

Журнал физической химии, 2019, T. 93, № 10, стр. 1470-1476
Реконструкция электронной плотности интермедиатов реакции гидролиза N-ацетиласпартата аспартоацилазой
М. Г. Хренова a, b, *, Е. Д. Коц a, А. М. Кулакова a, А. В. Немухин a, c
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Российская академия наук, Федеральный исследовательский центр Биотехнологии
Москва, Россия
c Российская академия наук, Институт биохимической физики имени Н.М. Эмануэля
Москва, Россия
* E-mail: wasabiko@lcc.chem.msu.ru
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
На примере гидролиза N-ацетиласпартата аспартоацилазой человека показано, что при описании интермедиатов реакций ферментативного катализа недостаточно использовать промолекулярную электронную плотность (суперпозицию плотностей сферических атомов). Необходимо учитывать деформацию электронной плотности, обязанную образованию ковалентных и координационных связей внутри системы. Это диктует необходимость развития новых подходов в белковой кристаллографии для более адекватной интерпретации наблюдаемой электронной плотности и более точному определению геометрических параметров реакционных интермедиатов.
Изучение каталитических процессов, осуществляемых ферментами, необходимое для важнейших биотехнологических и биомедицинских приложений, преимущественно проводится инструментальными методами. Однако с развитием теории строения молекул и стремительным ростом возможностей информационных и компьютерных технологий молекулярное моделирование оказывает все более значимую поддержку экспериментальным исследованиям деталей реакций ферментативного катализа. Практическая реализация метода квантовой механики/молекулярной механики (КМ/ММ) [1] позволила проводить вычисления энергетических профилей химических превращений, используя надежные квантово-химические подходы для участников реакции в активных центрах ферментов и с учетом влияния всей белковой макромолекулы и молекул растворителя на частицы в активном центре. Компьютерные расчеты обеспечивают необходимые данные по интермедиатам, которые могут быть использованы для управления процессами ферментативного катализа на основании установленного механизма реакции.
В лаборатории химической кибернетики кафедры физической химии Химического факультета МГУ имени М.В. Ломоносова в сотрудничестве с исследователями других подразделений МГУ моделирование реакций ферментативного катализа методами КМ/ММ проводится с начала XXI столетия (например, [2–6]). В каждом конкретном случае на основании полученных в результате расчетов энергетических диаграмм формулируются заключения о механизме сложной многостадийной реакции, определяются структуры фермент-субстратных комплексов и продуктов, реакционных интермедиатов (локальных минимумов на энергетической поверхности) и переходных состояний (седловых точек на энергетической поверхности).
Эта важнейшая информация редко доступна прямым экспериментальным наблюдениям – ферментативные реакции проходят настолько быстро, что зафиксировать и проанализировать строение интермедиатов можно только специальными приемами. Один из таких подходов – рентгеноструктурный анализ (РСА) кристаллов комплексов нативных ферментов с аналогами участников реакции или мутированных ферментов с субстратами или их аналогами [7]. В том случае, когда сопоставление результатов компьютерного моделирования и данных РСА по структурам изучаемых систем оказывается успешным, исследование механизма ферментативного процесса получает значительную всестороннюю поддержку.
Напомним, что важнейшей характеристикой и при моделировании реакционных интермедиатов методами на основе квантовой химии, и при рентгеноструктурных исследованиях является распределение электронной плотности в молекулах. Трехмерные карты электронной плотности белковых систем, определяемые в РСА, позволяют реконструировать трехмерную структуру, то есть найти координаты атомов (практически всегда за исключением атомов водорода). При расчетах, напротив, для каждого интермедиата реакции координаты всех атомов, включая и атомы водорода, известны по результатам расчетов геометрических конфигураций минимумов энергетической поверхности, и распределение электронной плотности можно построить с также известными из расчетов решениями уравнений квантовой химии.
Проблемы, возникающие на пути прямого сопоставления электронной плотности интермедиатов ферментативных реакций, восстанавливаемой в экспериментах РСА и рассчитываемой при молекулярном моделировании методами КМ/ММ, известны. Базовая модель, широко применяющаяся для расшифровки трехмерной структуры белков с лигандами по данным РСА, – модель сферических атомов [8]. Такая модель является вполне успешной при расшифровке стабильных структурных мотивов биологической макромолекулы. Однако для изучения структуры реакционных частиц в активном центре фермента приближения на основе модели сферических атомов может оказаться недостаточно. Распределение электронной плотности в молекулах деформировано по сравнению с распределением совокупности составляющих молекулу и гипотетически невзаимодействующих атомов. Для ферментативных реакций это особенно важно, поскольку активный центр должен быть существенно поляризован для направленного проведения химической реакции, что сказывается на распределении электронной плотности.
Проблема учета деформации электронной плотности, т.е. учета анизотропии электронной плотности около атомов, вызванной межатомными взаимодействиями, известна в кристаллографии [9–11]. Для обработки данных РСА предложены разнообразные мультипольные модели, позволяющие уточнять результаты для кристаллов небольших молекул и ионов. Однако несмотря на развитие биокристаллографии, связанное с появлением синхротронов нового поколения, лазеров на свободных электронах [12–15], позволяющими добиться достаточно высокого разрешения, а также с усовершенствованием экспериментальных установок, способных фиксировать интермедиаты реакции и проводить измерения электронной плотности с временным разрешением [16–20], методы обработки данных РСА в рамках стандартных компьютерных пакетов остаются прежними.
Целью настоящей работы является анализ деформации электронной плотности в реакционных интермедиатах и оценка применимости модели сферических атомов для конкретной ферментативной реакции – реакции гидролиза N‑ацетиласпартата в активном центре аспартоацилазы человека. Анализ основан на полученных ранее результатах моделирования каталитического цикла аспартоацилазы [5, 6] методами КМ/ММ. В нашем распоряжении имеются полноатомные геометрические конфигурации фермент-субстратного комплекса, первого интермедиата реакции гидролиза, а также системы с негидролизующимся аналогом интермедиата. Для этих структур проведена реконструкция электронной плотности с использованием результатов квантово-химических расчетов и различных моделей обработки этих данных.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все расчеты электронной плотности выполнены для фермент-субстратного комплекса и первого интермедиата реакции гидролиза N-ацетиласпартата (NAA) аспартоацилазой, структуры которых были получены ранее [6]. Кроме того, в данной работе была построена модель комплекса аспартоацилазы с негидролизуемым аналогом субстрата – N-фосфонометил-L-аспартатом (NPA). Протокол расчета аналогичен использованному в работе [6]. Полноатомная модель строилась на основании кристаллической структуры PDB ID 2O4H [21], содержащей комплекс аспартоацилазы с NPA. Равновесные геометрические конфигурации рассчитывались методом КМ/ММ, энергии и силы в КМ-подсистеме рассчитывались методом функционала электронной плотности в варианте Кона–Шэма с гибридным функционалом PBE0 [22] с дисперсионной поправкой D3 [23] и двухэкспонентным базисом с поляризационными функциями на всех атомах 6‑31G**. В квантово-механическую подсистему входил субстрат или его негидролизуемый аналог, каталитический остаток глутаминовой кислоты, каталитическая молекула воды (в случае комплекса с NAA), катион цинка с координационной сферой, а также ряд аминокислотных остатков, формирующих активный центр (рис. 1). ММ‑ подсистема, включающая оставшуюся часть белка и сольватную оболочку молекул воды, описывалась в рамках классического силового поля AMBER [24]. Для всех локализованных равновесных геометрических конфигураций, рассматриваемых в работе, проводился расчет электронной плотности квантовой подсистемы для последующего анализа. При этом учитывалась поляризация квантово-механической подсистемы со стороны ММ-части за счет вкладов от зарядов атомов, предписанных силовым полем, в одноэлектронную часть гамильтониана КМ-подсистемы. Расчеты методом КМ/ММ выполнялись в программном пакете NWChem [25], анализ электронной плотности – в программе Multiwfn [26].

ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Механизм реакции гидролиза N-ацетиласпартата в активном центре аспартоацилазы подробно описан в работе [6]. В данной работе для анализа выбраны участники только первой стадии реакции от фермент-субстратного комплекса к первому интермедиату, а также комплекс фермента с негидролизуемым аналогом субстрата. Реакция начинается с нуклеофильной атаки атома кислорода каталитической молекулы воды, что приводит к образованию тетраэдрического интермедиата и протонированию каталитического остатка глутаминовой кислоты. К настоящему моменту нет экспериментальных данных РСА по трехмерным структурам, содержащим в активном центре субстрат или интермедиаты гидролиза, однако известны рентгеноструктурные данные для комплекса аспартоацилазы с негидролизуемым аналогом субстрата NPA (рис. 2).
Рис. 2.
Механизм первой стадии реакции гидролиза N-ацетиласпартата в активном центре аспартоацилазы (а) и негидролизуемый аналог интермедиата реакции NPA (б).

Далее будут представлены результаты моделирования деформации электронной плотности для трех выбранных стационарных точек на энергетической поверхности. Мы начнем рассмотрение с фермент-субстратного комплекса. Согласно результатам расчетов нуклеофильная атака проходит с низким энергетическим барьером, что свидетельствует о существенном влиянии белкового окружения на взаимодействующие молекулы в активном центре фермента. Действительно, молекула воды поляризована катионом цинка, а также образует водородные связи с отрицательно заряженной карбоксильной группой аминокислотного остатка глутамата и пептидной группой основной цепи белка. Карбонильная группа субстрата координирована аминокислотным остатком аргинина, а также катионом цинка, что дополнительно поляризует С=О-связь.
Проанализируем, как изменяется картина электронной плотности при переходе от промолекулы (молекулы, электронная плотность которой состоит из суммы атомарных электронных плотностей) к реальной электронной плотности, получаемой из квантово-химического расчета. Для этого рассмотрим карты деформационной электронной плотности, получаемые вычитанием промолекулярной электронной плотности из рассчитанной. На рис. 3 представлена деформационная электронная плотность в плоскости Zn–O(Wat)–O(NAA). Видно, что неподеленные электронные пары атома кислорода каталитической молекулы воды ориентированы на катион цинка, а также в сторону карбонильного кислорода, о чем свидетельствует положительная область деформационной электронной плотности. Неподеленная пара, ориентированная на карбонильный углерод субстрата лучше просматривается на рис. 4. Другим важным наблюдением является уменьшение электронной плотности на карбонильном атоме углерода субстрата с той стороны, где происходит нуклеофильная атака. Наличие координационной связи карбонильного кислорода субстрата с катионом цинка, а также водородной связи с аминокислотным остатком аргинина, приводит к значительной поляризации атома кислорода и сильной анизотропии его электронной плотности (рис. 5).
Рис. 3.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Zn–O(Wat)–O(NAA) фермент-субстратного комплекса. Стрелки показывают направление нуклеофильной атаки. Здесь и далее сплошные изолинии соответствуют положительным значениям электронной плотности в диапазоне 0.05–1.35 а.е. с шагом 0.05 а.е., пунктирные изолинии – отрицательным значениям в диапазоне от –1.30 до –0.05 а.е. с шагом 0.05 а.е.; расстояния по осям сечений электронной плотности приведены в Å. Молекулярная модель фермент-субстратного комплекса (в).
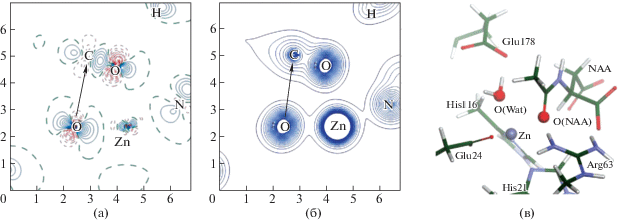
Рис. 4.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости O(Wat)–C(NAA)–O(NAA) фермент-субстратного комплекса. Стрелки показывают направление нуклеофильной атаки. Короткая пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель фермент-субстратного комплекса (в).

Рис. 5.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Zn–O(NAA)–N(ARG) фермент-субстратного комплекса. Короткая пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель фермент-субстратного комплекса (в).
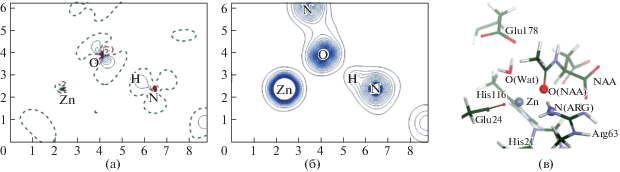
На картах разностной электронной плотности хорошо видна деформация электронной плотности в результате межатомных взаимодействий, в то время как на промолекулярной плотности четко наблюдается суперпозиция сферически симметричных атомарных электронных плотностей.
Такой же анализ проведен для первого интермедиата реакции и его негидролизуемого аналога, содержащего PO2-фрагмент вместо образующейся частицы СО2 (рис. 2). Из деформационных карт (рис. 6, 7) видно, что в случае интермедиата гидролиза NAA оба атома кислорода ориентированы на катион цинка, тогда как в случае комплекса с NPA деформационная плотность показывает взаимодействие цинка только с одним атомом кислорода.
Рис. 6.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Zn–O(Wat)–O(NAA) для интермедиата. Длинная пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель интермедиата (в).

Рис. 7.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Zn–O(NPA)–O(NPA) для комплекса с негидролизуемым аналогом субстрата. Длинная пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель комплекса с негидролизуемым аналогом субстрата (в).

Анализ взаимодействий каталитического остатка глутаминовой кислоты с негидролизуемым аналогом субстрата и первым интермедиатом гидролиза NAA показывает существенные различия. В случае интермедиата выделяется неподеленная электронная пара на атоме кислорода, ориентированная на протонированную карбоксильную группу Glu178, являющуюся донором водородной связи. Второй атом кислорода образует водородную связь с Arg63 и координационную связь с цинком, как и в фермент-субстратном комплексе. В случае комплекса с NPA, PO2-группа отвернута от глутаминовой кислоты из-за отсутствия на ней протона и, как следствие, невозможности формировать водородную связь. Это приводит к тому, что один из кислородов PO2-группы образует координационную связь с катионом цинка, а другой – водородную связь с боковой цепью аргинина. Эти наблюдения находят отражения на соответствующих картах деформационной электронной плотности (рис. 8, 9).
Рис. 8.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Oε1(Glu)–O(Wat)–C(NAA) для интермедиата. Длинная пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель интермедиата (в).

Рис. 9.
Деформационная (а) и промолекулярная электронная плотность (б) в плоскости Oε1(Glu)–O(Wat)–P(NPA) для комплекса с негидролизуемым аналогом субстрата. Длинная пунктирная линия соответствует нулевому значению электронной плотности. Молекулярная модель комплекса с негидролизуемым аналогом субстрата (в).

Таким образом, совместное использование метода квантовой механики/молекулярной механики для расчетов равновесных геометрических конфигураций и соответствующих волновых функций КМ-подсистемы и методов анализа электронной плотности КМ-подсистемы позволило выделить особенности электронного строения активного центра фермента. Мы продемонстрировали на примере комплекса аспартоацилазы с N-ацетиласпартатом и его негидролизуемым аналогом, что в активном центре происходит существенная поляризация лиганда, что можно наблюдать на картах деформационной электронной плотности. Эти данные диктуют необходимость развития методов обработки данных рентгеноструктурного эксперимента белковых макромолекул. Активно применяемые в настоящее время модели сферических атомов хорошо описывают основные черты белковой макросистемы, однако для анализа электронной плотности в активном центре фермента требуются более точные модели, например, основанные на мультипольном разложении.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 18-29-13006). Работа выполнена с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова.
Список литературы
Warshel A., Levitt M. // J. Mol. Biol. 1976. V. 103. P. 227.https://doi.org/10.1016/0022-2836(76)90311-9
Nemukhin A.V., Grigorenko B.L., Lushchekina S.V. et al. // Russ. Chem. Rev. 2012. V. 81. № 11. P. 1011.https://doi.org/10.1070/RC2012v081n11ABEH004311
Kots E.D., Khrenova M.G., Nemukhin A.V. et al //Ibid. 2019. V. 88. № 1. P. 1.https://doi.org/10.1070/RCR4842
Grigorenko B.L., Khrenova M.G., Nilov D.K. et al. // ACS Catal. 2014. V. 4. № 8. P. 2521.https://doi.org/10.1021/cs5002898
Khrenova M.G., Kots E.D., Varfolomeev S.D. et al. // J. Phys. Chem. B. 2017. V. 121. № 40. P. 9389.https://doi.org/10.1021/acs.jpcb.7b08759
Kots E.D., Khrenova M.G., Lushchekina S.V. et al. // Ibid. 2016. V. 120. № 18. P. 4221.https://doi.org/10.1021/acs.jpcb.6b02542
Wolfenden R. // Acc. Chem. Res. 1972. V. 5. № 1. P. 10. https://doi.org/10.1021/ar50049a002
Emsley P., Lohkamp B., Scott W.G. et al. // Acta Crystallogr. Sect. D Biol. Crystallogr. 2010. V. 66. № 4. P. 486.https://doi.org/10.1107/S0907444910007493
Guillot B., Jelsch C., Podjarny A. et al. // Ibid. 2008. V. 64. № 5. P. 567.https://doi.org/10.1107/S0907444908006082
Zarychta B., Pichon-Pesme V., Guillot B. et al. // Acta Crystallogr. Sect. A Found. Crystallogr. 2007. V. 63. № 2. P. 108. https://doi.org/10.1107/S0108767306053748
Carson M. Electron density fitting and structure validation. Oxford University Press: Macromolecular Crystallography, 2007. P. 191.
Levantino M., Yorke B.A., Monteiro D.C. et al. // Curr. Opin. Struct. Biol. 2015. V. 35. P. 41.https://doi.org/10.1016/j.sbi.2015.07.017
Johansson L.C., Stauch B., Ishchenko A. et al. // Trends Biochem. Sci. 2017. V. 42. № 9. P. 749.https://doi.org/10.1016/j.tibs.2017.06.007
Liu H., Spence J.C.H. // Quant. Biol. 2016. V. 4. № 3. P. 159.https://doi.org/10.1007/s40484-016-0076-z
Keedy D.A., Kenner L.R., Warkentin M. et al // Elife. 2015. V. 4. P. e07574.https://doi.org/10.7554/eLife.07574
Kovalevsky A.Y., Chumanevich A.A., Liu F. et al. // Biochemistry. 2007. V. 46. № 51. P. 14854.https://doi.org/10.1021/bi700822g
Das A., Mahale S., Prashar V. et al. // J. Am. Chem. Soc. 2010. V. 132. № 18. P. 6366.https://doi.org/10.1021/ja100002b
Song H., Dong C., Qin M. et al. // Ibid. 2016. V. 138. № 23. P. 7244.https://doi.org/10.1021/jacs.6b03437
Weber I.T., Agniswamy J., Fu G. et al. Reaction intermediates discovered in crystal structures of enzymes. Academic Press: Advances in protein chemistry and structural biology, 2012. P. 57–86.https://doi.org/10.1016/B978-0-12-398312-1.00003-2
Do H., Lee C.W., Lee S.G. et al. // J. Microbiol. 2016. V. 54. № 2. P. 114.https://doi.org/10.1007/s12275-016-5549-2
Le Coq J., Pavlovsky A., Malik R. et al. // Biochemistry. 2008. V. 47. № 11. P. 3484.https://doi.org/10.1021/bi702400x
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. № 13. P. 6158.https://doi.org/10.1063/1.478522
Grimme S., Antony J., Ehrlich S. et al. // Ibid. 2010. V. 132. № 15. P. 154104.https://doi.org/10.1063/1.3382344
Cornell W.D., Cieplak P., Bayly C.I. et al. // J. Am. Chem. Soc. 1995. V. 117. № 19. P. 5179.https://doi.org/10.1021/ja00124a002
Valiev M., Bylaska E.J., Govind N. et al. // Comput. Phys. Commun. 2010. V. 181. № 9. P. 1477.https://doi.org/10.1016/j.cpc.2010.04.018
Lu T., Chen F. // J. Comput. Chem. 2012. V. 33. № 5. P. 580.https://doi.org/10.1002/jcc.22885
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии