

Журнал физической химии, 2019, T. 93, № 3, стр. 385-391
Корреляция между термодинамическими и кинетическими параметрами реакций окисления замещенных моноядерных аренов в присутствии N-гидроксифталимида
М. Е. Соловьев a, Е. А. Курганова a, *, А. С. Фролов a, Г. Н. Кошель a, **
a Ярославский государственный технический университет
Ярославль, Россия
* E-mail: kurganovaea@ystu.ru
** E-mail: koshelgn@ystu.ru
Поступила в редакцию 16.05.2018
Аннотация
Исследовано влияние структуры замещенных моноядерных аренов на термодинамические функции реакций, протекающих при их окислении в присутствии N-гидроксифталимида. Изучена корреляция между свободной энергией реакций окисления кумола, изопропил-м-ксилола, изопропил-о-ксилола, изопропил-п-ксилола, м-цимола, п-цимола, циклогексилбензола и скоростью процесса при температурах 363–413 K. Установлена высокая корреляция между свободной энергией реакции отрыва водорода у третичного атома углерода N-оксифталимидным радикалом и константой скорости данной реакции, а также скоростью окисления для различных изомеров одного и того же углеводорода, что позволяет прогнозировать их относительную окисляемость.
Жидкофазное инициированное окисление замещенных моноядерных аренов является промышленным способом получения соответствующих гидропероксидов, находящих практическое применение во многих областях. Один из способов повышения эффективности данного процесса, позволяющих увеличить конверсию исходных углеводородов до гидропероксидов при сохранении высоких показателей селективности их образования, – использование в качестве гомогенного катализатора N-гидроксифталимида (N-ГФИ) [1–3]. Установлено, что применение N-ГФИ и его алкилзамещенных аналогов в процессе окисления позволяет повысить конверсию углеводорода в 2–3 раза по сравнению со стандартными условиями при сохранении высокой селективности образования гидропероксидов [4].
Практический интерес представляет прогнозирование способности углеводородов к окислению в присутствии N-ГФИ на основании их химического строения. В связи с существованием корреляций между кинетическими и термодинамическими параметрами реакций инициированного радикального окисления углеводородов [5] такая предварительная оценка может быть сделана на основании квантово-химического расчета термодинамических функций реакций передачи цепи, в которых принимает участие N-ГФИ. Цель настоящей работы – исследование влияния структуры замещенных моноядерных аренов на термодинамические функции реакций, протекающих при их окислении в присутствии N-ГФИ, и оценка их корреляции с кинетическими параметрами окисления.
В качестве объектов исследования в настоящей работе выбраны следующие углеводороды: кумол, изопропил-м-ксилол, изопропил-о-ксилол, изопропил-п-ксилол, м-цимол, п-цимол, циклогексилбензол.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1,3-Диметил-5-изопропилбензол (изопропил-м-ксилол), 1,2-диметил-4-изопропилбензол (изопропил-о-ксилол), 1,4-диметил-2-изопропилбензол (изопропил-п-ксилол) получали реакцией алкилирования изопропиловым спиртом соответствующих углеводородов в присутствии концентрированной серной кислоты и последующим переалкилированием полученных смесей изомеров с безводным алюминием хлористым (кроме изопропил-п-ксилола). Циклогексилбензол синтезировали гидродимеризацией бензола. Полученные углеводороды, а также изопропилбензол (кумол), 1-метил-3-изопропилбензол (м-цимол) и 1-метил-4-изопропилбензол (п-цимол) марки “х.ч.” очищали от ингибирующих примесей последовательно раствором NaOH и дистиллированной водой до нейтральной среды. Высушенные над безводным хлористым кальцием углеводороды перегоняли в вакууме.
В качестве катализатора использовали N-гидроксифталимид, полученный взаимодействием фталевого ангидрида (0.1 моль) и гидроксиламина солянокислого (0.11 моль) в среде пиридина при температуре 42°С. Осадок N-ГФИ отделяли, промывали на фильтре 0.01 н. раствором уксусной кислоты и сушили в вакууме. Чистоту полученного продукта определяли по его температуре плавления (Тпл = 231.5°С, литературные данные: Тпл = 233°С [6]).
