

Журнал эволюционной биохимии и физиологии, 2023, T. 59, № 1, стр. 20-31
ИЗУЧЕНИЕ ЗАЩИТНОГО И АНТИОКСИДАНТНОГО ДЕЙСТВИЯ ИНСУЛИНА НА НЕЙРОНЫ КОРЫ МОЗГА КРЫС ПРИ ДЕПРИВАЦИИ ГЛЮКОЗЫ И КИСЛОРОДА IN VITRO
И. О. Захарова 1, И. И. Зорина 1, Л. В. Баюнова 1, А. О. Шпаков 1, Н. Ф. Аврова 1, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: avrova@iephb.ru
Поступила в редакцию 03.10.2022
После доработки 17.11.2022
Принята к публикации 18.11.2022
- EDN: GXXUDU
- DOI: 10.31857/S0044452923010096
Аннотация
Инсулин при интраназальном введении является одним из наиболее перспективных протекторов для лечения нейродегенеративных и других болезней, связанных с поражениями мозга. Интересно, что при этих болезнях уровень инсулина в мозге (в противоположность его уровню в крови), как правило, сильно снижается, что, наряду с развитием резистентности к нему, приводит к нарушению инсулинового сигналинга в нейронах. При изучении in vitro механизмов защитного эффекта нейропротекторов при ишемии и реперфузии мозга используются разные модели. Целью настоящей работы является изучение защитного эффекта инсулина на нейроны коры мозга в культуре и его механизма действия при депривации глюкозы и кислорода (ДГК) и последующем восстановлении снабжения нейронов этими соединениями. Показано, что воздействие на нейроны ДГК в течение 1 или 3 ч с последующей инкубацией в полной ростовой среде с глюкозой и кислородом приводит к снижению жизнеспособности нейронов и увеличению образования активных форм кислорода, а преинкубация нейронов с инсулином в микромолярных концентрациях оказывает нейропротекторный и антиоксидантный эффекты. Найдено, что воздействие на нейроны ДГК в течение 1 ч и затем инкубация в полной ростовой среде приводит к падению активности протеинкиназы B – Akt (снижению отношения pAkt (Ser473)/Akt) и активации киназы гликогенсинтазы-3бета (GSK-3beta), одной из основных мишеней Akt, что выражается в снижении отношения pGSK-3beta (Ser9)/GSK-3beta. Преинкубация с инсулином, напротив, активирует Akt и инактивирует GSK-3beta. Эти эффекты инсулина, очевидно, вносят существенный вклад в его нейропротекторный эффект, т.к. активация GSK-3beta приводит к нарушению функций митохондрий и гибели нейронов. Показано также увеличение активности протеинкиназы, регулируемой внеклеточными сигналами (ERK1/2), при действии инсулина на нейроны, которая снижалась при действии ДГК с последующей инкубацией в полной ростовой среде.
Инсулин является одним из наиболее перспективных нейропротекторов среди тех, которые могут быть использованы в будущем в клинике для лечения ряда болезней, связанных с поражениями мозга. Во многих экспериментах показано, что при интраназальном введении инсулина животным с нейродегенеративными или диабетическими поражениями мозга, он повышает жизнеспособность нейронов и улучшает когнитивные функции мозга [1–4]. Наряду с этим клинические испытания инсулина при болезни Альцгеймера были достаточно успешными [5–8]. Дело в том, что при интраназальном введении инсулина он попадает непосредственно в мозг, используя структуры обонятельного и тройничного нервов и минуя гематоэнцефалический барьер [9, 10]. Интересно, что при таких болезнях, как сахарный диабет 2-го типа, метаболический синдром, болезни Альцгеймера и Паркинсона, уровень инсулина заметно увеличен в крови, а в мозге он, напротив, сильно снижается. Уменьшение уровня инсулина и резистентность клеток к инсулину при этих болезнях приводят к нарушению инсулинового сигналинга в нейронах разных районов мозга [11, 12]. При интраназальном введении инсулина он способен нормализовать метаболические процессы в мозге, в том числе в гипоталамусе, и, как следствие, также и в периферических органах [12, 13]. При этом эффекты инсулина изучены пока недостаточно. В наибольшей мере это относится к ишемии и реперфузии мозга, при которой эффекты интраназального введения инсулина только начинают исследоваться. В отличие от нейродегенеративных и диабетических патологий, при исследовании процессов ишемии и реперфузии мозга ученые сосредоточились преимущественно на изучении эффектов инсулин-подобного фактора роста (IGF-1) [см., например, 14–16], а не инсулина. И это, несмотря на то, что инсулин, вводимый интраназально, является, очевидно, более перспективным нейропротектором, чем IGF-1 [17]. В литературе нам не встретилось работ, посвященных изучению эффектов интраназального введения инсулина животным с ишемией и реперфузией мозга. Исключение составляют лишь работы нашей лаборатории. Показано, что инсулин при его интраназальном введении крысам с глобальной ишемией и реперфузией переднего мозга обладает антиоксидантным и нейропротекторным эффектом, предотвращает накопление в коре мозга различных продуктов перекисного окисления липидов и окислительную инактивацию Na+, K+-АТФазы. Также были выявлены различия метаболизма у старых и молодых крыс при ишемии и реперфузии мозга [18, 19].
При изучении in vitro механизмов защитного эффекта инсулина и других протекторов при ишемии и реперфузии мозга используются разные модели. С одной стороны, это модели, объясняющие модуляция активности каких сигнальных систем тем или иным протектором, включая инсулин, способна снижать образование активных форм кислорода (АФК) и нормализовать метаболизм в ткани мозга. С другой стороны, это модели, пытающиеся показать, каким образом нейропротекторы противостоят повреждающему действию ДГК и последующему нарушению метаболизма, связанному с прекращением депривации этих соединений. Экспериментальный подход с использованием депривации глюкозы и кислорода (ДГК) в культурах нейронов in vitro представляет собой попытку смоделировать ограничение или прекращение кровоснабжения, которое наблюдается при ишемии нервной ткани, и приводит к резкому снижению уровня кислорода и глюкозы в мозге. Восстановление уровня глюкозы и оксигенации клеток соответствует восстановлению кровотока, что соотносится с реперфузионным повреждением. Следует отметить, что исследования механизма нейропротекторного действия инсулина на нейроны в культуре, подвергнутые депривации глюкозы и кислорода [20, 21], являются пока единичными и неполными.
Целью настоящей работы является изучение способности инсулина оказывать нейропротекторный и антиоксидантный эффекты, предотвращая гибель нейронов коры мозга и метаболические нарушения в них при депривации глюкозы и кислорода и последующем восстановлении снабжения клеток этими соединениями. Проведенное исследование направлено на выяснение механизма нейропротекторного эффекта инсулина при ишемии и реперфузии мозга.
МЕТОДЫ ИССЛЕДОВАНИЯ
Материалы. Использовали перекись водорода, бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (МТТ), додецилсульфат натрия, дихлородигидрофлуоресцеин-диацетат, NADH и инсулин, приобретенные у фирмы (Sigma, США). Среду инкубации Neurobasal medium и ростовые добавки B-27 и B-27 без инсулина закупали у фирмы Gibco (Великобритания). В работе использовались диметилформамид фирмы Вектон (Россия) и раствор Хенкса от фирмы Биолот (Россия).
Выделение и культивирование нейронов коры мозга. Выделение нервных клеток проводили из коры мозга эмбрионов крыс линии Wistar на 17–18-й день развития согласно модифицированному методу Дичтера [22], как это описано ранее [23]. Полученные клетки 2 раза промывали раствором Хенкса и осторожно ресуспендировали в 3 мл среды Neurobasal medium, содержащей 2% ростовой добавки B-27, 2 мМ глутамина, 100 МЕ/мл пенициллина, 100 мкг/мл стрептомицина (полная ростовая среда). Подсчет клеток для оценки плотности их посадки проводили в камере Фукса-Розенталя после окрашивания клеток трипановым синим. Первичную культуру нейронов выращивали в полной ростовой среде при 37°С, 5% СО2 и влажности 100% в инкубаторе “Binder С-150” (Германия). Каждые 3 дня половину клеточной среды заменяли свежей средой.
Модель депривации глюкозы и кислорода in vitro. Опыты начинали на 11–12-й дни культивирования клеток в полной ростовой среде. До начала опытов проводили деаэрацию фосфатного буфера, использующегося для промывки клеток, и депривационной среды, представляющей собой Neurobasal medium без глюкозы и пирувата, газовой смесью, содержащей 95% N2 и 5% CO2 в течение 30 мин. Затем нейроны промывали деаэрированным фосфатным буфером, переводили в деаэрированную депривационную среду и вносили инсулин в разных концентрациях. Контрольные клетки промывали фосфатным буфером и переводили в среду, аналогичную по составу депривационной, но с добавлением глюкозы до ее концентрации, присутствующей в полной ростовой среде. Клетки содержали в инкубаторе при 37°С, 5% СО2 и влажности 100%. Для окончания депривации среду заменяли на полную ростовую среду. Через 24 ч оценивали жизнеспособность нейронов коры мозга с помощью МТТ-метода. Исследование антиоксидантного эффекта инсулина проводили через 40 мин после окончания депривации. Для анализа механизмов, лежащих в основе проявляемого инсулином нейропротекторного действия, проводили отбор проб сразу после окончания одночасовой ДГК и через 3 и 24 ч после последующего содержания нейронов в полной ростовой среде. Пробы затем анализировали с помощью иммуноблоттинга, определяя отношение фосфорилированных и исходных протеинкиназ, что позволяло судить об активности этих протеинкиназ на разных этапах проведения экспериментов.
Определение жизнеспособности нейронов коры мозга с помощью МТТ метода. Для определения жизнеспособности нейронов использовали колориметрический МТТ-метод. Он основан на восстановлении митохондриями жизнеспособных клеток МТТ в окрашенный МТТ-формазан [24]. Опыт начинали на 11-й день инкубации in vitro. Среда инкубации при проведении этих опытов описана в предыдущем разделе. Для проведения МТТ-теста 5 × 106 клеток на лунку высеивали в предварительно покрытые поли-D-лизином 24-луночные планшеты, МТТ-тест проводили, как это было описано ранее [25]. К пробам добавляли МТТ в конечной концентрации 0.05%, инкубировали в течение 2 ч, затем к пробам добавляли лизирующий раствор (в количестве 167 мкл на лунку) и проводили измерение экстинкций проб. Для приготовления лизирующего раствора использовали 36 г додецилсульфата натрия, 90 мл диметилформамида и 90 мл 0.1 N соляной кислоты. Экстинкцию контрольных проб принимали за 100%, экстинкции других проб выражали в процентах от контроля. При обобщении данных всех поставленных опытов вычисляли в каждом опыте процент клеток, гибель которых была предотвращена в результате преинкубации с инсулином в разных концентрациях (“rescue rates”). Для этого вычисляли разницу между экстинкциями проб при действии на нейроны перекиси водорода после преинкубации с инсулином и при действии одной перекиси водорода. Эту величину делили на разницу между экстинкциями контрольных проб и проб, подвергнутых действию перекиси водорода, и умножали на 100. При отсутствии защитного эффекта инсулина числитель равен 0, как и процент клеток, гибель которых была предотвращена преинкубацией с инсулином.
Для оценки влияния инсулина на жизнеспособность клеток нейроны инкубировали с разными концентрациями инсулина в диапазоне от 50 нМ до 10 мкМ (либо без него) на стадии ДГК и с теми же концентрациями инсулина на стадии “реперфузии” – при последующей инкубации в полной ростовой среде. К пробам добавляли МТТ в конечной концентрации 0.05% за 2 ч до окончания инкубации, а затем – лизирующий раствор (20%-ный раствор додецилсульфата натрия в 50%-ном диметилформамиде в 0.05 N соляной кислоте). Экстинкцию контрольных проб принимали за 100%, экстинкции других проб выражали в процентах от контроля [25].
При обобщении данных всех опытов вычисляли в каждом из них процент клеток, гибель которых была предотвращена в результате преинкубации с инсулином в разных концентрациях (“rescue rates”), как это описано ранее [26].
Определение образования активных форм кислорода в нейронах коры мозга крыс. При изучении антиоксидантного эффекта инсулина в условиях депривации глюкозы и кислорода использовали раствор Рингера-Локка без глюкозы в качестве депривационной среды. Клетки промывали деаэрированным буфером Дальбекко и переводили в деаэрированный раствор Рингера-Локка (154 мM NaCl, 5.6 мM KCl, 2.3 мM CaCl2, 1 мM MgCl2, 3.6 мM NaHCO3, 5 мM HEPES, pH 7.25), в который вносили инсулин до конечной концентрации 50, 500 нМ, 1, 5 и 10 мкМ. В раствор для контрольных клеток добавляли глюкозу до конечной концентрации 25 мМ. После одночасовой ДГК к экспериментальным клеткам добавляли глюкозу до конечной концентрации 25 мМ, а к контрольным клеткам – воду. Нейроны инкубировали с 10 мкМ дихлородигидрофлуоресцеин диацетатом в течение 40 мин. Затем клетки 2 раза промывали раствором Хенкса. После этого флуоресценцию продукта реакции АФК с дихлордигидрофлуоресцеином измеряли на планшетном флуориметре “Fluoroscan FL” (“Thermo Fisher Scientific”, Финляндия) при длине волны возбуждения 485 нм и длине волны эмиссии 538 нм. Содержание АФК выражали в условных единицах, отражающих интенсивность флуоресценции в клетках.
Определение влияния инсулина и ДГК с последующей инкубацией в полной ростовой среде на уровень фосфорилированных протеинкиназ и их экспрессию. Для анализа механизмов, лежащих в основе проявляемого инсулином защитного действия, проводили отбор проб сразу после окончания депривации и через 3 и 24 ч после их инкубации в полной ростовой среде, которые затем анализировали с помощью иммуноблоттинга. Для иммуноблоттинга нейроны коры мозга высевали в 35 мм чашки Петри в количестве 2 × 106 клеток на чашку. Клетки инкубировали в депривационной среде (или в соответствующей контрольной среде) с разными концентрациями инсулина или без него. Сразу же после окончания воздействия ДГК или через последующие 3 или 24 ч содержания нейронов в полной ростовой среде (с добавлением тех же концентраций инсулина, что и на стадии ДГК) нейроны промывали 2 раза охлажденным фосфатным буфером и лизировали в буфере, содержащем фосфатазные и протеазные ингибиторы: 50 мМ трис рН 8.0, 150 мМ NaCl, 1% Тритон Х-100, 5 мМ ЭДТА, 10 мМ бета-глицерофосфата натрия, 10 мМ NaF, 1 мМ Na3VO4, 1 мМ фенилметансульфонил фторида и протеазный ингибиторный коктейль (Roche, Швейцария). Концентрацию белка в пробах определяли по модифицированному методу Лоури [26]. Из каждой пробы брали аликвоту, содержащую 25 мкг белка и разделяли методом электрофореза в 10%-ном полиакриламидном геле. Затем белки переносили на нитроцеллюлезные мембраны (Amersham, GE, Healthcare, UK). Неспецифическое связывание мембран блокировали, как это было описано ранее [26]. Блоты затем в течение ночи подвергали действию моноклональных антител, специфичных для тех соединений, уровень которых подлежал определению. В опытах использовали первичные антитела против pAkt (Ser473) (1:1000), Akt (1:1000), pGSK-3beta (Ser9) (1:1000), GSK-3beta (1:1000), ERK1/2 (1:1000) от Cell Signaling Technology(США). Антитела против pERK1(Thr202/Tyr204), ERK2 (Thr185/Tyr187) (1:2000), альфа-тубулина (1:2000) были произведены фирмой Sigma-Aldrich (США).
Блоты промывали три раза 0.1%-ным Tween 20 в солевом растворе Трис-буфера и инкубировали либо с HRP-IgG антимышиным, либо с HRP-IgG антикроличьим вторичным антителом (Cell Signaling Technology, США) в течение 1 ч при комнатной температуре. Для определения изменения экспрессии общих ERK1/2, Akt и GSK-3beta мембраны после окраски на фосфорилированные формы этих протеинкиназ подвергались стриппингу и затем окрашивали, используя антитела для этих протеинкиназ. Процедура стриппинга проводилась так, как это было описано ранее [26]. Для того, чтобы нормализовать полученные данные, в качестве референсного белка использовался альфа-тубулин (1 : 2000 Sigma-Aldrich, США). Определяли отношение протеинкиназ к альфа-тубулину и принимали его за 1.0 в контрольных нейронах коры мозга. Определяли отношения pERK1/2/ERK1/2, pAkt (Ser473)/Akt, pAkt (Thr308)/Akt, pGSK-3beta (Ser9)/GSK-3beta, их значения в контрольных клетках принимая за 1.0.
Сигнал вторичных антител HRP-IgG (1 : 1000, Cell Signaling) усиливали с помощью реактива Novex ECL HRP Reagent Kit (Invitrogen, Waltham, Massachusetts, США). Фотопленки сканировали на сканере Canon (CanoScan 8800F). Денситометрическую обработку данных проводили с помощью программы Bio 7.
Статистическая обработка данных. Статистическую достоверность различий определяли, применяя t критерий Стьюдента, используя преимущественно метод попарных сравнений. Различия считали достоверными при р < 0.05. Все данные представлены как среднее арифметическое значение ± стандартная ошибка среднего.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Экспериментальный подход с использованием депривации глюкозы и кислорода нейронов коры мозга в культуре в опытах in vitro позволяет смоделировать ограничение кровоснабжения нервной ткани при ишемии мозга. Оно сопровождается резким снижением уровня глюкозы и кислорода в крови и вызывает нарушения обмена веществ в ткани мозга. При этом восстановление уровня глюкозы в среде инкубации нейронов коры мозга и их оксигенации в нашем случае соответствует восстановлению кровотока в ткани мозга, что соотносится с реперфузионным повреждением нервной ткани.
При изучении защитного действия инсулина при ДГК инсулин добавляли непосредственно перед ДГК, которую осуществляли путем переноса клеток в депривационную среду Neurobasal без глюкозы и пирувата, деаэрированную газовой смесью следующего состава – 95% N2, 5% CO2. Затем клетки инкубировали в течение 1 или 3 ч в вышеуказанной жидкой среде, соответственно пробы все это время находились в среде, не содержащей глюкозу и кислород.
Через 1 или 3 ч проводили замену депривационной среды на полную ростовую среду, содержащую не только Neurobasal, глюкозу и кислород, но и набор нейротрофических факторов, кроме инсулина, входящих в добавку B-27. При переводе в полную ростовую среду в пробы добавляли инсулин в исследуемых концентрациях. На этой стадии среда инкубации контрольных нейронов и нейронов в опыте не различалась между собой. Анализ жизнеспособности нейронов осуществляли через 24 ч после перевода нейронов в полную ростовую среду. Эта стадия соответствует стадии реперфузии при ишемическом воздействии in vivo. Исследовали также влияние на нейроны коры мозга ДГК, которое не сопровождалось инкубацией в полной ростовой среде в присутствии глюкозы и кислорода (0 ч).
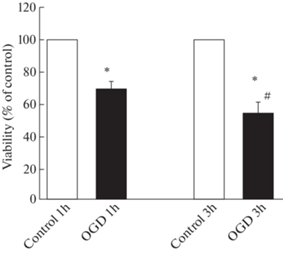
Нами показано, что ДГК в течение 1 ч и 3 ч приводила к значительной гибели нейронов, если после этого воздействия нейроны инкубировали 24 ч в полной ростовой среде (p < 0.01 по сравнению с контрольными клетками) (рис. 1). Трехчасовая ДГК оказывала более выраженное воздействие на гибель нейронов по сравнению с одночасовой ДГК. При 1 ч ДГК жизнеспособными оставались примерно 70% нейронов, а при 3 ч ДГК – лишь около 50% нейронов (рис. 1).
Рис. 1.
Влияние депривации глюкозы и кислорода (ДГК) в течение 1 и 3 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, содержащей глюкозу и кислород, на жизнеспособность нейронов коры мозга в культуре.
Данные представляют собой среднее ± SEM из 8 поставленных опытов. OGD – oxygen and glucose deprivation. Различия достоверны по t критерию Стьюдента по сравнению: * – с контролем, p < 0.001, # – с воздействием ДГК в течение 1 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, p < 0.05.
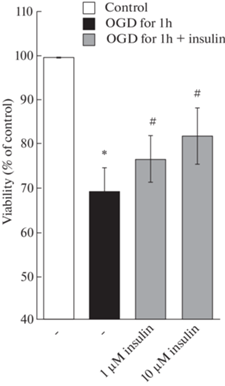
Изучение нейропротекторных свойств инсулина было проведено в диапазоне его концентраций 500 нМ – 10 мкМ. Инкубация нейронов коры мозга крыс с инсулином в микромолярных концентрациях (1 и 10 мкМ), достоверно повышала жизнеспособность нейронов, которые были подвергнуты ДГК в течение 1 ч (рис. 2), тогда как не удалось показать достоверного эффекта наномолярных концентраций этого нейропротектора. При этом величина “rescue rates”, определяющая процент клеток, гибель которых была предотвращена инкубацией с инсулином, при добавлении в среду инкубации 1 мкМ инсулина составляли 26.2 ± 7.4%, а при добавлении 10 мкМ инсулина – 45.2 ± 13.5% (эффект инсулина в обоих случаях достоверен, p < 0.02).
Рис. 2.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 1 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, содержащей глюкозу и кислород, на жизнеспособность нейронов коры мозга в культуре.
Данные представляют собой среднее ± SEM из 7–8 поставленных опытов. OGD – oxygen and glucose deprivation. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.001, # – с воздействием ДГК в течение 1 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, p < 0.05.
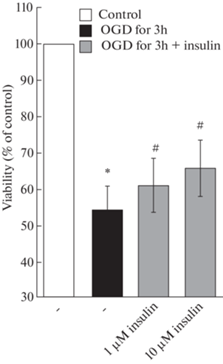
При воздействии ДГК в течение 3 ч защитное действие инсулина также наблюдалось в его концентрациях 1 и 10 мкМ, при этом процент нейронов, гибель которых была предотвращена преинкубацией с 1 и 10 мкМ инсулина, составлял 22.1 ± 8.9 и 32.3 ± 10.1% соответственно (рис. 3). Эффект инсулина в обеих концентрациях достоверен, p < 0.05. Как при одночасовой, так и при трехчасовой ДГК имела место тенденция к более выраженному эффекту 10 мкМ инсулина, чем 1 мкМ этого протектора (p < 0.1), но различия не достигали достоверности.
Рис. 3.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 3 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, содержащей глюкозу и кислород, на жизнеспособность нейронов коры мозга в культуре.
Данные представляют собой среднее ± SEM из 7–8 поставленных опытов. OGD – oxygen and glucose deprivation. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.001, # – с воздействием ДГК в течение 3 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, p < 0.05.
Изучение антиоксидантных свойств инсулина было проведено в диапазоне его концентраций 500 нМ–10 мкМ. Достоверное снижение образования АФК наблюдалось при добавлении к пробам инсулина в его микромолярных концентрациях – 1 и 10 мкМ (рис. 4). При инкубации инсулина с контрольными нейронами не обнаружено достоверных изменений в образовании АФК ни в одной из исследованных концентраций, на рисунке представлены данные о влиянии 10 мкМ инсулина на этот показатель в контрольных нейронах (рис. 4).
Рис. 4.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 1 ч с последующей инкубацией в течение 40 мин в среде Neurobasal на образование активных форм кислорода (АФК) в нейронах коры мозга в культуре.
Данные представляют собой среднее ± SEM из результатов поставленных опытов (n = 8 для контроля и воздействия ДГК, n = 4 для данных по эффектам разных концентраций инсулина на нейроны при ДГК). OGD – oxygen and glucose deprivation. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.001, # и ## – с воздействием депривации глюкозы и кислорода в течение 1 ч с последующей инкубацией в течение 40 мин в среде Neurobasal, # – p < 0.05, ## – p < 0.02.
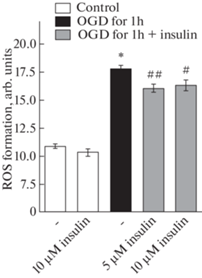
ДГК в течение 1 ч с последующей инкубацией в течение 24 ч в полной ростовой среде, содержащей глюкозу и кислород, приводила к значительному увеличению уровня АФК в нейронах (p < 0.01). Достоверное снижение накопления АФК в нейронах в условиях ДГК отмечалось только при инкубации с 5 и 10 мкМ инсулином, причем инсулин в обеих концентрациях действовал с примерно одинаковой эффективностью. При этом уровень АФК оставался сильно повышенным по сравнению с контролем (рис. 4).
Для выяснения механизмов защитного и антиоксидантного действия инсулина нами исследовалось его влияние на активность ряда протеинкиназ и сигнальных путей, от активности которых зависит жизнеспособность, либо, напротив, гибель нейронов. Для проведения этих исследований нами была выбрана концентрация инсулина, равная 10 мкМ, как наиболее эффективная. В этой серии опытов применялась ДГК продолжительностью 1 ч, как модель с более мягким воздействием на нейроны, чем модель с трехчасовой ДГК. Отбор проб осуществляли как сразу после воздействия ДГК, так и через 3 и 24 ч после инкубации нейронов в полной ростовой среде.
Показано, что ДГК приводила к достоверному снижению уровня фосфорилированной формы Akt в нейронах коры мозга, о чем судили по снижению отношения [pAkt(Ser473)/Akt] (рис. 5с). Оно наблюдалось сразу же после воздействия ДГК и через 3 ч после последующей инкубации нейронов в полной ростовой среде (рис 5с). Если же инкубация в полной ростовой среде после воздействия ДГК происходила в течение 24 ч, то активность Akt (pAkt (Ser 473)/Akt) сильно падала по сравнению с начальным контрольным значением, принятым за 1.0. Эти данные показывают, что ДГК приводит к достоверному снижению активности этой протеинкиназы. Инсулин достоверно увеличивал активность протеинкиназы Akt в нейронах после ДГК и инкубации нейронов в полной ростовой среде в течение 24 ч (рис. 5a, c), а также через 24 ч в контрольных нейронах (рис. 5b). Но при инкубации в течение 0 и 3 ч в полной ростовой среде инсулин не вызывал достоверного увеличения отношения pAkt(Ser473)/Akt в опытных и контрольных нейронах, наблюдалась лишь тенденция к такому эффекту (рис. 5).
Рис. 5.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 1 ч с последующей инкубацией в полной ростовой среде, содержащей глюкозу и кислород, на активность протеинкиназы B (Akt) в нейронах коры мозга в культуре [pAkt(Ser473)/Akt].
Данные представляют собой среднее ± SEM из 4 поставленных опытов. OGD – oxygen and glucose deprivation. (a) – иммуноблотты. (b) – влияние инсулина на активность Akt в контрольных нейронах. (c) – влияние ДГК и инсулина на активность Akt. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.05, & – с первоначальным контролем, принятым за 1.0, p < 0.01, # – с действием ДГК в течение 1 ч, с последующей инкубацией в полной ростовой среде, p < 0.05.
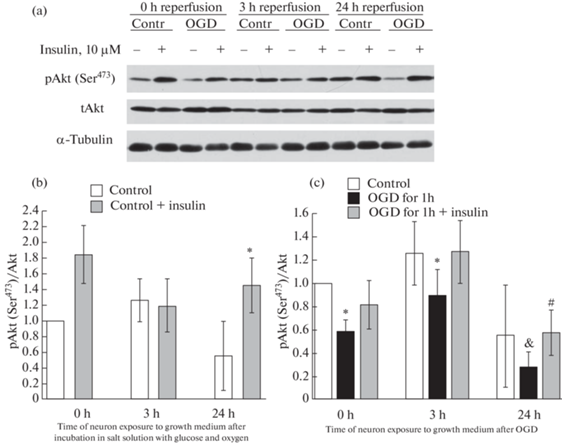
Следует отметить, что контрольные нейроны не испытывают недостатка в глюкозе и кислороде ни на одной из стадий опыта. Но они подвержены недостатку нейротрофических факторов при их содержании в солевой среде, тогда как в этот же период инкубации опытные нейроны страдают не только от недостатка в депривационной среде нейротрофических факторов, но и от недостатка глюкозы и кислорода.
Одной из основных мишеней протеинкинзы Akt является киназа гликогенсинтазы-3бета (GSK-3beta). Протеинкиназа B (Akt) усиливает фосфорилирование GSK-3beta по Ser9, что приводит к инактивации этой протеинкиназы. Этот эффект благоприятно сказывается на функциях митохондрий и приводит к увеличению жизнеспособности нервных клеток, в частности, в результате предотвращения их апоптотической гибели.
Под действием ДГК наблюдалось достоверное (p < 0.02) уменьшение уровня pGSK-3beta (Ser9)/GSK-3beta в нейронах коры мозга как сразу после этого воздействия, так и через 24 ч инкубирования в полной ростовой среде, содержащей глюкозу и кислород (рис. 6a, c). Эти данные показывают, что ДГК противодействует процессам инактивации GSK-3beta и способствует активации этой протеинкиназы и, как следствие, вызывает ухудшение функций митохондрий в нейронах коры мозга.
Рис. 6.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 1 ч с последующей инкубацией в полной ростовой среде, содержащей глюкозу и кислород, на инактивацию GSK-3beta в нейронах коры мозга в культуре [pGSK-3beta (Ser9)/GSK-3beta].
Данные представляют собой среднее ± SEM из 4 поставленных опытов. OGD – oxygen and glucose deprivation. (a) – Иммуноблотты. (b) – Влияние инсулина на активность GSK-3beta в контрольных нейронах. (c) – Влияние ДГК и инсулина на активность GSK-3beta в нейронах. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.02, & – с первоначальным контролем, принятым за 1.0, p < 0.01, # – с действием ДГК в течение 1 ч, с последующей инкубацией в полной ростовой среде, p < 0.05.
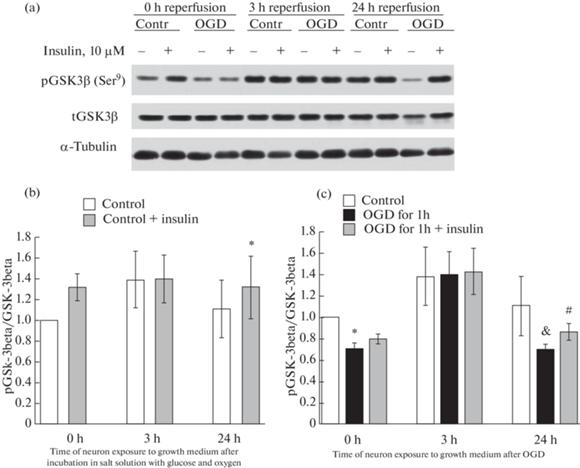
Инсулин же, напротив, способствовал инактивации GSK-beta. Так, как видно из данных (рис. 6), он достоверно увеличивал образование ее фосфорилированной по Ser9 формы в контрольных и подвергшихся воздействию ДГК нейронах через 24 ч после их инкубации в полной ростовой среде (p < 0.01 и p < 0.05 соответственно). Кроме того, в его присутствии не наблюдалось достоверного снижения уровня pGSK-3beta (Ser9) при действии ДГК без последующей инкубации нейронов в полной ростовой среде, которое имело место в отсутствие инсулина в среде (рис. 6c).
Нами было также изучено влияние ДГК и инсулина на активность ERK1/2 в нейронах коры мозга. Не выявлено влияние инсулина на активность фермента в контрольных нейронах (рис. 7a,b), а также в нейронах, подвергнутых ДГК без последующей инкубации в полной ростовой среде.
Рис. 7.
Влияние инсулина и депривации глюкозы и кислорода (ДГК) в течение 1 ч с последующей инкубацией в полной ростовой среде, содержащей глюкозу и кислород, на активность киназы, регулируемой внеклеточными сигналами (ERK1/2) (отношение pERK1/2 / ERK1/2) в нейронах коры мозга в культуре.
Данные представляют собой среднее ± SEM из 4 поставленных опытов. OGD – oxygen and glucose deprivation. (a) – Иммуноблотты. (b) – Влияние инсулина на активность ERK1/2 в контрольных нейронах. (c) – Влияние ДГК и инсулина на активность ERK1/2 в нейронах. Различия достоверны по t критерию Стьюдента при оценке методом попарных сравнений по сравнению: * – с контролем, p < 0.05, & – с первоначальным контролем, принятым за 1.0, p < 0.02, # – с действием ДГК в течение 1 ч, с последующей инкубацией 24 ч в полной ростовой среде, p < 0.05.
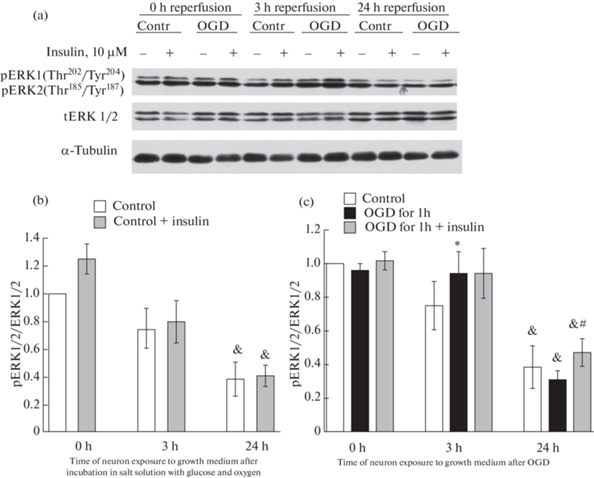
Наиболее разительные изменения активности ERK1/2 в нейронах коры мозга выявлены через 24 ч инкубации контрольных нейронов и нейронов, подвергнутых ДГК, в полной ростовой среде. В этом случае активность ERK1/2 (о которой судили по отношению pERK1/2 / ERK1/2) в контрольных нейронах и нейронах, подвергнутых ДГК, была снижена примерно в 3 раза. По-видимому, это снижение является результатом влияния инкубации нейронов в течение 1 ч в солевой среде, лишенной нейротрофических факторов. Наличие инсулина в пробах достоверно увеличивало активность ERK1/2 в нейронах коры мозга, подвергнутых ДГК в течение 1 ч, и затем 24 ч инкубации в полной ростовой среде, однако активность фермента продолжала оставаться сниженной по сравнению с исходным контролем (рис. 7).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Мы исследовали влияние депривации глюкозы и кислорода и влияние инсулина на жизнеспособность нейронов коры мозга в культуре. Основные нарушения обменных процессов и повреждение нейронов в опытах in vivo наступают, как правило, после реперфузии. Поэтому мы определяли жизнеспособность нейронов после ДГК продолжительностью 1 и 3 ч и последующей “реперфузии”, т.е. восстановления воздействия глюкозы и кислорода на нейроны, инкубируя их в течение 24 ч в полной ростовой среде. Полученные данные показали гибель значительной части нейронов при таких воздействиях. Особенно ярко она была выражена при воздействии ДГК на нейроны в течение 3 ч. Добавление инсулина в микромолярных концентрациях в среду инкубации клеток оказывало нейропротекторный эффект и достоверно повышало жизнеспособность нейронов. Наши данные о токсическом эффекте ДГК на нейроны в культуре и защитном эффекте инсулина согласуются с литературными данными, полученными на нейронах гиппокампа и зубчатой извилины [20, 21]. Было найдено, что наибольшей чувствительностью к защитному действию против ДГК инсулина, использовавшегося, как и в наших опытах, в микромолярных концентрациях, обладают органотипические нейроны срезов С2 района гиппокампа и зубчатой извилины, тогда как нейроны С1 и С3 районов гиппокампа менее чувствительны к действию этого гормона [21]. Как будет показано далее в тексте обсуждения, защитный эффект инсулина согласно данным этой работы (как и согласно нашим данным) определялся прежде всего его способностью активировать протеинкиназу B (Akt). Интересно, что в другой работе [20] было найдено, что, если подвергнуть нейроны гиппокампа ДГК и последующему восстановлению содержания глюкозы и кислорода в среде, то это приводит к значительному уменьшению количества GABA рецепторов на поверхности нейронов при практически неизменном количестве экспрессированных суммарных рецепторов. При действии же на нервные клетки инсулина он противодействовал уменьшению числа GABA рецепторов, по-видимому, снижая их эндоцитоз, возможно благодаря модуляции протеинкиназы С [20]. Воздействие инсулина приводило к восстановлению баланса между возбуждающими и ингибирующими сигналами на нейроны [20]. В литературе есть и другие свидетельства того, что эксайтотоксичность может быть одной из причин гибели нейронов при воздействии на них ДГК [см., например, 27], так что эти данные об эффекте инсулина представляют интерес.
Но в более поздней публикации [21] и в нашей работе было показано, что защитный эффект инсулина связан с активацией протеинкиназы B (Akt) и последующих сигнальных путей, что характерно для механизма нейропротекторного эффекта инсулина при разных токсических воздействиях. Это не исключает возможности противодействия инсулина к эксайтотоксическим воздействиям при ДГК.
Одной из клеточных моделей, пригодных для изучения механизма защитного действия нейропротекторов при ряде патологических состояний мозга, включая его нейродегенеративные и ишемические поражения, являются нейроны в состоянии окислительного стресса, вызванного действием прооксиданта. Это объясняется тем, что одной из основных причин гибели нейронов при этих поражениях мозга является чрезмерная активация свободнорадикальных процессов. В роли такого природного прооксиданта нами ранее была использована перекись водорода. Были проведены исследования, посвященные механизму защитного действия инсулина, используя эту модель повреждающего действия на клетки мозга [28–30]. Было показано, что инсулин повышает жизнеспособность нейронов в условиях окислительного стресса, обладает хорошо выраженным антиоксидантным действием.
При сравнении результатов, полученных ранее на модели окислительного стресса [28–30], для индукции которого в нейронах коры мозга применялось воздействие 100 мкМ перекиси водорода в течение 6 ч, и в настоящей работе, можно сделать ряд предположений и выводов. По-видимому, ДГК обладает более выраженным токсическим эффектом на нейроны, чем воздействие перекиси водорода. Так, для защиты нейронов от ДГК потребовались относительно высокие микромолярные концентрации инсулина – 1–10 мкМ (рис. 2–4). А при защите от перекиси водорода инсулин был эффективным уже в концентрации 10–100 нМ [28–30], при этом дальнейшее увеличение концентрации инсулина не приводило к достоверному увеличению его защитного эффекта по сравнению со 100 нМ инсулина. А на модели ДГК инсулин обладал защитным эффектом лишь в микромолярных концентрациях.
Нами найдено, что ДГК снижает активность протеинкиназы B (Akt) в нейронах коры мозга сразу после ДГК. Снижение имеет место и после последующей инкубации клеток в полной ростовой среде в течение 24 ч. Инсулин же активирует протеинкиназу Akt в нейронах коры мозга как непосредственно после ДГК, так и при последующей инкубации в полной ростовой среде в течение 24 ч (рис. 5). Эти результаты согласуются с данными, ранее полученными с использованием ингибиторов протеинкиназ [21], о том, что нейропротекторный эффет инсулина на модели ДГК определяется прежде всего его способностью активировать протеинкиназу B (Akt).
Как известно, активация протеинкиназы Akt инсулином может приводить к уменьшению апоптотической гибели нейронов благодаря увеличению фосфорилирования GSK-3beta по Ser9, что приводит к инактивации этой протеинкиназы (GSK-3beta является одной из основных мишеней Akt). Наряду с этим активация Akt способствует усилению синтеза антиапоптотического белка Bcl-2. При активации GSK-3beta имеет место нарушение функций митохондрий, падение мембранного потенциала митохондрий, открытие mitochondrial permeability transition pores и усиление апоптотической гибели нервных клеток. Ингибирование этой протеинкиназы, напротив, способствует увеличению жизнеспособности нервных клеток.
В настоящей работе нами показано, что ДГК приводит к активации GSK-3beta как непосредственно после этого воздействия, так и через 24 ч после последующей инкубации в полной ростовой среде (рис. 6). Инсулин способен достоверно уменьшать активность фермента в нейронах после действия ДГК и последующей их инкубации в полной ростовой среде в течение 24 ч (т.е. после “ишемии” и “реперфузии”), как и в контрольных нейронах (рис. 6). По-видимому, активация инсулином Akt и последующая инактивация GSK-3beta играют важную роль в механизме его нейропротекторного эффекта при поражении мозга в результате ишемии и последующей его реперфузии.
Аналогичные данные о механизме защитного действия инсулина были получены нами на модели воздействия на нейроны коры мозга окислительного стресса, вызванного действием такого природного прооксиданта, как перекись водорода [28–30]. Было показано, что окислительный стресс приводит к инактивации Akt, активации GSK-3beta и увеличению отношения Bax/Bcl-2, тогда как инсулин, напротив, активирует Akt и инактивирует GSK-3beta, нормализуя активность этих протеинкиназ и отношение Bax/Bcl-2 в условиях окислительного стресса [28–30]. При этом найдено, что нейропротекторный эффект инсулина достоверно снижается в присутствии ингибиторов активации протеинкиназы B (Akt) и инактивации GSK-3beta [30]. Таким образом, используя обе модели ишемического поражения мозга (ДГК и окислительный стресс), выявлен сходный механизм защитного действия инсулина, заключающийся в активации протеинкиназы B (Akt) и инактивации GSK-3beta.
Сложнее объяснить данные, полученные при изучении влияния ДГК и последующего восстановления содержания глюкозы и кислорода в среде на активность ERK1/2 в нейронах коры мозга. Происходило очень значительное снижение активности фермента после воздействия ДГК (или солевого раствора в случае контрольных клеток) и последующей инкубации в течение 24 ч в полной ростовой среде. Инсулин достоверно повышал активность фермента, но она оставалась сильно сниженной по сравнению с исходными значениями. Аналогичного снижения активности ERK1/2 мы не наблюдали в динамике развития окислительного стресса в нейронах коры мозга, вызванного перекисью водорода ни на одной из его стадий. Можно высказать предположение о том, что активность этой протеинкиназы наиболее чувствительна к временной инкубации в солевой среде, лишенной нейротрофических факторов, что при последующей инкубации нейронов в полной ростовой среде проявляется на относительно поздних этапах после воздействия.
Список литературы
Chen Y, Guo Z, Mao YF, Zheng T, Zhang B (2018) Intranasal insulin ameliorates cerebral hypometabolism, neuronal loss, and astrogliosis in streptosotocin-induced Alzheimer’s rat model. Neurotox Res 33: 716–724. https://doi.org/10.1007/s12640-017-9809-7
Song Y, Ding W, Bei Y, Xiao Y, Tong HD, Wang LB, Ai LY (2018) Insulin is a potential antioxidant for diabetes-associated cognitive decline via regulating Nrf2 dependent antioxidant enzymes. Biomed Pharmacother 104: 474–484. https://doi.org/10.1016/j.biopha.2018.04.097
Fine JM, Stroebel BM, Faltesek KA, Terai, K; Haase L, Knutzen K.E, Kosyakovsky J, Bowe TJ, Fuller A.K, Frey WH, Hanson LR (2020) Intranasal delivery of low-dose insulin ameliorates motor dysfunction and dopaminergic cell death in a 6-OHDA rat model of Parkinson’s Disease. Neurosci Lett 714: 134567. https://doi.org/10.1016/j.neulet.2019.134567
Milstein JL, Ferris HA (2021) The brain as an insulin-sensitive metabolic organ. Mol Metab. 52: 101234. https://doi.org/10.1016/j.molmet.2021.101234
Claxton A, Baker LD, Hanson AJ, Trittschuh EH, Collerton B, Morgan A, Callaghan M, Arbuckle M, Behl C, Craft S (2015) Long-acting insulin Detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis 44: 897–906. https://doi.org/10.3233/JAD-141791
Craft S, Claxton A, Baker LD, Hanson AJ, Collerton B, Trittschuh EH, Dahl D, Caulder E, Neth B, Montine TJ, Jung Y, Maldjian J, Whitlow C, Friedman S (2017) Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: A pilot clinical trial. J Alzheimers Dis 57: 1325–1334. https://doi.org/:10.3233/JAD-161256
Avgerinos KI, Kalaitzidis G, Malli A, Kalaitzoglou D, Myserlis PG, Lioutas VA (2018) Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment. A systematic review. J Neurol 265: 1497–1510. https://doi.org/10.1007/s00415-018-8768-0
Novak P, Maldonado DAP, Novak V (2019) Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS One 14: e0214364. https://doi.org/10.1371/journal.pone.0214364
Lochhead JJ, Kellohen KL, Ronaldson PT, Davis TP (2019) Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci Rep 9: 2621. https://doi.org/10.1038/s41598-019-39191-5
Fan LW, Carter K, Beatt A, Pang Y (2019) Rapid transport of insulin to the brain following intranasal administration in rats. Neural Regen Res 14: 1046–1051. https://doi.org/10.4103/1673-5374.250624
Hallschmid M (2021) Intranasal insulin. J Neuroendocrinol 33: e12934. https://doi.org/10.1111/jne.12934
Shpakov AO, Derkach KV, Berstein LM (2015) Brain signaling systems in the Type 2 diabetes and metabolic syndrome: promising target to treat and prevent these diseases. Future Sci OA 1: FSO25. https://doi.org/10.4155/fso.15.23
Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428: 569–574. https://doi.org/10.1038/nature02440
Rizk NN, Myatt-Jones.J, Rafols J, Dunbar JC (2007) Insulin-like growth factor-1 (IGF-1) decreases ischemia-reperfusion induced apoptosis and necrosis in diabetic rats. Endocrine 31: 66–71. https://doi.org/10.1007/s12020-007-0012-0
Jiang LH, Yuan XL, Yang NY, Ren., Zhao. M, Luo BX, Bian YY, Xu JY, Lu DX, Zheng YY, Zhang CJ, Diao YM, Xia BM, Chen GJ (2015) Daucosterol protects neurons against oxygen-glucose deprivation/reperfusion-mediated injury by activating IGF1 signaling pathway. Steroid Biochem Mol Biol 152: 45–52. https://doi.org/10.1016/j.jsbmb.2015.04.007
Gong P, Zou Y, Zhang W, Tian Q, Han S, Xu Z, Chen Q, Wang X, Li M (2021) The neuroprotective effects of Insulin-like growth factor-1 via the Hippo/YAP signaling pathway are mediated by the PI3K/AKT cascade following cerebral ischemia/reperfusion injury. Brain Res Bull 177: 373–387. https://doi.org/10.1016/j.brainresbull.2021.10.017
Lioutas VA, Alfaro-Martinez F, Bedoya F, Chung CC, Pimentel DA, Novak V (2015) Intranasal insulin and insulin-like growth factor-1 as neuroprotectants in acute ischemic stroke. Transl Stroke Res 6: 264–275. https://doi.org/10.1007/s12975-015-0409-7
Zorina II, Galkina OV, Bayunova LV, Zakharova IO (2019) Effect of insulin on lipid peroxidation and glutathione levels in a two-vessel occlusion model of rat forebrain ischemia followed by reperfusion. J Evol Biochem Physiol 35: 333–335. https://doi.org/10.1134/S0022093019040094
Zorina II, Fokina EA, Zakharova IO, Bayunova LV, Shpakov AO (2020) Characteristics of changes in lipid peroxidation and Na+/K+-ATPase activity in the cortex of old rats in conditions of two-vessel cerebral ischemia/reperfusion. Adv Geront 10: 156–161. https://doi.org/10.1134/s2079057020020162
Mielke JG, Wang YT (2005) Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABA receptors following oxygen-glucose deprivation in cultured cortical neurons. J Neurochem 92: 103–113. https://doi.org/10.1111/j.1471-4159.2004.02841.x
Sun X, Yao H, Douglas RM, Gu XQ, Wang J, Haddad GG (2010) Insulin/PI3K signaling protects dentate neurons from oxygen-glucose deprivation in organotypic slice cultures. J Neurochem 112: 377–388. https://doi.org/10.1111/j.1471-4159.2009.06450.x
Dichter MA (1978) Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res 149: 279–293. https://doi.org/10.1016/0006-8993(78)90476-6
Mironova EV, Evstratova AA, Antonov SM (2007) A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J Neurosci Methods 163: 1–8. https://doi.org/10.1016/j.jneumeth.2007.02.010
Hansen MB, Nielsen SE, Berg K (1989) Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 119: 203–210. https://doi.org/10.1016/0022-1759(89)90397-9
Zorina II, Bayunova LV, Zakharova IO, Avrova NF (2018) The dependence of the protective effect of insulin on its concentration and modulation of ERK1/2 activity under the conditions of oxidative stress in cortical neurons. Neurochem J 10: 111–116. https://doi.org/0.1134/S1819712417040110
Zakharova I, Sokolova T, Vlasova Y, Bayunova L, Rychkova M, Avrova N (2017) α-Tocopherol at nanomolar concentration protects cortical neurons against oxidative stress. Int J Mol Sci. 18: 216. https://doi.org/10.3390/ijms18010216
Bonde C, Noraberg J, Noer H, Zimmer J (2005) Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen-glucose deprivation of hippocampal slice cultures. Neuroscience 136: 779–794. https://doi.org/10.1016/j.neuroscience.2005.07.020
Zakharova IO, Sokolova TV, Zorina I, Bayunova LV, Rychkova MP, Avrova NF (2018) Protective effect of insulin on rat cortical neurons in oxidative stress and its dependence on modulation of protein kinase B (Akt) activity. J Evol Biochem Physiol 54: 192–204. https://doi.org/10.1134/S0022093018030043
Bayunova LV, Zorina II, Zakharova IO, Avrova NF (2018) Insulin increases viability of neurons in rat cerebral cortex and normalizes Bax/Bcl-2 atio under conditions of oxidative stress. Bull Exp Bio Med 165: 14–17. https://doi.org/10.1007/s10517-018-4088-8
Zakharova, IO, Sokolova TV, Bayunova LV, Zorina II, Rychkova MP, Shpakov AO, Avrova NF (2019) The protective effect of insulin on rat cortical neurons in oxidative stress and its dependence on the modulation of Akt, GSK-3beta, ERK1/2, and AMPK activities. Int J Mol Sci 20 (15): E3702. https://doi.org/10.3390/ijms20153702
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии