

Известия РАН. Физика атмосферы и океана, 2022, T. 58, № 5, стр. 554-565
О динамике образования сульфатов в капельной влаге атмосферы с участием ионов переходных металлов
А. Н. Ермаков a, *, А. Е. Алоян b, **, В. О. Арутюнян b
a Институт энергетических проблем химической физики им. В.Л. Тальрозе ФИЦ ХФ им. Н.Н. Семенова РАН
119334 Москва, Ленинский пр., 38, к. 2, Россия
b Институт вычислительной математики РАН
8119333 Москва, ул. Губкина, Россия
* E-mail: polclouds@yandex.ru
** E-mail: ezmakr2010@yandex.ru
Поступила в редакцию 28.03.2022
После доработки 07.04.2022
Принята к публикации 09.06.2022
- EDN: PDFBKW
- DOI: 10.31857/S0002351522050030
Аннотация
Применительно к моделированию образования сульфатов в атмосфере проводится обобщение данных лабораторных опытов по кинетике жидкофазного окисления SO2 растворенным кислородом в присутствии суммы ионов Mn/Fe. Найдено, что имеющиеся в литературе эмпирические выражения для скорости этого процесса не воспроизводят данных физического моделирования. Обсуждается механизм жидкофазного окисления SO2 с участием ионов Mn/Fe, удовлетворительно описывающий экспериментальные данные. Этот цепно-каталитический процесс осуществляется с участием свободных ион-радикалов и характеризуется разветвлением цепей. В этих рамках находит естественное толкование явление синергизма – эффекта известного неаддитивного усиления каталитического действия пары этих ионов. В работе приводятся предварительные оценки динамики образования сульфатов в капельной влаге атмосферы, указавшие на существенный вклад каталитического (нефотохимического) формирования сульфатов даже при невысокой кислотности капельной влаги и относительно низких концентрациях ионов Mn/Fe.
ВВЕДЕНИЕ
Окисление диоксида серы в атмосферной капельной влаге, сопровождающееся формированием сульфатов ([S(VI)] = [${\text{HSO}}_{4}^{ - }$] + [${\text{SO}}_{4}^{{2 - }}$]), играет ключевую роль в самоочищении атмосферы (кислотные дожди) [1]. Важную роль этот процесс играет и в образовании сульфатов в аэрозоле [2, 3]. Их формирование в атмосфере рассматривается обыкновенно с участием захватываемых из воздуха ОН, O3 и Н2O2, возникающих в фотохимических реакциях [2]. Вместе с тем в качестве оксидантов SO2 могут вовлекаться также NO2, HONO, HOCl, перекисные соединения (CH3OOH и др.) и молекулярный кислород в присутствии ионов Mn/Fe, что указывает на существование нефотохимических источников сульфатов в атмосфере [3]. На недооценку их роли в высоких широтах ранее указывалось по результатам моделирования глобального распределения сульфатов в атмосфере [4]. В последние годы участились, однако, сообщения о масштабном образовании нефотохимических сульфатов (десятки мкг м–3 ч–1) и в средних широтах (severe haze episodes) [5–8]. Для воспроизведения динамики формирования сульфатов в атмосфере, в том числе и в экстремальных условиях в [5–8], использовались WRF-Chem, WRF-Chem-AWAC [8, 9] и другие модели. При этом расчеты скорости образования сульфатов с участием ионов переходных металлов выполнялись с применением эмпирических выражений [10, 11], встречающих, однако серьезные возражения [12]. Для определения действительного участия ионов переходных металлов в формировании сульфатов в атмосфере необходимо детальное рассмотрение данных по кинетике каталитических реакций и существующих выражений для скорости реакции. Цель настоящей работы – опираясь на результаты обобщения известных данных лабораторного моделирования жидкофазного окисления SO2 в присутствии переходных металлов и многолетние исследования авторов механизма этого процесса [12–14], выявить особенности его динамики в характерных условиях атмосферы.
ПРЕДВАРИТЕЛЬНЫЕ СВЕДЕНИЯ
О происхождении сульфатов в атмосфере с участием ионов переходных металлов свидетельствуют данные прямого контроля в холодное время изотопного состава атомарного кислорода (Δ17O) в сульфатах частиц не морского происхождения (non-sea salt, NSS) [15, 16]. В соответствии с [17] до ∼200 нг/м3 в январе, например, обязано их присутствию в Арктической дымке (Arctic Haze) в Alert (Canada) именно каталитическому процессу. В [6] сообщалось и о совпадении по времени пиковых концентраций сульфатов (≈30 мкг/м3) и ионов марганца (≈70 нг/м3) в аэрозольных частицах (Baoding, апрель, 2015) при формировании дымки в Китае, что косвенно подтверждает участие этих ионов в образовании сульфатов. В [18] по результатам глобального моделирования образования сульфатов с участием различных оксидантов указывалось, что на долю каталитического процесса приходиться от 9 до 17% их содержания в атмосфере. При этом в [3] и других работах подчеркивалось, что вклад этого процесса значим лишь при достаточно высокой кислотности капель (рН ≤ 3.5), что связывалось с ухудшением растворимости соединений железа при более высоких рН.
Сам факт катализа ионами переходных металлов жидкофазного окисления диоксида серы установлен уже более века назад. Наиболее активными в их ряду принято считать ионы марганца и железа [2, 12, 18]. Несмотря на обширную библиографию [19, 20] и многочисленные ссылки в этих обзорах, механизм действия ионов Mn и Fe, как и явление синергизма парного их действия, остаются неясными [12–14]. Отчасти об этом говорит и факт использования в модели WRF-Chem эмпирических выражений для скорости реакции с участием этих ионов. Сомнения вызывает сам подход к нахождению этих выражений. В [10, 11] вклад неаддитивного усиления действия пары Mn/Fe (wMn_Fe) находили, вычитая из наблюдаемой скорости реакции (wнабл) найденные в независимых опытах скорости процессов с участием ионов марганца (wMn) и железа (wFe): wMn_Fe = wнабл – wMn – wFe. В [10] выражение для рассчитанного таким образом вклада неаддитивного усиления действия пары Mn/Fe (синергизма) при рН ≤ 4.2 имеет вид: wMn_Fe = 3.7 × × 107[Mn(II)][Fe(III)][S(IV)]/(10–pH)–0.74 моль л–1 с–1. Похожее выражение wMn_Fe = 1010 × × [Mn(II)][Fe(III)][S(IV)] моль л–1 с–1 приводится в [11] и для рН 3. Здесь [S(IV)] = [SO2] + [${\text{HSO}}_{3}^{ - }$] + + [${\text{SO}}_{3}^{{2 - }}$] – суммарная концентрация (моль/л) компонентов диоксида серы в растворе, а римские цифры при символах химических элементов обозначают валентное их состояние в растворе, но не заряд ионов.
В основе подхода к нахождению wMn_Fe [10, 11] лежит неоправданное допущение о независимости каталитической активности ионов железа и марганца. Оно находится в противоречии с активированным характером реакций инициирования окисления SO2 с участием ионов марганца [21], что делает эти ионы каталитически неактивными в рассматриваемой реакции [12]. Наблюдаемое вопреки этому быстрое окисление S(IV) в присутствии добавок этих ионов (т.н. марганцевый “катализ”), как отмечалось в [12, 22], следует приписывать инициированию с участием неустранимых (unavoidable) ионов примесного железа (∼10–8 моль/л). Здесь кавычки указывают, что явления катализа жидкофазного окисления SО2 только ионами марганца, по-видимому, вовсе не существует [12, 22]. Поэтому нельзя считать корректным и приводимое в [10, 11] выражение для коэффициента синергизма S = wнабл/(wMn + wFe), характеризующего эффект неаддитивного ускорения реакции в присутствии ионов марганца и железа. При этих вычислениях в [10] и других работах из виду упускается различие концентраций примесных ионов железа ([Fe*]) по ходу “катализа” ионами марганца и ионами Fe, вводимыми в раствор при “железном” катализе.
О МЕХАНИЗМЕ КАТАЛИЗА ИОНАМИ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Отмеченные трудности в интерпретации природы эффекта синергизма пары ионов Mn/Fe и выделении его вклада в наблюдаемую скорость реакции удается обойти, рассматривая данный жидкофазный процесс в рамках цепного механизма с участием в качестве переносчиков цепи радикалов ${\text{S}}{{{\text{O}}}_{{3 - {{5}^{ - }}}}}$ и Mn(III) [12] (см. табл. 1). Каталитическая активность ионов марганца связывается при этом с их активацией ионами железа, находящимися в растворах даже в следовых концентрациях [12, 22] (см. реакцию (1) в табл. 1). Вместе они образуют синергическую пару, в которой неактивные сами по себе ионы марганца многократно усиливают каталитические свойства ионов Fe. Явление их синергизма вызвано не только снижением энергетического барьера инициирования, связанного с вовлечением ионов железа в (1), но и с ускорением лимитирующей стадии продолжения цепи за счет быстрых реакций (10, 11) (табл. 1). Одновременно с этим в присутствии ионов марганца растет также скорость инициирования реакции (1), что вызвано смещением распределения ионов железа по зарядовым формам (ζMn = [Fe(III)]/[Fe(II)]) в пользу Fe(III) [12]. Как показано в этой публикации, распределение их концентраций регулируется в основном конкуренцией реакций (6) и разветвления цепи (9) с участием ${\text{HSO}}_{5}^{ - }$. При этом взамен исчезающего в реакции разветвления ${\text{HSO}}_{5}^{ - }$ возникает два новых переносчика цепи: Fe(III) и ${\text{SO}}_{4}^{ - }$. Это влечет за собой рост их концентраций, сопровождающийся подъемом ζMn и увеличением скорости образования сульфатов [14]. Все сказанное находится в согласии с результатами независимого прямого контроля распределения валентных форм ионов железа в аэрированных растворах и скорости образования сульфатов (рН 3.8, [S(IV)] = 2 × 10–4, [Fe]о = 1.8 × 10–6 моль/л) при возрастании в растворах содержания ионов марганца: (0.18–1.8) × × 10–6 моль/л [23]. Здесь и далее [Fe]о = [Fe(II)] + + [Fe(III)] – концентрация ионов железа в растворе.
Таблица 1.
Механизм катализа ионами марганца и железа окисления S(IV) (μ ≈ 0) [14]
| № | Реакция | Константа скорости |
|---|---|---|
| 1 | Fe(OH)SO3H+ → Fe2+ + ${\text{SO}}_{3}^{ - }$ + H2O | 0.2* |
| 2 | ${\text{SO}}_{3}^{ - }$ + O2 → ${\text{SO}}_{5}^{ - }$ | 2.5 × 109 |
| 3а | ${\text{SO}}_{5}^{ - }$ + HSO3 → H${\text{SO}}_{5}^{ - }$ + ${\text{SO}}_{3}^{ - }$ | 3.4 × 103 |
| 3b | ${\text{SO}}_{5}^{ - }$ + H${\text{SO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{4}^{ - }$ + H+ | 2 × 102 |
| 4 | ${\text{SO}}_{4}^{ - }$ + H${\text{SO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{3}^{ - }$ + H+ | 7.5 × 108 |
| 5a | ${\text{SO}}_{5}^{ - }$+ ${\text{SO}}_{5}^{ - }$ → ${\text{SO}}_{4}^{ - }$ + ${\text{SO}}_{4}^{ - }$ + O2 | 8.7 × 107 |
| 5b | ${\text{SO}}_{5}^{ - }$ + ${\text{SO}}_{5}^{ - }$ → ${{{\text{S}}}_{{\text{2}}}}{\text{O}}_{8}^{{2 - }}$ + O2 | 1.3 × 107 |
| 6 | H${\text{SO}}_{5}^{ - }$ + H${\text{SO}}_{3}^{ - }$ + H + → 2${\text{SO}}_{4}^{{2 - }}$ + 3H+ | 107** |
| 7 | Fe2+ + ${\text{SO}}_{5}^{ - }$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Fe3+ + H${\text{SO}}_{5}^{ - }$ | 3.2 × 106 |
| 8 | Fe2+ + ${\text{SO}}_{4}^{ - }$ → Fe3+ + ${\text{SO}}_{4}^{{2 - }}$ | 3.0 × 108 |
| 9 | Fe2+ + H${\text{SO}}_{5}^{ - }$ → Fe3+ + ${\text{SO}}_{4}^{ - }$ + OН– | 3.5 × 104 |
| 10 | Mn2+ + ${\text{SO}}_{5}^{ - }$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Mn(III) + H${\text{SO}}_{5}^{ - }$ | 1.0 × 108 |
| 11 | Mn(III) + H${\text{SO}}_{3}^{ - }$ → Mn2+ + ${\text{SO}}_{3}^{ - }$ + H+ | 1.3 × 106 |
| 12 | Mn(III) + ${\text{SO}}_{5}^{ - }$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Mn(IV) + H${\text{SO}}_{5}^{ - }$ | ≈1 × 108 |
| 13 | Mn(III) + Mn(III) → Mn2+ + Mn(IV) | ≈1 × 105 |
О КИНЕТИКЕ КАТАЛИТИЧЕСКОЙ РЕАКЦИИ
В северном полушарии концентрации переходных металлов в атмосфере характеризуют колебания от <1 до 1000 нг/м3 [24–26]. В атмосфере Европы содержание Fe и Mn разнится в десятки раз: 65–82 и 3.7–4.4 нг/м3, соответственно, а содержание диоксида серы колеблется в диапазоне 1.13–2.52 мкг/м3 [27]. При этом бóльшая часть железа в атмосферной влаге находится в нерастворимом состоянии [18]. При рН ≈ 4.5 (Т = 298 К), например, нижняя граница содержания растворенного железа не опускается ниже уровня ≈4 × × 10–7 моль/л, отвечающего равновесию трудно растворимого гидроксида 3-валентного железа (Fe(OH)3): K10 = 2.6 × 10–38 моль4/л4 [24] (см. табл. 2). С учетом этого содержание растворимого железа при заданном уровне загрязнения им воздуха в Европе и типичном содержании капельной влаге в атмосфере (L = 3 × 10–4 л/м3 [28]) будем иметь: [Fe3+] ≈ 4 × 10–7 моль/л (7 × 10–3 мкг/м3). Для концентрации ${\text{HSO}}_{3}^{ - }$ – основной реакционной формы S(IV) в атмосфере, концентрации ионов марганца в каплях, а также соотношения (α = = [Mn(II)]/[${\text{HSO}}_{3}^{ - }$]), определяющих режим каталитической реакции [14], находим: [${\text{HS}}{{{\text{O}}}_{{{{3}^{ - }}}}}$] ≈ 3.5 × × 10–7, [Mn(II)] ≈ 2.5 × 10–7 моль/л и α ≤ 1. Оцененные уровни концентраций этих компонентов в атмосфере близки к таковым в лабораторных опытах по кинетике реакции в “пробирках” [10, 11, 22, 23], а также в других экспериментах [29–31]. Ниже проводится обобщение их результатов и детальное рассмотрение на этой основе динамики каталитического окисления диоксида серы в атмосфере.
Таблица 2.
Равновесие ионизации, гидролиза и комплексообразования компонентов растворов при каталитическом окислении сульфита (Т = 298 К, μ ≈ 0 [19])
| № | Равновесие | Константа равновесия KI |
|---|---|---|
| 1 | Н2О ⇆ H++ ОН- | 1.8 × 10–16 моль2/л2 |
| 2 | SO2(aq) ⇆ ${\text{HSO}}_{3}^{ - }$ + H+ | 1.3 × 10–2 моль/л |
| 3 | ${\text{HSO}}_{3}^{ - }$ ⇆ ${\text{SO}}_{3}^{{2 - }}$ + H+ | 6.24 × 10–8 моль/л |
| 4 | [Fe(H2O)6]3+ ⇆ [Fe(H2O)5(OH)]2+ + H+ | 6 × 10–3 л/моль |
| 5 | [Fe(H2O)5(OH)]2+ + ${\text{HSO}}_{3}^{ - }$ ⇆ [Fe(H2O)4(HSO3)(OH)]+ | 6 × 102 л/моль |
| 6 | [Fe(H2O)5(OH)]2+ ⇆ [Fe(H2O)4(OH)2]+ + H+ | 7 × 10–5 моль/л |
| 7 | [Fe(H2O)6]3+ + ${\text{HSO}}_{3}^{ - }$ ⇆ [Fe(H2O)5(HSO3)]2+ + H2O | 72 л/моль |
| 8 | [Fe(H2O)6]3+ + ${\text{SO}}_{3}^{{2 - }}$ ⇆ [Fe(H2O)5(SO3)]+ + H2O | 7.3 × 106 л/моль |
| 9 | [Fe(H2O)5(OH)]2+ + ${\text{SO}}_{3}^{{2 - }}$ ⇆ [Fe(H2O)4(SO3)] + H2O | 2 × 107 л/моль |
| 10 | Fe(OH)3 ⇆ Fe3+ + 3ОН– | 2.6 × 10–38 моль4/ л4 [22] |
Из представленного в табл. 1 механизма каталитического процесса следует, что образование ключевого для реакции промежуточного продукта ${\text{HSO}}_{5}^{ - }$ в рассматриваемых условиях осуществляется в цепочке реакций (10, 11), а бóльшая его часть расходуется в реакции (6), так как [${\text{HSO}}_{3}^{ - }$]/[Fe(II)] $ \gg $ 1 (ζMn$ \gg $ 1) [12–14]. Гибель цепей осуществляется главным образом в реакции рекомбинации ${\text{SO}}_{5}^{ - }$ + Mn(III) (12). Так, даже при минимальных [Mn(II)] (α = 5 × 10–3) в опытах [10], например, w12/w5b = (k10k12/k5bk11)α ≈ 3. В оценке w12/w5b принимали, что процесс формирования ${\text{НSO}}_{5}^{ - }$ протекает по цепному механизму с длиной цепи (ν ≈ w10/w12) превышающей, по крайней мере, 102 звеньев, т.е. (w10–w11) $ \ll $ (w1 – ‒ (w5b + w12 + w13)) и [${\text{SO}}_{5}^{ - }$]/[Mn(III)] = = k11[${\text{HSO}}_{3}^{ - }$]/k10[Mn(II)]. В подтверждение в [12] по данным [22] в опытах с добавками Mn(III) (α ≈ 1) нами сообщалось что ν ≈ 104.
С учетом вышесказанного в стационарных условиях получаем: [${\text{S}}{{{\text{O}}}_{{{{5}^{ - }}}}}$] = k9k11[Fe(II)]/k6k12 и [${\text{НSO}}_{5}^{ - }$] = k9k10k11α[Fe(II)]/$k_{6}^{2}$k12. Концентрацию Fe(II) найдем из уравнения ζMn = k9[${\text{HSO}}_{5}^{ - }$]/k1χ полагая, что [Fe(III)] ≈ [Fe]o, т.е. ζMn $ \gg $ 1. Это приближение находится в согласии с данными опытов [31]; замена ионов Fe(II) на Fe(III) не повлекла за собой изменений скорости гетерогенного окисления SO2 в этих опытах. Здесь χ = = [Fe(OH)SO3H+]/[Fe(III)] – доля каталитически активного комплекса ионов трехвалентного железа Fe(OH)SO3H+ в общем содержании ионов железа в виде: Fe3+, FeOH2+ и др. [19] (см. табл. 2):
В оригинальных работах, за исключением [32], где приведено аналитически определенное значение [Fe*], данных о содержании примесных ионов железа в растворах нет. Об их концентрации в опытах по “катализу” ионами марганца можно однако судить, отождествляя найденные в этих экспериментах численные значения наблюдаемые константы скорости этого процесса (kMn) с таковыми по ходу совместного действия ионов Mn/Fe при контролируемом уровне [Fe]о. Приравнивая (kMn[Mn], с–1) к величине kнабл применительно к условиям опытов [10] будем иметь: [Fe*] = (kMn)2k12[Mn(II)]/(4$\chi _{{{\text{pH}} = 4.2}}^{*}$k1k10k11). Здесь kMn ≈ 1.6 × 103 л моль–1 с–1 (рН 4.2) [10], а ki – фигурирующие в выражении для kнабл (II) константы скорости реакций (1, 10–12) в табл. 1. При [Mn(II)] = 10–6 моль/л [10], например, приходим к [Fe*] ≈ 1.5 × 10–8 моль/л, что близко к оцениваемым нами ранее уровням содержания Fe* в аэрированных растворах S(IV) [12], косвенно подтверждая участие ионов примесного железа в “катализе” ионами марганца.
Возвращаясь к обсуждению характеристики неаддитивного действия ионов Mn/Fe, для величины синергетического коэффициента в этих рамках будем иметь: S = wнабл/$w_{{{\text{Mn\_Fe*}}}}^{{}}$ = = ([Fe]o/[Fe*])1/2. Скорость “железного” катализа в расчетах S в соответствии с [10] можно при этом не учитывать. Данный канал образования сульфатов нельзя считать, во-первых, независимым. Во-вторых, образование сульфатов в реакциях радикалов ${\text{SO}}_{5}^{ - }$ по каналам (5а, 5b) оказывается подавленной в присутствии ионов Mn(II) из-за более быстрого расходования этих радикалов в параллельной реакции с Mn(III) (12): w12/w5а$ \gg $ 1. Из найденного нами выражения для S следует, что величина синергетического коэффициента не зависит от содержания ионов марганца, а определяется лишь концентрациями ионов вводимого и примесного железа в растворах. Это говорит о том, что быстрые реакции с участием ионов марганца не являются лимитирующими звеньями образования сульфатов. Принимая [Fe]o = 10–6 [10] и [Fe*] ≈ 10–8 моль/л для величины эффекта неаддитивного ускорения реакции находим: S ≈ 10. Это примерно в полтора раза ниже приводившегося в [10] максимального значения S при этой концентрации ионов трехвалентного железа (14.4), но примерно вдвое выше приводимого в цитируемой публикации S при минимальной их концентрации (1.25 × 10–7 моль/л).
ДАННЫЕ ЛАБОРАТОРНОГО МОДЕЛИРОВАНИЯ
Как отмечалось, к их рассмотрению привлекались данные опытов в “пробирках”, концентрационные условия которых близки к атмосферным (0.05 ≤ α ≤ 1) [10, 11, 22, 23]. Часть данных заимствовали также из результатов динамических экспериментов (проточные условия [29], барботаж [30]). В дополнение к динамическим экспериментам, которые исключают тормозящее влияние подвода в зону реакции кислорода, к рассмотрению привлекались и результаты опытов по гетерофазному окислению SO2 в каплях малых размеров, находившихся в контакте c воздухом, содержащем примесь диоксида серы [31]. Сформированный таким образом массив данных, охватывает диапазон концентраций ионов металлов (≈10–7–10–3 моль/л) и S(IV) (от ~2 × 10–6 до ~1 × × 10–2 моль/л).
Ввиду различия условий (рН, [S(IV)]) в [10, 11, 22, 23, 29–31] приводимые на Рис. 1 данные о зависимости kнабл = wнабл/[${\text{HSO}}_{3}^{ - }$] (с–1) от [Mn(II)][Fe(III)] пересчитывались к рН 3 и [${\text{HSO}}_{3}^{ - }$] = 10–5 моль/л: kнабл = kнабл_i($\chi _{{{\text{pH}} = 3}}^{*}/\chi _{i}^{*}$)–1/2, см. выражение (I). Здесь kнабл_i и $\chi _{i}^{*}$ – наблюдаемые константы реакции, а $\chi _{i}^{*}$ – доля в этих опытах каталитически активного комплекса ионов Fe(III) в рассматриваемых источниках. Внимание привлекает неплохое согласие при близких [Mn(II)][Fe(III)] приведенных к одинаковым условиям данных опытов по гетерофазному окислению [31] и результатов экспериментов по кинетике реакции в “пробирках” [10, 11, 22, 23], что указывает на малую роль диффузионных ограничений [33]. Вопреки сообщаемым в [10, 34] высоким активационным барьерам для каталитической реакции (≈16 ккал/моль) приведенные к одинаковым условиям данные не обнаруживают и слишком заметного влияния температуры (Т = 281–323 К) на kнабл в опытах [30, 31].
Рис. 1.
Катализ ионами марганца и железа кислородного окисления SO2. Зависимость наблюдаемой константы скорости реакции kнабл, с–1 от [Mn(II)][Fe(III)]. Данные лабораторных опытов в растворах. Т = 298 К, рН 3 и [${\text{HSO}}_{3}^{ - }$] = = 10–5 моль/л. ◻ [10], △ [11], ⚪ [22], ▽ [23], ◇ [29]. Серым цветом показаны △ по гетерофазному окислению SO2 в каплях [31]. На половину закрашенными в серый цвет показаны данные при температурах отличных от Т = 298К: ◻ [30] (323 К), △ [31] (281 К). На вставке показаны зависимости kнабл (сплошная линия) и пунктир kMn_Fe([Mn(II)][Fe(III)]), c–1 при низких [Mn(II)][Fe(III)]: ☆ и ◻ [10]; △ [11].
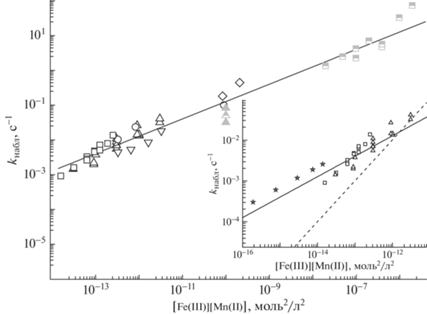
ПОРЯДОК РЕАКЦИИ ПО ИОНАМ ПМ
Приведенные на рис. 1 данные хотя и обнаруживают разброс, но группируются вокруг линии: kнабл ∼ ([Mn(II)][Fe(III)])1/2. Этот результат служит доказательством половинного порядка реакции из произведения [Mn(II)][Fe(III)], см. (I), но находится в противоречии с данными [10, 11] о первых порядках реакции по Mn(II) и Fe(III). Иллюстрация различия отклика реакции на рассматриваемые здесь различия порядков по ионам металлов приводится на вставке рисунка. Здесь представлены данные о зависимости kнабл и kMn_Fe = = wMn_Fe/[S(IV)] [10] от [Mn(II)][Fe(III)] (сплошная и пунктирная линии соответственно). При этом данные о kнабл при самых низких [Mn(II)][Fe(III)] (≤10–13 моль2/л2) рассчитывались по данным опытов по “катализу” ионами марганца. Полученные таким образом данные естественным образом продолжают ход зависимости kнабл в область экстремально низких [Mn(II)][Fe(III)], вновь подтверждая участие ионов примесного железа в марганцевом “катализе”. Из данных вставки следует, что kMn_Fe [10] при низких [Mn(II)][Fe(III)] заметно уступают по величине kнабл, что связано с ошибочным представлением о wнабл как суммы независимых каталитических каналов рассматриваемой реакции: wMn, wFe и wMn_Fe.
ПОРЯДОК РЕАКЦИИ ПО S(IV)
Подтверждение сообщавшемуся первому порядку каталитической реакции по S(IV) в [10, 11, 22, 23, 29–31] находим в данных Рис. 2. Здесь показано, как сказывается влияние [${\text{HSO}}_{3}^{ - }$] на наблюдаемую константу скорости рассматриваемой реакции. Для нивелирования различий концентраций ионов Mn/Fe в этих публикациях в качестве наблюдаемых констант скорости реакции рассматривались пересчитанные к одинаковым условиях величины: $k_{{{\text{набл}}}}^{*}$ = kнабл_pH = 3/($\chi _{{{\text{pH}} = 3}}^{*}$[Mn(II)][Fe(III)])–1/2. Каждая из точек усреднялась по массиву данных о wнабл и [S(IV)] приводимых в цитируемых публикациях. Так, массив данных в [10] включает 30 индивидуальных значений wнабл при разных [Mn(II)] и [Fe(III)]. Значительное их число приводится также в других публикациях, что было принято во внимание при усреднении $k_{{{\text{набл}}}}^{*}$.
Рис. 2.
Зависимость наблюдаемой константы скорости реакции $k_{{{\text{набл}}}}^{*}$ = kнабл/(χ*[Mn(II)][Fe(III)])1/2 от [HSO3]. Данные в растворах: T ≈ 281–323 K, рН 3. ◻ [10], △ [11], ⚪ [22], ▽ [23], ◇ [29] Серым цветом показаны △ по гетерофазному окислению SO2 в каплях: △ [31]. Закрашенные в серый цвет наполовину точки показывают данные при температурах отличных от 298К: ◻ [30] (323 К), △ [31] (281 К). На вставке показаны рН зависимости kнабл (сплошная линия) и kMn_Fe([Mn(II)][Fe(III)]) (пунктир).
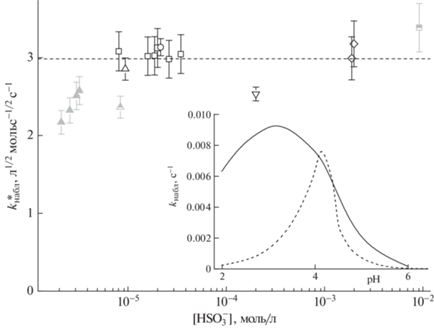
Несмотря на разброс, данные рис. 2 свидетельствуют о близости к постоянству $k_{{{\text{набл}}}}^{*}$ в диапазоне концентраций ${\text{HSO}}_{3}^{ - }$ от ≈ 2 × 10–6 до 10–2 моль/л, что указывает на первый порядок реакции по ${\text{HSO}}_{3}^{ - }$. Усредненное по всем данным абсолютное значение $k_{{{\text{набл}}}}^{*}$ = 980 л1/2моль–1/2 с–1 этой константы скорости неплохо согласуется с найденным по выражению для $k_{{{\text{набл}}}}^{*}$ = (4k1k10k11.1/k12)1/2 = (4 × 0.2 × × 1.3 × 106 × 108/108) ≈ ≈ 103 л1/2 моль–1/2 с–1, используя для вычислений значения констант скорости реакций (1, 10–12), приведенных в табл. 1. Это согласие найденных при усреднении и вычислениях величин $k_{{{\text{набл}}}}^{*}$ дополнительно свидетельствует о корректности рассматриваемой картины механизма каталитического процесса.
ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ ОТ КИСЛОТНОСТИ
На вставке рис. 2 показана рН зависимость kн-абл для рассматриваемых в [10] концентрационных условиях ([S(IV)] ≈ 10–5, [Mn(II)] = 2.5 × 10–7 и [Fe(III)] = 7.5 × 10–7 моль/л). Ее вид демонстрирует колоколообразный характер. Подобный характер обнаруживает и рН зависимость наблюдаемой константы скорости kMn_Fe[Mn(II)] ⋅ [Fe(III)], рассчитанной нами по данным [10] (пунктирная линия). При этом просматривается сужение максимума и его смещение к бóльшим рН, Причина – ошибочное представление о wнабл как о сумме независимых каталитических каналов рассматриваемой реакции: wMn, wFe и wMn_Fe и необоснованный вычет wMn + wFe из wнабл при нахождении kMn_Fe.
В рассматриваемых концентрационных условиях максимум kнабл наблюдается при рН ≈ 3.5. Такое поведение зависимости kнабл от рН указывает на то, что доминирующая часть ионов железа в экспериментах [10, 11, 22, 23] находится в форме Fe(III), т.е. ζMn$ \gg $ 1 и [Fe(III)] ≈ [Fe]o, так как вид этой зависимости в деталях воспроизводит вид рН зависимости χ [12–14]. Распределение концентраций Fe(III) по формам (χ*) при низком содержании S(IV) в растворах (10–5 моль/л) в зависимости от рН регулируют в основном процессы гидролиза Fe(III) по первой (3) и второй ступеням (5) [17] (см. табл. 2). Отметим, что процессы гидролиза благоприятствуют росту растворимости трехвалентного железа в форме FeOH2+ и ${\text{Fe(OH}})_{2}^{ + }$, что игнорируется в [10, 11] и других публикациях. В отсутствие учета этих форм ионов железа, т.е. рассматривая ионы Fe3+ в качестве единственного компонента Fe(III), заданное содержание железа в [10] оказывается в растворимой форме лишь при рН ≤ 3.5. Последующий рост рН (≥3.5) приводит к смещению влево равновесия (10, табл. 2), что ведет к формированию практически нерастворимого Fe(OH)3. В результате концентрация трехвалентного железа в растворе драматически спадает. Вместе с этим снижаются и скорость инициирования (1) и wнабл. С этим и связана значимость каталитического формирования сульфатов в атмосфере лишь при невысоких рН. Учет же в растворе содержания FeOH2+ и ${\text{Fe(OH}})_{2}^{ + }$ наряду с Fe3+ показывает, что вводимое в растворы железо (7.5 × 10–7 моль/л) оказывается в растворенном состоянии и при рН 4.4. В силу одновременно возрастающей при этом растворимости диоксида серы, в каплях усиливается также образование комплексов ${\text{FeHSO}}_{3}^{{2 + }}$, ${\text{FeSO}}_{3}^{ + }$ и др. [19] (см. табл. 2). Их образование способствует не только дальнейшему росту растворимости железа, но и росту концентрации каталитически активной (Fe(OH)SO3H+, табл. 2) формы ионов железа. С учетом сказанного можно ожидать, что нефотохимический процесс окисления растворенного диоксида серы с участием ионов Mn/Fe в атмосфере оказывается, по-видимому, значимым в гораздо более широком диапазоне рН, чем рассматривалось ранее [3, 5, 35].
О КАТАЛИТИЧЕСКОМ ОБРАЗОВАНИИ СУЛЬФАТОВ В АТМОСФЕРЕ
Как проявляет себя рассматриваемая каталитическая реакция с участием ионов марганца и железа при формировании сульфатов в капельной влаге в атмосфере, в том числе и при повышенных рН? На рис. 3 в качестве примера приводится рассчитанные нами скорости каталитическом образовании сульфатов (мкг м–3 ч–1) в атмосферной капельной влаге (1 г/м3) в зависимости от кислотности капель (рН 2–6) для приводимых в [10] концентрационных условий (см. кривую 1). Здесь wS(VI) = wнабл$L{{{\text{M}}}_{{{\text{SO}}_{4}^{{2 - }}}}}$ × 3600 × 106 = = $k_{{{\text{набл}}}}^{*}$(χ*[Mn(II)][Fe(III)])1/2 [${\text{HS}}{{{\text{O}}}_{{{{3}^{ - }}}}}$]$L{{{\text{M}}}_{{{\text{SO}}_{4}^{{2 - }}}}}$ × × 3600 × 106. При вычислениях учитывали зависимость от рН распределения ионов трехвалентного железа, растворимости железа
Рис. 3.
Зависимость скорости образования сульфатов (мкг м–3 ч–1) от кислотности капель при катализе ионами марганца и железа кислородного окисления диоксида серы в капельной фазе атмосферы, Т = 298 К, Содержание диоксида серы 5 ppb, концентрации марганца железа: 1.7 и 5 нг/м3, объем жидкой влаги в воздухе 10–3 л/м3 (1 г/м3) [10]. Сплошная кривая 1 – расчет wS(VI) с использованием $k_{{{\text{набл}}}}^{*}$, кривая 2 с использованием kMn_Fe.
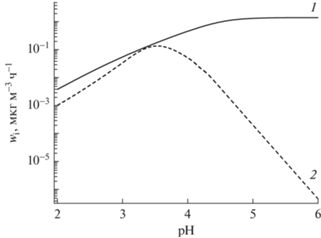
Из данных рис. 3 видно, что рост рH (≤3.5) сопровождается увеличением и wS(VI), и $w_{{{\text{S(VI)}}}}^{*}$. Причиной служит отмеченный выше рост в капельной фазе содержания ${\text{HSO}}_{3}^{ - }$, вызванный смещением равновесия растворимости диоксида серы вправо: см. (2) в табл. 2. При этом темпы роста $w_{{{\text{S(VI)}}}}^{*}$ с увеличением рН выглядят несколько более высокими. Причиной служит уже отмечавшийся спад растворимости трехвалентного железа по мере увеличения рН и связанное с этим уменьшение wFe. В результате wMn_Fe с увеличением рН приближается к wнабл, а вместе с этим $w_{{{\text{S(VI)}}}}^{*}$ становится ближе к wS(VI). Их величины практически сравниваются при рН ≈ 3.5, свидетельствуя о пренебрежимо малой роли wMn в образовании сульфатов.
При рН > 3.5 поведение расчетных кривых 1 и 2 кардинально изменяется. Ход кривой 2 указывает при этом на драматический спад $w_{{{\text{S(VI)}}}}^{*}$. Причиной служит уменьшение скорости инициирования (1, табл. 1) с увеличением рН вследствие уменьшения концентрации Fe(III). Результатом является снижение скорости образования сульфатов. В противовес кривая 1 обнаруживает близость к насыщению скорости формирования сульфатов wS(VI) ≈ 1.4 мкг м–3 ч–1, несмотря на высокие рН. Такое поведение реакции обязано исключительно связыванию ионов трехвалентного железа в комплексы (FeOH2+, см. табл. 2). Оставшееся в растворе благодаря этому бóльшее содержание ионов трехвалентного железа, в том числе и в виде Fe(OH)SO3H+ поддерживает скорость инициирования реакции (1) на уровне достаточном, чтобы обеспечить близость к постоянству wS(VI) с ростом рН. Это происходит за счет одновременного прироста с увеличением рН скорости продолжения цепи (10, 11, табл. 1), вызванного ростом растворимости диоксида серы. Все это вновь подтверждает потенциальную значимость каталитической (нефотохимической) реакции с участием переходных металлов в образовании сульфатов в атмосфере при гораздо более высоких рН в сравнении с рассматриваемыми ранее [3, 5, 35].
Для абсолютной величины константы скорости конверсии SO2 в сульфаты в расчете на газовую фазу находим: kнабл_SO2_газ = wнаблRTL/${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = = $k_{{{\text{набл}}}}^{*}$(χ*[Mn][Feр × 10–9/MMn)1/2K298HSO2_298R × × TL1/2/10–pH, с–1. Здесь ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ – парциальное давление диоксида серы, атм, [Mn] – концентрация марганца в атмосфере нг/м3, [Feр] – концентрация растворенного трехвалентного железа, MMn = = 55 г/моль – атомный вес марганца, HSO2_298 = = 1.23 моль л–1 атм–1 [36] – константа Генри, характеризующая физическую растворимость диоксида серы при Т = 298 К, R = 8.2 × 10–5 м3 атм моль–1 град–1 – универсальная газовая постоянная и 10–9 г/нг – размерный коэффициент. Абсолютная величина ${{k}_{{{\text{набл\_S}}{{{\text{O}}}_{{\text{2}}}}{\text{\_газ}}}}}$ определяется при этом в основном кислотностью капель, которая определяет в свою очередь содержание растворенного железа. При характерных для атмосферы Европы концентрационных условиях рН 4.5, [Mn(II)] ≈ 3 нг/м3 и [Feр] ≈ 4 × 10š7 моль/л, находим ${{k}_{{{\text{набл\_S}}{{{\text{O}}}_{{\text{2}}}}{\text{\_газ}}}}}$ ≈ 1.5 × 10–5 с–1. Эта величина почти вчетверо уступает по абсолютной величине эффективной константы скорости образовании сульфатов в реакции с озоном (${{k}_{{{\text{набл\_газ\_}}{{{\text{O}}}_{{\text{3}}}}}}}$ = 1.5 × × 109HO3_298${{P}_{{{{{\text{O}}}_{{\text{3}}}}}}}$HSO2_298KIKIIRTL/(10–pH)2 ≈ 4 × × 10‒6 с–1) даже при близкой к максимальной концентрации озона в тропосфере (30 ppb): O3 + + ${\text{SO}}_{3}^{{2 - }}$ → ${\text{SO}}_{4}^{{2 - }}$ + O2. Здесь 1.5 × 109 л моль–1 с–1 – константа скорости реакции растворенного озона с ${\text{SO}}_{3}^{{2 - }}$ [37], ${{{\text{P}}}_{{{{{\text{O}}}_{{\text{3}}}}}}}$ – парциальное давление озона (атм), а HO3_298 = 1.1 × 10–2 моль л–1 атм–1 [36] – константа Генри, характеризующая растворимость озона при Т = 298 К. Проведенные оценки показывают, что скорость реакции окисления диоксида серы молекулярным кислородом в присутствии ионов Mn/Fe не слишком разнится от скорости реакции с участием присутствующего в воздухе в высокой концентрации озона даже при рН 5, что подчеркивает значимость этого процесса в атмосфере и при невысокой кислотности капель. На недооценку этого и других нефотохимических источников сульфата в атмосфере указывалось и в [38]. Результаты моделирования и натурных измерений в цитируемой публикации удалось согласовать только за счет допущения нефотохимического превращения SO2 в сульфаты в атмосфере с усредненной по году константой скорости (1–2) × 10–6 с–1.
ЗАКЛЮЧЕНИЕ
В работе обобщены данные по кинетике катализа ионами Mn/Fe жидкофазного окисления SO2 молекулярным кислородом. Приведен механизм жидкофазного окисления SO2 с участием ионов переходных металлов, удовлетворительно описывающий эти данные. Этот процесс, развивающийся по цепному механизму, характеризуется разветвлением цепей с участием нарабатываемого по ходу промежуточного продукта ${\text{HSO}}_{5}^{ - }$. В этих рамках естественным образом удается истолковать происхождение явления синергизма, т.е. эффекта неаддитивного усиления каталитического действия пары ионов Mn/Fe в образовании сульфатов. Приведены результаты предварительных расчетов участия ионов переходных металлов в этом процессе в атмосфере. По результатам этих расчетов сделан вывод о существенности вклада каталитического действия пары ионов Mn/Fe в образовании сульфатов даже при невысокой кислотности капель и низких концентрациях ионов переходных металлов. Полученные в работе выражения для скорости образования сульфатов можно использовать для расчетов динамики накопления сульфатов в атмосфере в WRF-Chem и других моделях высокого уровня.
Список литературы
Langner J., Rodhe H. A global three-dimensional model of the tropospheric sulfur cycle // J. Atmos. Chem. 1991. V. 13. № 3. P. 225–263.
Warneck P., Mirabel P., Salmon G.A., et al. Review of the activities and achievements of the EUROTRAC subproject HALIPP (Ed. P. Warneck), Heterogeneous and liquid phase processes, Springer-Verlag Berlin Heidelberg, 1996. P. 7.
Seinfeld J. H., Pandis S.N., Atmospheric Chemistry and Physics, from Air Pollution to Climate Change. John Wiley & Sons, Hoboken, New Jersey, USA, 2016. 1152 P.
Feichter J., Kjellstrom E., Rodhe H., et al., Simulation of the tropospheric sulfur cycle in a global climate model // Atmos. Environ. 1996. V. 30. № 10–11. P. 1693–1707.
Cheng Y.F., Zheng G., Way Ch., Mu Q. Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China // Science Advances. 2016. V. 2. № 12. e1601530.
Wang G.H. Zhang R.Y., Gomes M.E., et al. Persistent sulfate formation from London fog to Chinese haze // Proc. Natl. Acad. Sci. U.S.A. 2016. V. 113. № 48. P. 13 630–13 635.
Xie Y.Z., Liu Z.R., Wen T.X. et al. Characteristics of chemical composition and seasonal variations of PM2.5 in Shijiazhuang, China: impact of primary emissions and secondary formation // Sci. Total Environ. 2019. V. 677. P. 215–229.
Zheng G.J., Duan F.K., Su H., et al. Exploring the severe winter haze in Beijing: the impact of synoptic weather, regional transport and heterogeneous reactions.// Atmos. Chem. Phys. 2015. V. 15. № 6. P. 2969–2983.
Grell G.A. Peckham S., Schmitz R., McKeen S.A., Frost G.J., Eder B. Fully coupled “online” chemistry within the WRF model // Atmos. Environ. 2005. V. 39. № 37. P. 6957–6975.
Ibusuki T., Takeuchi K. Sulfur dioxide oxidation by oxygen catalyzed by mixtures of manganese(II) and iron(III) at environmental reaction conditions // Atmos. Envir. 1987. V. 21. № 7. P. 1555–1560.
Martin L.R., Good T.W. Catalyzed oxidation of sulfur dioxide in solution: the iron-manganese synergism // Atmos. Envir. 1991. V. 25A. № 10. P. 2395–2399.
Ermakov A.N., Purmal A.P. Catalysis of ${\text{HSO}}_{3}^{ - }{\text{/SO}}_{3}^{{2 - }}$ oxidation by manganese ions // Kinetics and Catal. 2002. V. 43. № 2. P. 249–260.
Yermakov A.N., Purmal A.P. Iron-catalyzed oxidation of sulfite: From established results to a new understanding. Progr. React. Mech. 2003. V. 28. P. 189–255.
Ермаков А.Н. О катализе ионами марганца окисления сульфита. Кинетика реакции в избытке ионов металла // Кинетика и катализ. 2021. Т. 62. № 5. С. 518–526.
Matsuhisa Y., Goldsmith J.R., Clayton R.N. Mechanisms of hydrothermal crystallization of quartz at 250 C and 15kbar // Geochim. Cosmochim. Acta. 1978. V. 42. № 2. P. 173–182.
Farquhar J., Savarino J., Jackson T.L., Thiemens M.H. Evidence of atmospheric sulphur in the Martian regolith from sulphur isotopes in meteorites // Nature. 2000. V. 404. P. 50– 52.
Mc-Cabe J.R., Savarino J., Alexander B., Gong S. Isotopic constraints on non-photochemical sulfate production in the Arctic winter // Geophys. Res. Lett. 2006. V. 33. № 5. L05810.
Alexander B., Park R.J., Jacob, D.J., Gong, S.L. Transition metal-catalyzed oxidation of atmospheric sulfur: global implications for the sulfur budget // J. Geophys. Res. Atmos. 2009. V. 114. D02309.
Brandt Ch., van Eldik R. Transition Metal-Catalyzed Oxidation of Sulfur(lV) Oxides. Atmospheric-Relevant Processes and Mechanisms // Chem. Rev. 1995. V. 95. P. 119–190.
Kuo D.T.F., Kirk D.W., Jia C.Q. The chemistry of aqueous S(IV)–Fe–O2 system: State of the art // J. Sulfur. Chem. 2006. V. 27. № 5. P. 461–530.
Stanbury D.M. Reduction potentials involving inorganic free radicals in aqueous solution // Adv. Inorg. Chem. 1989. V. 33. P. 69–138.
Berglund J., Fronaeus S., Elding L.I. Kinetics and mechanism for manganese-catalyzed oxidation of sulfur(IV) by oxygen in aqueous solution // Inorg. Chem. 1993. V. 32. № 21. P. 4527–4537.
Grgić I., Berčič G. A Simple Kinetic model for autoxidation of S(IV) oxides catalyzed by iron and/or manganese ions // J. Atmos. Chem. 2001. V. 39. № 2. P. 155–170.
Graedel T.E., Weschler C.J. Chemistry within aqueous atmospheric aerosols and raindrops // Rev. Geophys. 1981. V. 19. № 4. P. 505–539.
Matthijsen J., Builtjes P.J.H., Sedlak D.H. Cloud model experiments of the effect of iron and cooper on tropospheric ozone under marine and continental conditions // Meteorol. Atmos. Phys. 1995. V. 57. № 1. P. 43–60.
Sedwick P.N., Sholkovitz E.R., Church T.M. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea, Geochem // Geophys. Geosyst. 2007. V. 8. № 10. Q10Q06.
www.emep.int.
Knollenberg R.G. Techniques for probing cloud microstructure. In: Clouds: Their formation. Optical properties and Effects, Hobbs P.V., Deepak A. eds. P. 15–91, Academic Press, New-York, 1981.
Coughanowr D.R., Krause F.E. The reaction of SO2 and O2 in aqueous solutions of MnSO4 // Ind. Eng. Chem. Fund. 1965. V. 4. № 1. P. 61–66.
Ulrich R.K, Rochelle G.T., Prada R.E. Enhanced oxygen absorption into bisulfite solutions containing transition metal ion catalysts // Chem. Engng Sci. 1986. V. 41. № 8. P. 2183–2191.
Barrie L.A., Georgii H.W. An experimental investigation of the absorption of sulphur dioxide by water drops containing heavy metal ions // Atmos. Environ. 1976. V. 10. № 9. P. 743–749.
Huss A.Jr., Lim P.K., Eckert C.A. Oxidation of aqueous sulfur dioxide. 1. Homogeneous manganese(II) and iron(III) catalysis at low pH // J. Phys. Chem. 1982. V. 86. № 21. P. 4224–4228.
Schwartz S.E., Freiberg J.E. Mass-transport limitation to the rate of reaction of gases in liquid droplets: Application to oxidation of SO2 in aqueous solutions // Atmos. Environ. 2007. V. 15. № 7. P. 1129–1144.
Grgić I., Hudnik V., Bizjak M. Levec J. Aqueous S(IV) oxidation—I. Catalytic effects of some metal ions. Atmos. Envir. 1991. V. 25A. № 8. P. 1591–1597.
Liu T., Clegg S.L., Abbatt J.P.D. Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles // Proc. Natl. Acad. Sci. U.S.A. 2020.V. 117. № 3. P. 1354–1359.
Herrmann H., Ervens B., Jacobi H.-W., Wolke R., Nowacki P., Zellner R. CAPRAM2.3: A chemical aqueous phase radical mechanism for troposheric chemistry // J. Atmos. Chem. 2000. V. 36. № 3. P. 231–284.
Hoffman M.R. On the kinetics and mechanism of oxidation of aquated sulfur dioxide by ozone // Atmos. Envir. 1986. V. 20. № 6. P. 1145–1154.
Kasibhatla P., Chameides W.L., and St. John J., A three-dimensional global model investigation of seasonal variations in the atmospheric burden of anthropogenic sulfate aerosols, J. Geophys. Res., 1997. 102 (D3). P. 3737–3759.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Физика атмосферы и океана