Кинетические закономерности процесса изучали в ряде экспериментов по окислению углеводородов в среде воздуха при температурах 363–413 K в течение 90 мин в присутствии 0.105–0.116 моль/л N-ГФИ и в его отсутствие (автоокисление). Количество поглощенного в процессе окисления кислорода контролировали волюметрическим методом, накопление гидропероксидов – методом йодометрического титрования. Подробная методика проведения эксперимента и описание установки приведены в работе [7].
Расчет электронных энергий и термодинамических функций участвующих в реакциях соединений производили квантово-химическим методом функционала плотности [8, 9] c гибридным обменно-корреляционным функционалом Беке (Becke) [10], Ли, Янга и Парра (Lee, Yang and Parr) [11] DFT B3LYP/cc-pvdz с открытыми оболочками [12] с использованием программного комплекса NWChem [13]. Выбор метода DFT B3LYP/cc-pvdz был обусловлен удовлетворительным соотношением точность/скорость вычислений для энергий атомизации углеводородов, а также азот- и кислородсодержащих соединений, близких к рассматриваемым в настоящей работе [14]. В процессе вычислений для индивидуальных соединений и радикалов выполняли оптимизацию геометрии, при этом достижение минимумов на поверхностях потенциальной энергии контролировали по условию положительной определенности матрицы Гессе (вторых производных потенциальной энергии). В конформации с минимальной энергией рассчитывали термодинамические функции (энтальпия и свободная энергия Гиббса) как сумму полной электронной энергии DFT в описанном выше приближении B3LYP/cc-pvdz и термодинамической составляющей в приближении “жесткий ротатор–гармонический осциллятор” при температуре реакции, вычисленной по стандартной методике на основе вибрационного анализа в состоянии идеального газа [12]. Свободные энергии реакций вычисляли как разности соответствующих величин для продуктов реакций и реагентов при температуре реакции.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Цикл кинетической цепи окисления в присутствии N-ГФИ, описанный в работах [1, 15–17] выглядит следующим образом:
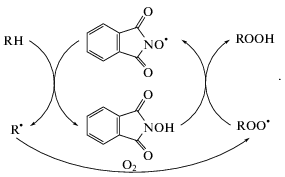
Процесс протекает путем повторяющихся циклов элементарных стадий, особенность которых в том, что участвующие в этих реакциях свободные радикалы, в том числе и N-оксифталимидный радикал (phthalimide-N-oxyl, PINO$_{{}}^{\centerdot }$), регенерируются в пределах каждого цикла [18].
Активным промежуточным продуктом реакции, обеспечивающим рост кинетической цепи, является PINO$_{{}}^{\centerdot }$, возникающий при отрыве атома водорода связи >NO–H в N-ГФИ. Существование PINO$_{{}}^{\centerdot }$ доказано методом ЭПР [17]. С учетом этого формальная схема окисления углеводорода в присутствии катализатора N-ГФИ (PINOH) может быть записана в следующем упрощенном виде:
(1)
${\text{RH}} + {{{\text{O}}}_{2}} \to {{{\text{R}}}^{\centerdot }} + {\text{HO}}_{2}^{\centerdot },$(2)
$2{\text{RH}} + {{{\text{O}}}_{{\text{2}}}} \to 2{{{\text{R}}}^{\centerdot }} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}},$(3)
${\text{PINOH}} + {{{\text{O}}}_{{\text{2}}}} \to {\text{PIN}}{{{\text{O}}}^{\centerdot }} + {\text{HO}}_{2}^{ \bullet },$(6)
${\text{RO}}_{2}^{ \bullet } + {\text{PINOH}} \to {\text{ROOH}} + {\text{PIN}}{{{\text{O}}}^{\centerdot }},$(7)
${\text{PIN}}{{{\text{O}}}^{\centerdot }} + {\text{RH}} \to {{{\text{R}}}^{\centerdot }} + {\text{PINOH}},$(8)
${\text{ROOH}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{O}}{{{\text{H}}}^{\centerdot }},$(9)
$2{\text{ROOH}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{RO}}_{2}^{\centerdot } + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$(10)
${\text{ROOH}} + {\text{RH}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {{{\text{R}}}^{\centerdot }} + {{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(12)
${\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{RH}} \to {{{\text{R}}}^{\centerdot }} + {\text{ROH}},$(13)
${\text{H}}{{{\text{O}}}^{\centerdot }} + {\text{RH}} \to {{{\text{R}}}^{\centerdot }} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}{\text{,}}$(14)
${\text{HO}}_{2}^{ \bullet } + {\text{RH}} \to {{{\text{R}}}^{\centerdot }} + {{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(16)
${\text{PIN}}{{{\text{O}}}^{\centerdot }} + {\text{PIN}}{{{\text{O}}}^{\centerdot }} \to {\text{п р о д у к т ы }}{\text{.}}$Реакции (1) и (2) представляют соответственно бимолекулярное и тримолекулярное автоинициирование [5], реакция (3) отвечает инициированию с участием N-ГФИ, реакции (4) и (5) – классическому продолжению цепи окисления, тогда как реакции (6) и (7) соответствуют продолжению цепи окисления с участием радикала PINO$_{{}}^{\centerdot }$. Реакции (8)–(10) представляют различные варианты вырожденного разветвления. Обрыв цепей окисления описывается реакцией (11), являющейся некоторой упрощенной моделью данного многостадийного процесса. Реакции (12)–(14) описывают продолжение цепей в результате взаимодействия углеводорода с оксидными и гидропероксидными радикалами. Реакция (15) моделирует процесс растворения молекулярного кислорода, находящегося над раствором в газообразном состоянии. Реакция (16) соответствует некоторому “непроизводительному” расходу катализатора N-ГФИ, учет которого необходим, как показал анализ экспериментальных кинетических кривых.
Хотя в целом приведенная кинетическая схема формальна и представляет лишь упрощенную модель сложного набора реакций, протекающих в реальной системе, тем не менее, она позволяет адекватно описать кинетические кривые поглощения кислорода и накопления гидропероксидов, наблюдавшиеся в эксперименте для изученных в данной работе объектов. Использование подобных упрощенных моделей имеет практическую ценность, как было показано в работах [19, 20].
Для формулировки системы дифференциальных уравнений химической кинетики, отвечающей схеме реакций (1)–(16) введем следующие обозначения для концентраций реагирующих веществ: x(1) – [RH], x(2) – [RH$_{{}}^{\centerdot }$], x(3) – [O2](раствор), x(4) – [RO$_{2}^{ \bullet }$], x(5) – [ROOH], x(6) – [PINOH], x(7) – [PINO$_{{}}^{\centerdot }$], x(8) – [RO$_{{}}^{\centerdot }$], x(9) – [OH$_{{}}^{\centerdot }$], х(10) – [OOH$_{{}}^{\centerdot }$], x(11) – [O2] (поглощенное в результате реакции), x0(3) – начальная (равновесная) концентрация О2 в растворе. Тогда, обозначив константы скоростей реакций символами k(1)–k(16), для скоростей изменения концентраций данных реагентов получим следующую систему дифференциальных уравнений:
(17)
$\begin{gathered} \frac{{dx(1)}}{{dt}} = - k(1)x(1)x(3) - 2k(2)x(1)x(1)x(3) - \\ - \;k(5)x(4)x(1) - k(7)x(7)x(1) - k(10)x(5)x(1) - \\ - \;k(12)x(8)x(1) - k(13)x(9)x(1) - k(14)x(10)x(1), \\ \end{gathered} $(18)
$\begin{gathered} \frac{{dx(2)}}{{dt}} = k(1)x(3)x(1) + 2k(2)x(1)x(1)x(3) - \\ - \;k(4)x(2)x(3) + k(5)x(4)x(1) + k(7)x(7)x(1) + \\ + \;k(10)x(5)x(1) + k(12)x(8)x(1) + \\ + \;k(13)x(9)x(1) + k(14)x(10)x(1), \\ \end{gathered} $(19)
$\begin{gathered} \frac{{dx(3)}}{{dt}} = - k(1)x(1)x(3) - k(2)x(1)x(1)x(3) - \\ - \;k(3)x(6)x(3) - k(4)x(2)x(3) + k(15)(x0(3) - x(3)), \\ \end{gathered} $(20)
$\begin{gathered} \frac{{dx(4)}}{{dt}} = k(4)x(2)x(3) - k(5)x(4)x(1) - \\ - \;k(6)x(4)x(6) - 2k(11)x(4)x(4) + k(9)x(5)x(5), \\ \end{gathered} $(21)
$\begin{gathered} \frac{{dx(5)}}{{dt}} = k(5)x(4)x(1) + k(6)x(4)x(6) - \\ - \;k(8)x(5) - 2k(9)x(5)x(5) - k(10)x(5)x(1), \\ \end{gathered} $(22)
$\begin{gathered} \frac{{dx(6)}}{{dt}} = - k(6)x(4)x(6) + \\ + \;k(7)x(1)x(7) - k(3)x(6)x(3), \\ \end{gathered} $(23)
$\begin{gathered} \frac{{dx(7)}}{{dt}} = k(6)x(4)x(6) - k(7)x(1)x(7) + \\ + \;k(3)x(6)x(3) - 2k(16)x(7)x(7), \\ \end{gathered} $(24)
$\begin{gathered} \frac{{dx(8)}}{{dt}} = k(8)x(5) + k(9)x(5)x(5) + \\ + \;k(10)x(5)x(1) - k(12)x(8)x(1), \\ \end{gathered} $(26)
$\begin{gathered} \frac{{dx(10)}}{{dt}} = k(1)x(1)x(3) + \\ + \;k(3)x(6)x(3) - k(14)x(10)x(1), \\ \end{gathered} $Кинетические константы реакций k(1)–k(16) оценивали методом наименьших квадратов путем минимизации суммы квадратов отклонений для экспериментальных и расчетных значений концентраций поглощенного кислорода и гидропероксидов. Расчетные значения концентраций вычисляли численным интегрированием системы дифференциальных уравнений кинетики (17)–(27), соответствующей схеме реакций (1)–(12) с начальными условиями, отвечающими заданной начальной концентрации углеводорода, N-ГФИ и растворенного кислорода, для которой было принято значение 7.5 × 10–3 моль/л. Для остальных реагентов начальную концентрацию принимали нулевой. При этом вычисление оценок кинетических констант производили в два этапа. Сначала по кинетическим кривым окисления в отсутствие N-ГФИ (автоокисление) вычисляли оценки констант скоростей реакций (1)–(4), (8)–(15), а затем данные оценки использовали при аппроксимации всей кинетической схемы по данным окисления в присутствии N-ГФИ. На рис. 1, 2 в качестве примера приведены расчетные и экспериментальные кривые поглощения кислорода при различных температурах для некоторых углеводородов. Как видно, наблюдается достаточно хорошее согласие экспериментальных и расчетных данных, что подтверждает возможность использования принятой кинетической схемы. Сравнение кинетических кривых показало, что исследованные углеводороды по скорости автоокисления располагаются в следующий ряд: кумол > п-цимол > м-цимол > циклогексилбензол > > изопропил-о-ксилол > изопропил-м-ксилол > > изопропил-п-ксилол.
Рис. 1.
Экспериментальные точки и расчетные кинетические кривые поглощения кислорода в процессе автоокисления п-цимола (а) и при окислении его в присутствии N-гидроксифталимида (б); Т = 90 (1), 100 (2), 110 (3) и 120°С (4); концентрация N-ГФИ – 0.1051 моль/л.
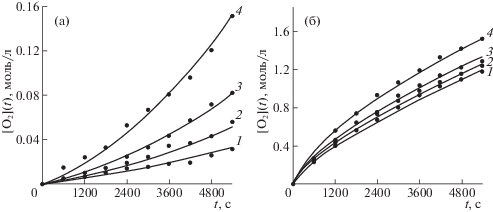
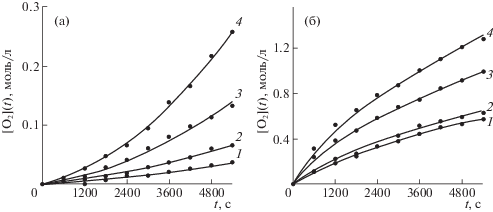
Рис. 2.
Экспериментальные точки и расчетные кинетические кривые поглощения кислорода в процессе автоокисления циклогексилбензола (а) и при окислении его в присутствии N-гидроксифталимида (б); Т = 100 (1), 110 (2), 120 (3) и 130°C (4); концентрация N-ГФИ – 0.1159 моль/л.
Каталитическое действие N-ГФИ состоит в генерировании радикалов PINO$_{{}}^{\centerdot }$ в реакциях (3), (6) и регенерировании PINOH в реакции (7), определяющей скорость генерирования углеводородных радикалов. Вычисленное на уровне теории, рассматриваемом в настоящей работе, значение энергии связи >NO–H в N-ГФИ составило 348 кДж/моль. Хотя эта величина близка к значениям энергии связи O–H в фенольных антиоксидантах, в отличие от них PINO$_{{}}^{\centerdot }$ почти не образует стабильных молекулярных продуктов и не обрывает кинетические цепи окисления, так что вклад реакции (16) достаточно мал по сравнению с обычными антиоксидантами. В результате в системе возрастает концентрация углеводородных радикалов, что приводит к увеличению скорости окисления. Исходя из описанной выше схемы реакций, можно заключить, что влияние структуры углеводорода на скорость окисления в присутствии катализатора N-ГФИ будет проявляться в реакции (7), определяющей скорость генерирования углеводородных радикалов, в результате чего скорость окисления существенно возрастает.
В табл. 1 приведены рассчитанные квантово-химическим методом термодинамические функции реагентов реакции (7) для исследованных углеводородов.
Таблица 1.
Рассчитанные квантово-химическим методом DFT B3LYP/cc-pvdz термодинамические функции реагентов реакции (7) при Т = 298.15 K
| Углеводороды и их радикалы | –Е, кДж/моль | Δ, кДж/моль | $C\text{v}$, Дж/моль | S, Дж/(моль K) |
|---|---|---|---|---|
| PINOH | 1 544 432.69 | 340.25 | 149.00 | 396.24 |
| PINO$_{{}}^{\centerdot }$ | 1 542 771.41 | 307.45 | 140.82 | 384.75 |
| Изопропил-м-ксилол | 1 125 927.64 | 658.85 | 191.15 | 454.59 |
| R$_{{}}^{\centerdot }$(изопропил-м-ксилол) | 1 124 240.18 | 623.95 | 190.33 | 443.18 |
| Изопропил-о-ксилол) | 1 125 926.59 | 658.78 | 189.97 | 436.19 |
| R$_{{}}^{\centerdot }$(изопропил-о-ксилол) | 1 124 240.18 | 623.95 | 190.33 | 443.18 |
| Изопропил-п-ксилол | 11 25 919.08 | 659.44 | 190.37 | 450.51 |
| R$_{{}}^{\centerdot }$(изопропил-п-ксилол) | 1 124 215.93 | 624.58 | 189.94 | 441.76 |
| п-Цимол | 1 022 693.62 | 587.78 | 164.57 | 418.63 |
| R$_{{}}^{\centerdot }$(п-цимол) | 1 020 999.95 | 552.51 | 165.07 | 424.24 |
| м-Цимол | 1 022 693.60 | 587.83 | 164.54 | 419.30 |
| R$_{{}}^{\centerdot }$(м-цимол) | 1 020 998.59 | 552.52 | 165.30 | 423.53 |
| Циклогексилбензол | 1 225 978.57 | 685.23 | 174.19 | 414.45 |
| R$_{{}}^{\centerdot }$(циклогексилбензол) | 1 224 285.89 | 651.58 | 174.71 | 417.87 |
| Кумол | 919 471.46 | 507.74 | 140.00 | 380.53 |
| R$_{{}}^{\centerdot }$(кумол) | 917 784.66 | 472.30 | 140.44 | 381.86 |
На основании данных табл. 1 вычислены изменения энергии Гиббса в результате реакции (7) при температурах реакции для всех исследованных углеводородов. Все вычисленные значения оказываются положительными. Это объясняется тем, что прочность связи R–H для данных углеводородов составляет более 380 кДж/моль, т.е.превышает вычисленное значение прочности связи >NO–H в N‑ГФИ. Аналогичная ситуация имеет место и для реакции передачи цепи (5), поскольку прочность связи ROO–H составляет 365–370 кДж/моль [5]. Таким образом, реакция (7) не протекает самопроизвольно, а играет роль лимитирующей стадии всего процесса, т.е. между изменением энергии Гиббса в результате реакции (7) и ее скоростью должна наблюдаться отрицательная корреляция.
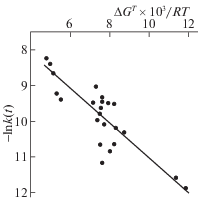
Действительно, как видно из данных, приведенных на рис. 3, наблюдается отрицательная корреляция между ln k(7) и изменением энергии Гиббса в результате реакции (7) при температуре реакции (ΔGT), отнесенным к RT. Для оценки близости линейной зависимости между исследуемыми параметрами вычисляли коэффициент корреляции [21] по формуле:
Рис. 3.
Корреляция между lnk(7) и изменением энергии Гиббса в результате реакции (7) при температуре реакции (ΔGT), отнесенным к RT.
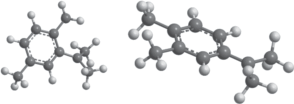
В то же время, для соединений близкой химической структуры, в частности изомеров, по величине ∆G реакции (7) можно сделать качественный вывод о соотношении их скоростей окисления. Это хорошо видно из табл. 2, если сравнивать скорости окисления и ∆G реакции (7) для изомеров цимола, а также изомеров изопропилксилола. В последнем случае привлекает внимание особенно низкая по сравнению с другими изомерами скорость окисления изопропил-п-ксилола. Причина этого становится понятной, если проанализировать геометрическую структуру радикалов R$_{{}}^{\centerdot }$ разных изомеров. В качестве примера на рис. 4 приведены геометрические модели радикалов изопропил-п-ксилола и изопропил-о-ксилола в минимуме энергии. В случае изопропил-о-ксилола (рис. 4б) в структуре, отвечающей минимуму энергии, все три атома углерода изопропильного фрагмента располагаются в плоскости бензольного кольца. Это обеспечивает наилучшие условия для делокализации электронной плотности неспаренного электрона и уменьшения, вследствие этого, энергии радикала. При этом вычисленное значение спиновой плотности на атоме углерода с неспаренным электроном составляет 0.71. Для изопропил-п-ксилола (рис. 4а) наличие у бензольного кольца метильного заместителя в непосредственной близости от изопропильного создает стерические затруднения при повороте изопропильного фрагмента. В результате в минимуме энергии он оказывается частично повернут относительно плоскости кольца на небольшой угол. Это ухудшает условия для делокализации электронной плотности неспаренного электрона (значение спиновой плотности на атоме углерода с неспаренным электроном составляет 0.74), и энергия радикала в результате возрастает, и как следствие, возрастает ∆G реакции (7).
Таблица 2.
Зависимость между количеством поглощенного кислорода (с) за 30 мин окисления при T = = 413 K и изменением энергии Гиббса реакции (7) для различных углеводородов
| Соединения | с, моль/л | ΔG413, кДж/моль |
|---|---|---|
| Кумол | 0.85 | 18.6 |
| п-Цимол | 0.74 | 23.8 |
| м-Цимол | 0.58 | 25.8 |
| Изопропил-о-ксилол | 0.43 | 16.4 |
| Изопропил-м-ксилол | 0.23 | 24.9 |
| Изопропил-п-ксилол | 0.10 | 39.6 |
| Циклогексилбензол | 0.47 | 25.4 |
ЗАКЛЮЧЕНИЕ
Таким образом, изучена корреляция между свободной энергией реакций окисления некоторых углеводородов из класса моноядерных аренов и кинетическими параметрами данных реакций, а также скоростью окисления при температурах 363–413 K. Установлено, что, как при автоокислении, так и при окислении в присутствии катализатора N-ГФИ, по скорости окисления изученные углеводороды располагаются в следующий ряд: кумол > п-цимол > циклогексилбензол > м-цимол > изопропил-о-ксилол > изопропил-м-ксилол > изопропил-п-ксилол.
Установлена высокая корреляция между свободной энергией реакции отрыва водорода у третичного атома углерода N-оксифталимидным радикалом и константой скорости данной реакции, а также скоростью окисления для различных изомеров одного и того же углеводорода. Показано, что квантово-химическая оценка свободной энергии данной реакции позволяет прогнозировать относительную окисляемость различных изомеров одного и того же углеводорода.
Список литературы
Yoshino Y., Hayashi Y., Iwahama T., Sakaguchi S. et al. // J. Org. Chem. 1997. V. 62. P. 6810.
Кошель Г.Н., Смирнова Е.В., Курганова Е.А., Плахтинский В.В. и др. // Хим.технология. 2009. Т. 10. № 12. С. 719.
Кошель Г.Н., Смирнова Е.В., Курганова Е.А. и др. // Катализ в промышленности. 2010. № 3. С. 26.
Курганова Е.А., Сапунов В.Н., Кошель Г.Н., Фролов А.С. // Изв. АН. Сер. хим. 2016. № 2. С. 2115.
Denisov E.T., Afanas’ev I.B. Oxidation and Antioxidants in Organic Chemistry and Biology. CRC Press. Taylor & Francis Group. 2005. 992 p.
Alfa Aesar Research Chemical Metals and Materials. A Johnson Matthey Company, 2008. 2741 p.
Frolov A.S., Kurganova E.A., Koshel’ G.N., Nesterova T.N. // European Journal of Analytical and Applied Chemistry. 2015. № 1. C. 16.
Hohenberg P., Kohn W. // Phys. Rev. 1964. V. 136. № 3B. P. B864.
Kohn W., Sham L. // Ibid. 1965. V. 140. № 4A. P. A1133.
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 7. P. 5648.
Miehlich B.A., Savin H.S., Preuss H. // Chem. Phys. Lett. 1989. V. 157. № 3. P. 200.
Levine I.N. Quantum Chemistry 5Th Ed. Prentice-Hall Of India Pvt. Limited, 2000. 739 p.
Valiev M., Bylaska E.J., Govind N. et al. // Comput. Phys. Commun. 2010. V. 181. P. 1477.
Computational Chemistry Comparison and Benchmark DataBase. [Электронный ресурс]: Режим доступа: http://cccbdb.nist.gov.
Iwahama T., Yoshima Y., Keitoku T. et al. // J. Org. Chem. 2000. V. 65. P. 6502.
Sakaguchi S., Nishiwaki Y., Kitamura T., Yshii Y. // Angew Chem. Int. Ed. 2001. V. 40. I. 1. P. 222.
Amorati R., Lucarini M., Mugnaini V., Pedulli G.F. // J. Org. Chem. 2003. V. 68. P. 5198–5204.
Опейда И.А., Плехов А.Л., Кущ О.В., Матвиенко А.Г. // Журн. физ. химии. 2011. Т. 85. № 7. С. 1223.
Базанов Т.А., Петров Л.В., Психа Б.Л. и др. // Нефтехимия. 2008. Т. 48. № 4. С. 295.
Базанов Т.А., Петров Л.В., Психа Б.Л. и др. // Хим. физика. 2009. Т. 28. № 7. С. 42.
Кобзарь А.И. Прикладная математическая статистика. Для инженеров и научных работников. М.: Физматлит, 2006. 816 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии