem 50: 888–896. https://doi.org/10.1139/o72-124
Kinboshi M, Ikeda A, Ohno Y (2020) Role of Astrocytic Inwardly Rectifying Potassium (Kir) 4.1 Channels in Epileptogenesis. Front Neurol 11: 1832. https://doi.org/10.3389/fneur.2020.626658
Larsen BR, MacAulay N (2014) Kir4.1-mediated spatial buffering of K+ : Experimental challenges in determination of its temporal and quantitative contribution to K + clearance in the brain. Channels 8: 544–550. https://doi.org/10.4161/19336950.2014.970448
Moroni RF, Inverardi F, Regondi MC, Pennacchio P, Frassoni C (2015) Developmental expression of Kir4.1 in astrocytes and oligodendrocytes of rat somatosensory cortex and hippocampus. Int J Dev Neurosci 47: 198–205. https://doi.org/10.1016/j.ijdevneu.2015.09.004
Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y (2004) Differential Assembly of Inwardly Rectifying K+ Channel Subunits, Kir4.1 and Kir5.1, in Brain Astrocytes. J Biol Chem 279: 44065–44073. https://doi.org/10.1074/jbc.M405985200
Steinhäuser C, Seifert G, Bedner P (2012) Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60: 1192–1202. https://doi.org/10.1002/glia.22313
Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Taubøll E, Gjerstad L, Spencer DD, Ottersen OP, Nagelhus EA, Lanerolle NC de (2012) Loss of Perivascular Kir4.1 Potassium Channels in the Sclerotic Hippocampus of Patients With Mesial Temporal Lobe Epilepsy. J Neuropathol Exp Neurol 71: 814–825. https://doi.org/10.1097/NEN.0b013e318267b5af
Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton S-A, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R (2010) KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A 107: 14490–14495. https://doi.org/10.1073/pnas.1003072107
Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG (2010) Molecular Mechanisms of EAST/SeSAME Syndrome Mutations in Kir4.1 (KCNJ10). J Biol Chem 285: 36040–36048. https://doi.org/10.1074/jbc.M110.163170
Nagao Y, Harada Y, Mukai T, Shimizu S, Okuda A, Fujimoto M, Ono A, Sakagami Y, Ohno Y (2013) Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine–induced temporal lobe epilepsy model. Front Cell Neurosci 7: 104. https://doi.org/10.3389/fncel.2013.00104
Chever O, Djukic B, McCarthy KD, Amzica F (2010) Implication of Kir4.1 Channel in Excess Potassium Clearance: An In Vivo Study on Anesthetized Glial-Conditional Kir4.1 Knock-Out Mice. J Neurosci 30: 15769–15777. https://doi.org/10.1523/JNEUROSCI.2078-10.2010
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55: 274–281. https://doi.org/10.1002/glia.20455
Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, Satlin LM, Meiler J, Weaver CD, Lindsley CW, Hopkins CR, Denton JS (2018) Discovery, Characterization, and Effects on Renal Fluid and Electrolyte Excretion of the Kir4.1 Potassium Channel Pore Blocker, VU0134992. Mol Pharmacol 94: 926–937. https://doi.org/10.1124/mol.118.112359
Aréchiga-Figueroa IA, Marmolejo-Murillo LG, Cui M, Delgado-Ramírez M, van der Heyden MAG, Sánchez-Chapula JA, Rodríguez-Menchaca AA (2017) High-potency block of Kir4.1 channels by pentamidine: Molecular basis. Eur J Pharmacol 815: 56–63. https://doi.org/10.1016/J.EJPHAR.2017.10.009
Ohno Y, Kunisawa N, Shimizu S (2021) Emerging Roles of Astrocyte Kir4.1 Channels in the Pathogenesis and Treatment of Brain Diseases. Int J Mol Sci 22: 10236. https://doi.org/10.3390/ijms221910236
Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y (2007) Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51. https://doi.org/10.1016/j.brainres.2007.08.018
Leo M, Schmitt L-I, Kutritz A, Kleinschnitz C, Hagenacker T (2021) Cisplatin-induced activation and functional modulation of satellite glial cells lead to cytokine-mediated modulation of sensory neuron excitability. Exp Neurol 341: 113695. https://doi.org/10.1016/j.expneurol.2021.113695
Leo M, Schmitt L-I, Steffen R, Kutritz A, Kleinschnitz C, Hagenacker T (2021) Modulation of Glutamate Transporter EAAT1 and Inward-Rectifier Potassium Channel Kir4.1 Expression in Cultured Spinal Cord Astrocytes by Platinum-Based Chemotherapeutics. Int J Mol Sci 22: 6300. https://doi.org/10.3390/ijms22126300
Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, Okuda A, Fujimoto M, Ito H, Ikeda A, Ohno Y (2018) Antiepileptic Drugs Elevate Astrocytic Kir4.1 Expression in the Rat Limbic Region. Front Pharmacol 9: 845. https://doi.org/10.3389/fphar.2018.00845
Wang D, Gao G (2014) State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med 18: 151–161.
Ingusci S, Cattaneo S, Verlengia G, Zucchini S, Simonato M (2019) A Matter of Genes: The Hurdles of Gene Therapy for Epilepsy. Epilepsy Curr 19: 38–43. https://doi.org/10.1177/1535759718822846
Wang D, Tai PWL, Gao G (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18: 358–378. https://doi.org/10.1038/s41573-019-0012-9
McCown TJ (2006) Adeno-associated Virus-Mediated Expression and Constitutive Secretion of Galanin Suppresses Limbic Seizure Activity in Vivo. Mol Ther 14: 63–68https://doi.org/10.1016/J.YMTHE.2006.04.004
Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A (2008) Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 131: 1506–1515https://doi.org/10.1093/BRAIN/AWN079
Cattaneo S, Verlengia G, Marino P, Simonato M, Bettegazzi B (2021) NPY and Gene Therapy for Epilepsy: How, When, and Y. Front Mol Neurosci 13: 608001. https://doi.org/10.3389/fnmol.2020.608001
Bernard C (2012) Treating epilepsy with a light potassium diet. Sci Transl Med 4: 161fs40. https://doi.org/10.1126/scitranslmed.3005297
Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, Kullmann DM (2012) Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 4: 161ra152. https://doi.org/10.1126/scitranslmed.3004190
Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, Hughes MP, Massaro G, Rahim AA, Hashemi KS, Kullmann DM, Walker MC, Schorge S (2019) Epilepsy Gene Therapy Using an Engineered Potassium Channel. J Neurosci 39: 3159–3169. https://doi.org/10.1523/JNEUROSCI.1143-18.2019
Walker MC, Kullmann DM (2020) Optogenetic and chemogenetic therapies for epilepsy. Neuropharmacology 168: 107751. https://doi.org/10.1016/j.neuropharm.2019.107751
Magloire V, Cornford J, Lieb A, Kullmann DM, Pavlov I (2019) KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat Commun 10: 1225. https://doi.org/10.1038/s41467-019-08933-4
Cheung DL, Cooke MJ, Goulton CS, Chaichim C, Cheung LF, Khoshaba A, Nabekura J, Moorhouse AJ (2022) Global transgenic upregulation of KCC2 confers enhanced diazepam efficacy in treating sustained seizures. Epilepsia 63: e15–e22. https://doi.org/10.1111/epi.17097
Han X, Boyden ES (2007) Multiple-Color Optical Activation, Silencing, and Desynchronization of Neural Activity, with Single-Spike Temporal Resolution. PLoS One 2: e299. https://doi.org/10.1371/journal.pone.0000299
Berndt A, Lee SY, Ramakrishnan C, Deisseroth K (2014) Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science 344: 420–424. https://doi.org/10.1126/science.1252367
Berndt A, Lee SY, Wietek J, Ramakrishnan C, Steinberg EE, Rashid AJ, Kim H, Park S, Santoro A, Frankland PW, Iyer SM, Pak S, Ährlund-Richter S, Delp SL, Malenka RC, Josselyn SA, Carlén M, Hegemann P, Deisseroth K (2016) Structural foundations of optogenetics: Determinants of channelrhodopsin ion selectivity. Proc Natl Acad Sci U S A 113: 822–829https://doi.org/10.1073/pnas.1523341113
Alfonsa H, Lakey JH, Lightowlers RN, Trevelyan AJ (2016) Cl-out is a novel cooperative optogenetic tool for extruding chloride from neurons. Nat Commun 7: 13495. https://doi.org/10.1038/ncomms13495
Malyshev AY, Roshchin MV, Smirnova GR, Dolgikh DA, Balaban PM, Ostrovsky MA (2017) Chloride conducting light activated channel GtACR2 can produce both cessation of firing and generation of action potentials in cortical neurons in response to light. Neurosci Lett 640: 76–80. https://doi.org/10.1016/j.neulet.2017.01.026
Messier JE, Chen H, Cai Z-L, Xue M (2018) Targeting light-gated chloride channels to neuronal somatodendritic domain reduces their excitatory effect in the axon. Elife 7: e38506. https://doi.org/10.7554/eLife.38506
Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17: 694–703. https://doi.org/10.1038/nn.3691
Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, Brignone MS, Lanciotti A, Moro F, Grottesi A, Catacuzzeno L, Baldini S, Hasan S, D’Adamo MC, Franciolini F, Molinari P, Santorelli FM, Pessia M (2016) Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep 6: 34325https://doi.org/10.1038/srep34325
Niday Z, Tzingounis AV (2018) Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox. Neurosci 24: 368–380. https://doi.org/10.1177/1073858418763752
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии