

Журнал физической химии, 2019, T. 93, № 6, стр. 946-955
Фотохимические реакции аминоазобензола в растворах по данным ультравысокоскоростной VIS-спектроскопии
Ю. А. Михеев a, *, Ю. А. Ершов b
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 10.09.2018
После доработки 10.09.2018
Принята к публикации 10.09.2018
Аннотация
На основе анализа литературных данных по транзитным VIS-спектрам при импульсном лазерном возбуждении λex = 400 нм определены фотоиндуцированные процессы (фотоника) в транс-аминоазобензоле (t-AAB). В основу анализа положены представления о ридимерном строении аминоазобензольных красителей и, в частности, t-AAB2 (основного состояния красителя). Показано, что фотохимическое появление и последующая трансформация транзитных спектров, а также изомеризация t-AAB2 в цис-аминоазобензол (с-AAB) обусловлены параллельно протекающими актами расщепления ридимеров t-AAB2 на мономеры; расщепление ридимеров индуцируется поглощением VIS-света катионами фениламинильного типа – хромогенами ридимеров t-AAB2. Приведен механизм процесса, включающий образование франк-кондоновских мономерных пар t-AAB двух типов. Показано, что пара одного типа состоит из двух одинаковых мономеров, она релаксирует в исходный ридимер t‑AAB2, пара другого типа содержит один мономер t-AAB и двукратно заряженный мономер с двумя катионами фениламинильного типа и VIS-поглощением в интервале длин волн 500–750 нм. Мономерный дикатион изомеризуется в c-AAB. Приведены формулы строения транзитных электронных конфигураций, объясняющие фотонику ридимеров t-AAB2.
Анализ UV–VIS-спектров аминоазобензольных красителей в работах [1–6] позволил обнаружить ранее не известное фундаментальное свойство: эти красители (в частности аминоазобензол AAB) в своем основном состоянии являются ридберговскими димерами (ридимерами). Ридимеры стабилизируются межмономерной ковалентной связью нового типа. Эта связь образуется в результате спаривания электронов, промотированных с sp2-орбиталей атомов N азогрупп на ридберговские 3s-орбитали азогрупп. При этом характерная для азокрасителей цветность связана не с их хиноидными мономерами, как полагали более 100 лет, а с катионами фениламинильного типа (CPhAT) ридимеров.
Результаты работ [1–6] позволяют корректировать достижения предшествующего длительного этапа накопления экспериментальных данных. Например, в [6] дан новый анализ спектров резонансной рамановской спектроскопии, опубликованных в работе [7]. В [6] впервые показано возрастание силы сопряженных кислот ридимеров (AAB+H)2 при фотовозбуждении, ведущее к их депротонированию даже в среде с достаточно высокой кислотностью.
В данной работе впервые объясняется природа транзитных спектров, наблюдавшихся в [8] при импульсном лазерном фотолизе транс-4-аминоазобензола (t-AAB, t-PhN=NPħNH2) с учетом того, что реально основным состоянием этого красителя являются не мономеры (t-AAB), а ридимеры t-AAB2.
Авторская [8] феноменология спектральных трансформаций транс-аминоазобензола
Авторы [8] использовали методику ультрабыстрой регистрации спектров поглощения транзитных состояний аминоазобензола при импульсном лазерном возбуждении раствора красителя в этаноле и гептаноле. Возбуждение осуществляли с помощью пучка видимого (VIS) света с длиной волны λex = 400 нм. Мощность этого излучения сделали меньше 200 мкДж/импульс с целью исключения многофотонного поглощения, частота повторения импульсов составляла 1000 Гц. Была установлена практическая идентичность спектральных изменений в обоих растворителях.
Авторы [8] преследовали цель детализировать закономерности транс–цис-изомеризации красителя, опираясь на обсуждаемые в литературе закономерности фотоизомеризации его родительского соединения – азобензола (AB). При этом они исходили, как теперь известно [1–6], из неверного представления, что имеют дело с индивидуальными молекулами t-AAB. Кроме того, опираясь на традиционное, но неверное отнесение n → π*-перехода в транс-азобензоле (t-AB) к слабой полосе при 440 нм [4, 9], они постулировали, что n → π*-переход молекул “t-AAB” находится там же, где и у t-AB, т.е. на длинноволновой стороне интенсивной полосы поглощения “t‑AAB” (λmax ∼ 390 нм), в виде “хвоста”.
Для демонстрации факта расходования исходного “t-AAB” и образования c-AAB авторы [8] сопоставили спектр VIS-полосы “t-AAB” (раствор в этаноле) с разностной кривой (рис. 1в [8]), полученной после облучения того же раствора в течение 20 мин ксеноновой лампой. К сожалению, они не привели спектр раствора с накопленным изомером c-AAB, из которого провели вычитание. В целях полноты такого сопоставления мы приводим более наглядный рис. 1 со спектрами, полученными сходным образом в работе [10, рис. 3 ]. На рис. 1 кривая 1 характеризует поглощение мнимого мономерного “t-AAB” в диэтиловом эфире до облучения. Кривая 2 на рис. 1 представляет спектр фотостационарного состояния транс → цис-изомеризации t-AAB в диэтиловом эфире, полученный под действием света ксеноновой лампы, а кривая 3 – разность кривых 2 и 1. (Отметим наличие полного качественного сходства кривой 1 работы [8, рис. 1] с кривой 1 на рис. 1, а также сходство разностных спектров, представленных в обеих сопоставленных работах [8] и [10]).
Рис. 1.
Спектры поглощения раствора t-AAB2 в диэтиловом эфире: 1 – до, 2 – после облучения светом ксеноновой лампы, 3 – результат вычитания спектра 1 из спектра 2. Данные [10].

Кажущаяся простота спектров 1–3 на рис. 1, полученных под действием непрерывного излучения ксеноновой лампы, казалось бы, свидетельствует об относительно простом механизме процесса транс → цис-фотоизомеризации “t‑AAB”. Между тем, в действительности этот процесс является достаточно сложным, о чем можно сделать вывод по картине спектральной трансформации промежуточных (транзитных) соединений, полученной с применением ультравысокоскоростной спектроскопии (рис. 2 в [8]) и представленной нами на рис. 2. Транзитные спектры на рис. 2 получены для растворенного в этаноле t-AAB2. Использовали проточную кварцевую ячейку, растворы облучали осциллирующим лазерным светом с частотой повторения 1 кГц и длиной волны λex = 400 нм, времена задержки для импульсов зондирующих лучей: 0.1, 0.5, 1.0 и 5.0 пс (ps) – кривые 1–4, соответственно.
Рис. 2.
Спектры транзитных состояний t-AAB2 в этаноле при импульсном облучении светом с λex = = 400 нм и временах задержки зондирующих импульсов: 1 – 0.1, 2 – 0.5, 3 – 1.0, 4 – 5 пс. Данные [8].

Напомним, что, по мнению авторов [8], поглощение света с λex = 400 нм вызывает π → π*-переход мнимой молекулы “t-AAB” на мнимый второй π*-уровень (S0 → S2), а поглощение на длинноволновом “хвосте“ этой полосы соответствует переходу n → π* (S0 → S1) (о природе “хвоста” см. ниже).
Наблюдавшееся в [8] транзитное поглощение имеет сложную природу. Сразу после возбуждения появляется спектр 1 (рис. 2), содержащий широкую полосу транзитного поглощения в области 410–750 нм и относительно долгоживущую полосу отбеливания (bleachihg) в области поглощения основного состояния t-AAB2 при 370–410 нм. В интервале длин волн 600–725 нм снижение интенсивности транзитного поглощения во времени описывается тремя экспоненциальными функциями с различными характеристическими временами τi. Наиболее быстро распадающаяся компонента соответствует τ1 = 0.2 пс. Наличие этой компоненты в области 500–600 нм практически не определяется, а в области 600–725 нм ее вклад становится пренебрежимо низким после десяти периодов полураспада 10T0.5 = 1.38 пс. Две другие компоненты распадаются с временами τ2 = 0.6 пс и τ3 = 1.9 пс. Оценка положений максимумов указанных полос привела к значениям 650 нм для компоненты с τ1 = 0.2 пс и 550 нм для двух других компонент.
Релаксация поглощения в области 500–600 нм соответствует экспоненциальным функциям двух транзитных состояний с τ2 = 0.6 пс и τ3 = 1.9 пс. В области 425–475 нм эти две компоненты налагаются на полосу отбеливания, возвратная эволюция которой соответствует моноэкспоненциальной функции с характеристической константой τ = 15 пс. В конце эволюции наблюдается спектр, соответствующий раствору, обедненному транс-формой t-AAB2 и обогащенному цис-формой AAB. Согласно (рис. 3 в [8]) доля восстановления t-AAB2 соответствует ∼75%, тогда как ∼25% превращается в c-AAB. Характерно также, что оптическая плотность (D) в области перекрывания транзитного поглощения с транзитной полосой отбеливания (410–470 нм) сначала увеличивается (рис. 2, кривая 2), а затем снижается в область отрицательных значений D.
Обсуждая результаты, авторы [8] приписывают быструю транзитную компоненту (0.2 пс, область 600–750 нм) процессу конверсии энергии с S2-уровня (π, π*-состояние) на S1-уровень (“n, π*-состояние”). Относительно двух компонент спада, идущих в спектральной области 410–750 нм с τ2 = 0.6 пс и τ3 = 1.9 пс, авторы подчеркивают наличие у них спектров с одинаковой формой и связывают их, не объясняя причины, с наличием двух разных путей спада “n,π*-состояния”.
Заканчивая рассмотрение авторского [8] описания трансформации спектра возбужденного t‑AAB, следует подчеркнуть несостоятельность использованной ими трактовки природы π → π*- и n → π*-полос у t-AB и t-AAB2. Так, согласно недавно опубликованной работе [4], действительное положение n → π*-полос у t-AB и t-AAB2 локализовано в УФ-области спектра, а полоса t‑AAB2 с λmax при 390 нм соответствует π → π*-переходу S0 → S1 в катионах CPhAT [1–6, 9].
Очевидно, что изложенное выше требует пересмотра экспериментальных результатов работы [8] с учетом ридимерной природы AAB2 и его хромогенов, в роли которых выступают индивидуальные и бинарные катионы CPhAT.
Феноменология катионов фениламинильного типа CPhAT
Существование CPhAT было установлено впервые при анализе VIS-спектров поглощения катионов t-AB в концентрированной серной кислоте [11]. Формула строения CPhAT у образующейся протонированной формы AB+H имеет вид

Интенсивный желто-оранжевый цвет AB+H обязан его VIS-полосе (λmax ∼ 430–440 нм), связанной с преимущественной локализацией обоих π-электронов азогруппы на pz-орбитали протонированного азота N+H и появлением индуцированного положительного заряда на pz-орбитали непротонированного азота. Этот положительный заряд частично распределяется на орто- и пара-положениях смежного фенильного кольца, и возникающий таким образом катион фениламинильного типа (CPhAT, ph+N+–) принимает на себя роль хромогена [11] подобно тому, как это происходит в бензильных и фениламинильных катионах.
Все указанные катионы имеют не только связывающие и разрыхляющие молекулярные орбитали (МО), но еще и вакантные несвязывающие орбитали (НСМО) с нулевой энергией. Именно на НСМО переходят возбуждаемые VIS-светом электроны с высших занятых МО, приводя к появлению в спектрах указанных катионов VIS-полос, сходных по форме и положению.
Хромогены CPhAT существуют в малом содержании также и в молекулах AB, при отсутствии такого индуктора поляризации как ковалентно присоединенный к азоту протон [9]. В молекуле AB в роли индуктора поляризации выступает несвязывающая ридберговская 3s-орбиталь (R3s-) азогруппы (локального хромофора N=N). Она частично перекрывается с несвязывающими атомными sp2-орбиталями атомов азота азогруппы и с невысокой вероятностью обратимо акцептирует электрон то с одного, то с другого атома N. В такой ситуации обратимые электронные (e-) переходы (:sp2) + $R_{{3s}}^{0}$ ↔ (•sp2) + $R_{{3s}}^{ \bullet }$ реализуют чередование форм специфического комплекса с переносом заряда, а остающаяся с одним электроном •sp2-орбиталь создает положительный заряд на родительском атоме N, как в ситуации с присоединенным протоном. Соответствующее электронно-таутомерное равновесие в AB имеет вид (схема 1):
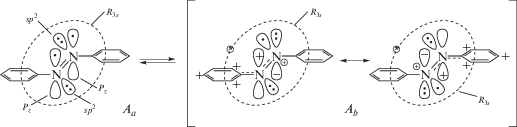
В схеме 1 Aa – преобладающая по содержанию неполярная структура phN=Nph с вакантной R3s-орбиталью, Ab – меньшая по содержанию структура полярного e-таутомера, в котором R3s-орбиталь содержит один электрон, причем в Ab чередуются акты обмена электронов с изменением электронных e-конфигураций CPhAT (ph+N+–): (+)ph…(+)N…N–ph)$R_{{3s}}^{{( - )}}$ ←→ (ph–N…N(+)…ph(+))$R_{{3s}}^{{( - )}}$. (Частота этих актов на многие порядки превосходит частоту колебаний атомов.) Согласно [9], именно катион CPhAT (ph+N+–) в непротонированном азобензоле ответственен за слабую π → π*-полосу при 420–440 нм, которую в течение многих лет приписывали n → π*-переходу. Что же касается абсорбционных полос непосредственно переходов n → π* в молекулах AB и ридимерах t-AAB2, то, согласно работе [4], они расположены в УФ-области спектра при 300–330 нм.
Характерная e-таутомерия (схема 1) наделяет молекулу t-AB двумя неодинаковыми системами электронных уровней. Рассмотренные выше e-структуры Ab имеют низкий статистический вес относительно структуры Aa с ее π → π*-полосой, расположенной в УФ-области при 320 нм. Согласно Симпсону [12], появление этой УФ-полосы несовместимо с теорией Хюккеля и объясняется механизмом делокализации электронного возбуждения между локальными субъединицами, в качестве которых выступают фенильные группы, испытывающие лишь некоторое возмущение со стороны групп N=N. По теории [12], в основном состоянии возмущенные азогруппами фенилы Aa не имеют π-сопряжения друг с другом, а рассматриваются как квантово-волновые резонаторы. Взятые в отдельности, эти резонаторы имеют одинаковые энергетические (вырожденные) π*- состояния (УФ-полоса отдельного резонатора PhN=N– находится при 40 000 см–1 (250 нм [12])). В условиях образования бинарных резонаторов вырождение снимается вследствие делокализации энергии возбуждения между ними с сопутствующим расщеплением их π*-уровней. (У ридимеров t-AAB2 тоже существуют e-таутомеры с бинарными резонаторами из катионов CphAT, они ответственны за тот “хвост” VIS-поглощения, который был принят в [8] за n → π*-полосу t‑AAB, см. ниже.)
Фемта-секундное абсорбционное исследование [13] показало, что кроме поглощения света в области слабой VIS-полосы t-AB (420–500 нм, переход электрона в катионе CPhAT с ВЗМО на НСМО) регистрируется появление такой же транзитной VIS-полосы. Одновременно наблюдалась широкая VIS-полоса в области 500–700 нм другого транзитного состояния. Согласно работе [9], эти результаты соответствуют последовательности актов C+ → C+,* → C++,* (схема 2).

Следует отметить, что в [9] первичный возбужденный катион C+,* представлен в виде хиноидной структуры с положительным зарядом в пара-положении кольца, что соответствует лишь частному случаю. Реально заряд делокализуется между пара- и орто-положениями кольца, как это изображено на схеме (2), фактически повторяя распределение заряда в катионе бензония C6${\text{H}}_{7}^{ + }$ c VIS-полосой при 417 нм [14]. Возбуждаемый светом переход C+ → C+,* первоначально ведет к локализации заряда на фенильном кольце при том, что уходящий из кольца электрон занимает место на pz-орбитали смежного атома N (схема 2). Вслед за этим соседний атом N передает один из своих двух $p_{z}^{2}$-электронов на ридберговскую 3s1-орбиталь, переводя ее в состояние 3s2 ($R_{{3s}}^{{ \bullet \bullet }}$).
Таким образом возникает дикатион C++,*. Далее происходит e-обмен: положительный заряд бензония аннулируется за счет переноса на него электрона с $R_{{3s}}^{{ \bullet \bullet }}$-орбитали, а возникшая при этом $R_{{3s}}^{ \bullet }$-орбиталь захватывает один из электронов sp2-орбитали от связанного с бензонием азота, восстанавливая состояние $R_{{3s}}^{{ \bullet \bullet }}$. При этом появившаяся неустойчивая структура C++,* с двумя положительно заряженными sp2-орбиталями на атомах азота претерпевает перемещение фенильных колец в плоскости AB навстречу друг другу [9]. Одновременно происходит перегибридизация атомов азота в sp-состояния, и в азогруппе возникают две pz- и две px-орбитали, стабилизирующие два катиона CPhAT (ph+N+–). Таким образом, дикатион C++,* получает прямолинейную плоскую структуру. Его электроны на двух pz- и двух px-орбиталях, соответствующих sp-состояниям азотов, соединяются в “нормальную” πx-связь и ослабленную πz-связь.
Наличие в дикатионе C++,* вставки NN (с двойной связью (σ, πx) и неполноценной πz-связью) стабилизирует на некоторое время бинарный резонатор CphAT с расщепленными по Симпсону возбужденными π**-уровнями ${\text{C}}_{1}^{{ + + ,**}}$ и ${\text{C}}_{2}^{{ + + ,**}}$ [9]. Это позволяет обнаруженную в [13] транзитную VIS-полосу с λtr = 500–700 нм, (λmax 589 нм) приписать переходу Sb,1 (C++,*) → Sb,2 (${\text{C}}_{1}^{{ + + ,**}}$) [9].
Согласно [9], возбужденный транзитный дикатион C++,* с прямолинейной структурой не имеет состояния S0 и прямого пути релаксации в состояние Sb,0 путем флуоресценции или внутренней конверсии. Дезактивация, как и образование C++,*, требует перемещения фенильных колец, которое ведет либо к возврату в t-AB, либо к образованию c-AB. В первом случае волна перемещения по схеме (2), сначала достигает e-конфигурации линейного дикатиона C++,*, затем включает релаксационное перемещение фенильных колец в прямом или обратном направлении (C++,* → C+,* → C+ + kT, hν) с сохранением плоской структуры AB. Второй случай соответствует транс–цис-изомеризации AB, которая идет с изменением плоской конфигурации дикатиона C++,*. При изомеризации сначала совершается поворот одного из фенильных колец вокруг связи С–N, затем в том же направлении вокруг другой связи С–N движется второе кольцо при сохранении возбуждения и $R_{{3s}}^{{ \bullet \bullet }}$-орбитали с двумя электронами [9].
Следует подчеркнуть, что именно реализация процесса с перемещением фенильных колец по схеме (2) (C+ → C+,* → C++,*) приводит к экспериментально наблюдаемому появлению полосы “отбеливания” (bleachihg) у AB (390–550 нм с λmax = 420 нм) [9]. При этом прямолинейный дикатион C++,* исполняет роль временного хранителя энергии возбуждения. В отсутствие C++,* “отбеливание” полосы CPhAT не может иметь места. Дело в том, что электрон, возбужденный с ВЗМО на НСМО без перемещения колец (в акте “C+ → C+,*”), может еще испытать возбуждение с НСМО на нижнюю разрыхляющую МО (НРМО). В таком случае потеря одного абсорбера компенсируется появлением такого же абсорбера (НСМО расположена посередине между ВЗМО и НРМО), что исключает возможность наблюдаемого “отбеливания”. Иначе говоря, именно e-конфигурационный переход “C+ → C+,* → C++,*” вызывает временное исчезновение НСМО-уровня и, соответственно, исчезновение поглощения с переходами ВЗМО → НСМО, НСМО → НРМО.
Характерно, что скорость обратимого процесса “C+ → C+,* → C++,* → C+,* → C+ + kT” зависит от величины энергии поглощенного фотона. Так, согласно данным фемтасекундной абсорбционной спектроскопии, прямое возбуждение азобензола в гексане VIS-светом с λвозб = 503 нм дает лишь транзитное состояние C+,* (т.е. Sb,1 с VIS- полосой 390–420 нм), которое затухает по экспоненциальному кинетическому уравнению (τ2 = = 2.5 пс) [13]. Между тем, более энергичный VIS-свет с λвозб = 390–420 нм обеспечивает затухание состояния C+,* по кинетической кривой, соответствующей сумме двух экспонент: τ2 = 2.5 пс и τ1 = = 0.6–0.8 пс (амплитуды сигналов относятся как 3 : 1) [13]. Сходным образом, возбуждение AB в этаноле VIS-светом с λвозб = 435 нм генерирует две транзитные абсорбционные VIS-полосы в области 440–750 нм, затухающие биэкспоненциально с τ1 = 0.32 и τ2 = 2.1 пс (амплитуды сигналов 2 : 1) [15]. Появление двух характеристических времен релаксации объясняют различием в энергиях колебательного возбуждения, вносимого с фотонами на разных участках общей полосы электронного возбуждения. Эту ситуацию, пользуясь схемой (2), можно связать с тем, что недостаточно сильное возбуждение приводит к франк-кондоновскому состоянию с относительно низкой колебательной температурой и соответственно низкой скоростью прохождения актов “C+ → C+,* → C++,* → C+,* → C+ + kT”. Более высокая колебательная температура ускоряет такой процесс, который, однако, замедляется вследствие снижения колебательной температуры молекулы AB.
e-Таутомерия ридимеров t-AAB2
Введение аминогруппы в качестве заместителя в молекулу AB придает орбитали R3s свойство фиксировать ридимеры [1–6] за счет образования межмономерных связей R3s–R3s. Таутомерное равновесие включает e-таутомеры (a)–(d), схема 3:
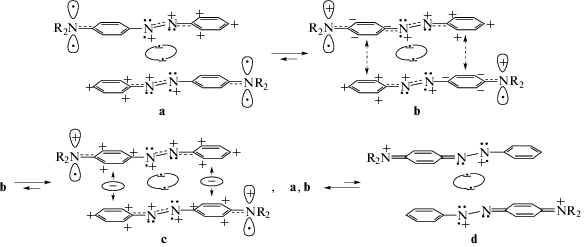
В схеме 3 R = H, точки под атомами N азогруппы обозначают электроны на sp2-орбиталях, а точки над N азогруппы – электроны на pz-орбиталях. Между азогруппами мономеров показаны ридберговские ковалентные связи из двух электронов, промотированных с sp2-орбиталей азогрупп на R3s-орбитали. В каждом мономере sp2-орбиталь, потерявшая электрон, наделяет свой атом N положительным зарядом. На pz-орбиталь этого атома N переходит электрон с pz-орбитали соседнего атома N (имеющего неподеленную электронную пару на sp2-орбитали). Данная ситуация имеет место в каждом мономере ридимера. Потерявшие sp2-электроны атомы N мономеров индуцируют положительные заряды на pz-орбиталях соседних атомов N, и последние поляризуют электронные pz-системы в сопряженных кольцах с образованием катионов CPhAT.
В схеме (3а) на аминных азотах (в группах R2N) обозначены pz-орбитали, возникающие в ридимерах вследствие изменения валентного состояния аминных атомов N. Эти атомы подают по электрону в соседние фениленовые кольца, в результате чего возникают e-таутомеры (3b). Фениленовые кольца в (3b), получив по электрону, становятся донорами электронов для катионов CPhAT (Ph+N+) в противолежащих мономерах. В итоге связь между мономерами в ридимере усиливается и возникают наиболее устойчивые e-таутомеры (3c). В них между противолежащими катионами обоих мономеров действуют одноэлектронные связи по типу связей, существующих в катионе ${\text{H}}_{2}^{ + }$ [3–5]. Эти связи фиксируют четыре катиона фениламинильного типа, обладающие VIS-поглощением в области 390–420 нм. Таким образом, именно электронодонорная функция аминогрупп раскрывает физическую сущность их ауксохромного влияния.
Кроме того, известно, что по своей волновой природе электроны перераспределяются “последовательно по всей мезомерной системе связей” [16]. В ридимерах потенциал для такого перераспределения неаддитивно возрастает по сравнению с мономерами вследствие высокой динамики химических связей R3s–R3s, и это тоже способствует стабилизации ридимеров.
Равновесие по схеме (3) оставляет наименьшую концентрацию для e-таутомеров (3a) и (3d). Их присутствие проявляется по очень слабым полосам при 320 нм [1]. е-Таутомеры (3d) существуют в небольшой концентрации вследствие того, что обратимые e-переходы (:sp2) + R3s ↔ (•sp2) + + R3s• в локальном хромофоре NN, попеременно меняя заряды на азотах, создают с некоторой частотой энергетически мало выгодные конфигурации с хиноидными мономерами. Они обнаруживаются также по крайне слабым сигналам хиноидных связей C=N в резонансных рамановских спектрах t-AAB2 [6].
Следует отметить, что таутомеры (3a) ответственны и за слабое поглощение в области при 500 нм. Они обладают двумя катионами фениламинильного типа CPhAT, образующими бинарный хромогенный резонатор [1, 9] (о бинарных резонаторах было отмечено выше). Именно с таутомерами (3a), имеющими низкую концентрацию, следует связывать длинноволновый “хвост” при VIS-полосе с λ = 390 нм на рис. 1 (кривая 1, λ > 420 нм), приписанный n → π*-переходу в молекуле t-AAB в [8].
Из изложенного выше следует, что использованное в [8] импульсное лазерное излучение с λex = 400 нм должно взаимодействовать преимущественно с e-таутомером (3с).
Главные направления фотоники t-AAB2
Из схемы (3) следует, что играющий роль главного абсорбера в VIS-области 390–420 нм e-таутомер (3с) должен обладать налагающимися друг на друга полосами от двух разных по строению хромогенов CPhAT, а именно R2N$^{{ \bullet + }}$PћN$^{{ \bullet \bullet }}$ (${\text{C}}_{1}^{ + }$) и Ph+N+ (${\text{C}}_{2}^{ + }$) (схема 3с) [8]. Этот факт приводит к выводу о необходимости рассмотрения раздельно фотоники каждого из катионов.
Фотоника ридимера 3с при возбуждении катиона ${\text{C}}_{1}^{ + }$
Результат возбуждения катиона ${\text{C}}_{1}^{ + }$ (λex = = 400 нм) можно выразить схемой 4.
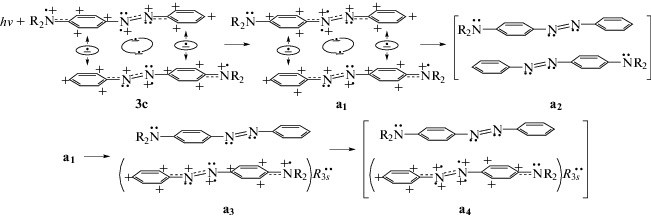
Здесь поглощение VIS-света вызывает перенос электрона в группе R2N$^{{ \bullet + }}$Pћ+${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$ (верхний мономер в ридимере 3с) от атома азота азогруппы (${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$) на азот аминогруппы с образованием структуры 4а1. (Этот процесс идет через промежуточное появление катиона фенония (R2N$^{{ \bullet \bullet }}$Pћ+${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$) путем переноса электрона из фениленового кольца на азот аминной группы, после чего феноний акцептирует электрон с pz-орбитали соседнего атома ${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$). При этом в азогруппе возникает атом ${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$ с двумя зарядами (на pz-орбитали и sp2-орбитали) и растет кулоновское возмущение ридберговской связи Rs–Rs, ведущее к ее разрыву. В результате ее разрыва возможны два варианта e-эволюции: 1) нейтрализация зарядов на обоих мономерах, 2) локализация зарядов на одном из мономеров.
В случае 1 освободившиеся ридберговские электроны возвращаются на родительские sp2-орбитали. При этом в конечном итоге образуется мономерная пара в франк-кондоновском состоянии (4a2). Один из вариантов e-переходов в этом случае можно описать следующим образом. В нижнем мономере (4a1) нейтрализуется левый катион Ph+${\text{N}}_{{ \bullet \bullet }}^{ + }$ вследствие возврата к нему pz-электрона с образовавшегося соседнего атома ${\text{N}}_{{ \bullet \bullet }}^{{ \bullet \bullet }}$. Одновременно освобождается левый электрон одноэлектронной связи, который нейтрализует катион R2${{{\text{N}}}^{{ \bullet + }}}$Pћ+${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$ верхнего мономера с появлением атома ${\text{N}}_{{ \bullet \bullet }}^{{ \bullet \bullet }}$ в его азогруппе. Этот атом ${\text{N}}_{{ \bullet \bullet }}^{{ \bullet \bullet }}$ отдает один из pz-электронов на образование двойной связи N=N, а второй электрон – на нейтрализацию верхнего катиона N+Ph+. При этом образуется мономер t-AAB (верхний в схеме 4а2) и освобождается электрон из второй одноэлектронной связи (справа в 4а1), который нейтрализует катион Ph+N+•R2 нижнего мономера. В итоге образуется мономерная пара (4а2) в франк-кондоновском состоянии.
Другой путь эволюции тоже начинается с разрыва связи R3s–R3s в (4a1) и появляющийся на одной из орбиталей $R_{{3s}}^{ \bullet }$ электрон возвращается на sp2-орбиталь атома ${\text{N}}_{{ + \bullet }}^{{ \bullet + }}$ с повышенным зарядом (верхний мономер в 4а1). В то же время вторая освободившаяся орбиталь $R_{{3s}}^{ \bullet }$ с одним электроном захватывает электрон из одноэлектронной связи (на правой стороне ридимера 4а1). Появляется ридберговская орбиталь с неподеленной электронной парой $R_{{3s}}^{{ \bullet \bullet }}$, которая стабилизирует на некоторое время поляризованную форму нижнего мономера (4а3). При этом освобождается электрон второй одноэлектронной связи, который нейтрализует катион R2${{{\text{N}}}^{{ \bullet + }}}$Pћ+${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$ и одновременно стимулирует нейтрализацию катиона ${\text{N}}_{{ \bullet \bullet }}^{ + }$Ph+ (верхний мономер в 4а1). Таким образом, формируется франк-кондоновская пара (4a3), состоящая из мономера t-AAB и поляризованного мономера с орбиталью $R_{{3s}}^{{ \bullet \bullet }}$.
Далее в поляризованном мономере (4а3) происходит обмен электронов с участием орбитали $R_{{3s}}^{{ \bullet \bullet }}$. Один электрон переходит с $R_{{3s}}^{{ \bullet \bullet }}$ на pz-орбиталь положительно заряженного атома ${\text{N}}_{{ \bullet \bullet }}^{ + }$, одновременно электрон с sp2-орбитали того же атома восстанавливает $R_{{3s}}^{{ \bullet \bullet }}$, что приводит к образованию атома ${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$ с двумя зарядами. Вслед за этим снова один электрон с $R_{{3s}}^{{ \bullet \bullet }}$ переходит на pz-орбиталь заряженной аминогруппы, аннулируя катион Pћ+N+•R2, но одновременно $R_{{3s}}^{{ \bullet \bullet }}$ восстанавливается путем захвата электрона с pz-орбитали правого атома ${\text{N}}_{{ + \bullet }}^{{ \bullet \bullet }}$ азогруппы. Появляется поляризованный мономер с сильно ослабленной связью ${\text{N}}_{{ \bullet + }}^{{ \bullet + }}$- -${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$ в азогруппе, чьи заряды уравновешиваются отрицательным зарядом $R_{{3s}}^{{ \bullet \bullet }}$ (нижний мономер в паре 4a4). В итоге возникает франк-кондоновская гетеропара из поляризованного и нейтрального мономеров.
Фотоника ридимера 3с при возбуждении катиона ${\text{C}}_{2}^{ + }$
Этому процессу соответствует схема 5:

Согласно схеме 5, поглощение ридимером (3с) VIS-света с λex = 400 нм осуществляется катионом ${\text{C}}_{2}^{ + }$ (Ph+${\text{N}}_{{ \bullet \bullet }}^{ + }$, нижний мономер), что приводит к перемещению в нем электрона из Ph+ кольца на pz-орбиталь атома ${{{\text{N}}}^{ \bullet }}$ и локализации положительного заряда на кольце в виде катиона фенония (ридимер 5b1). Затем ридимер (5b1), претерпев разрыв ридберговской связи R3s–R3s и ряд актов смещения электронов, превращается в франк-кондоновскую пару мономеров t-AAB (5b2).
Кроме того, ридимер (5b1) (после разрыва ридберговской связи R3s–R3s) может образовать франк-кондоновскую гетеропару из мономера t‑AAB и поляризованного мономера с ридберговской орбиталью $R_{{3s}}^{{ \bullet \bullet }}$ (пара 5b3). В поляризованном (верхнем) мономере пары (5b3) происходит e-обмен с участием орбитали $R_{{3s}}^{{ \bullet \bullet }}$.
В данном случае орбиталь $R_{{3s}}^{{ \bullet \bullet }}$ отдает один электрон катиону R2${{{\text{N}}}^{{ \bullet + }}}$Pћ+${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$, превращаясь в орбиталь $R_{{3s}}^{ \bullet }$. Орбиталь $R_{{3s}}^{ \bullet }$ акцептирует электрон с pz-орбитали атома ${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$, возвращая себе состояние $R_{{3s}}^{{ \bullet \bullet }}$. При этом образуется другой катион фениламинильного типа – R2${{{\text{N}}}^{{ \bullet \bullet }}}$Pћ+${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$. Затем один из электронов орбитали $R_{{3s}}^{{ \bullet \bullet }}$ локализуется на pz-орбитали другого атома азогруппы ${\text{N}}_{{ \bullet \bullet }}^{ + }$, создавая орбиталь $R_{{3s}}^{ \bullet }$ и атом ${\text{N}}_{{ \bullet \bullet }}^{ \bullet }$. Вслед за этим из этого же атома ${\text{N}}_{{ \bullet \bullet }}^{ \bullet }$ обратно возвращается на $R_{{3s}}^{ \bullet }$ один из электронов sp2-орбитали. В результате этого в азогруппе появляется второй атом ${\text{N}}_{{ + \bullet }}^{{ \bullet + }}$, и соответственно образуется франк-кондоновская гетеропара (5b4), которая фактически соответствует гетеропаре (4а4).
Превращения франк-кондоновских пар
Характер трансформации транзитных спектров, сопряженный с процессом t → c лазерной фотоизомеризации t-AAB [8], обнаружил достаточно сложную картину, которую нельзя объяснить, исходя из идеи молекулярного строения этого красителя. Между тем, сущность этой картины можно представить с учетом его ридимерного строения.
Изложенное выше позволяет сделать вывод, что установленный в работе [8] процесс наиболее быстрой эволюции с характеристическим временем τ1 = 0.2 пс связан с изомеризацией положительно заряженного мономера, образующегося путем расщепления t-AAB2. Действительно, полоса VIS-поглощения этого мономера находится в длинноволновой области спектра (600–750 нм, с λmax = 650 нм), как у дикатиона Ph+HN+–N+HPh+ протонированного азобензола [11] и дикатиона Ph+N+–N+Ph+ возбужденного AB [9] (эти дикатионы также обладают бинарными катионами фениламинильного типа).
Исчезновение VIS-полосы 600–750 нм в начале эволюции сочетается с сопутствующим усилением поглощения в области 410–470 нм [8] (рис. 2, кривая 2). Это можно связать с нарушением плоского транс-строения поляризованных мономеров в (4а4) и (5b4) при изомеризации и исчезновением бинарных хромогенов. При этом появляются транзитные индивидуальные катионы CPhAT (Pћ+${{{\text{N}}}^{{ \bullet + }}}$R2 и Ph+N+), которые поглощают свет в области 410–470 нм. Они исчезают на последних стадиях изомеризации в результате их нейтрализации электронами ридберговских орбиталей $R_{{3s}}^{{ \bullet \bullet }}$. В соответствии со схемами 4 и 5, доля образующихся молекул цис-изомеров (c-AAB) должна составлять всего около четверти от числа всех мономеров t-AAB2, так как претерпеть t – c‑изомеризацию способны только поляризованные мономеры. Изомеризация последних облегчается за счет наличия в них ослабленной азосвязи в сочетании с сильным кулоновским отталкиванием положительно заряженных атомов азота. Данное описание полностью соответствует результату эксперимента [8].
Что касается мономеров t-AAB и c-AAB, то у них должно быть очень слабое VIS-поглощение, как у молекул t-AB и c-AB, чем можно объяснить появление полосы отбеливания. Мономеры t‑AAB имеют нормальную двойную связь N=N, сильно затрудняющую t → c-изомеризацию. По этой причине для них остается только путь возвращения к ридимерам t-AAB2. Естественно ожидать, что ридимеризация франк-кондоновских мономерных пар будет идти значительно легче, чем одиночных t-AAB, образующихся в гетеропарах (схемы 4а4, 5b4). Одиночные t-AAB должны претерпеть более или менее длительные стадии самодиффузии перед образованием пар, что внесет замедленную стадию в исчезновение полосы “отбеливания”.
Вступая в ридимеризацию, каждый мономер t‑AAB отдает на создание межмономерной связи по электрону, промотированному на ридберговскую орбиталь (схема 6).

При этом образуются ридимеры, с бинарными резонаторами из катионов Ph+N+ (6a) и R2${{{\text{N}}}^{{ \bullet \bullet }}}$Pћ+${\text{N}}_{{ \bullet \bullet }}^{ + }$ (6b). В каждой паре таких резонаторов происходит расщепление π*-уровней, характерных для одиночного резонатора (механизм Симпсона), и это приводит к появлению суммарной широкой полосы VIS-поглощения в области 500–750 нм с λmax ∼ 550 нм [1, 4]. В момент своего образования каждая франк-кондоновская пара несет на себе некоторый избыток колебательной энергии. Сначала, когда этот избыток достаточно высок, наблюдается быстрая стадия исчезновения транзитного поглощения образующихся структур (6а) и (6b) вследствие перехода в термически неравновесное состояние (схема 3а) с константой τ2 = 0.6 пс. Снижение степени колебательного возбуждения исходной нейтральной пары приводит к более медленной стадии (с τ3 = 1.9 ps) перехода образующихся структур (6а), (6b) в термически неравновесное состояние (3а).
Согласно мнению авторов [8], обусловленная фотолизом “молекул” t-AAB полоса “отбеливания” исчезает в ходе релаксации колебательной энергии “молекул”, достигших основного e-состояния, с характеристической константой τ = = 15 пс. Теперь можно заключить, что образующие ридимеры сохраняют некоторый избыток колебательной энергии, который перераспределяется на осцилляторы среды. Этот процесс сначала создает среду с локальной неравновесной температурой, которая в конечном итоге достигает термического равновесия с наиболее устойчивой формой ридимеров (3с).
Таким образом, представление о ридимерном строении аминоазобензольных красителей позволило объяснить не только фотонику протонированной формы t-AAB2, как это сделано в [6], но и лазерную ультра-высокоскоростную фотонику t-AAB2, взамен неверной “молекулярной модели” [8]. Приведена расшифровка транзитных (быстро проходящих во времени) спектров в привязке к образующимся из ридимеров короткоживущим (транзитным) интермедиатам, каждый из которых обладает собственной электронной конфигурацией. Можно считать, что направление, давно обозначенное акад. А. Н. Терениным как “Фотоника молекул красителей и родственных органических соединений” [17], должно включать в себя и фотонику ридимеров.
Список литературы
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2017. Т. 91. № 4. С. 672.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2017. Т. 91. № 10. С. 1683.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 2. С. 267.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 1. С.
Matazo D.R.C., Ando R.A., Borin A.C., Santos P.S. // J. Phys. Chem. A. 2008. V. 112. № 19. P. 4437.
Hirose Ya., Yui H., Sawada Ts. // Ibid. 2002. V. 106. № 13. P. 3067.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 11. С. 1773.
Joshi N.K., Fuyuki M., Wada A. // J. Phys. Chem. B 2014. V. 118. № 7. P. 1891.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 2. С. 243.
Robin M.B., Simpson W.T. // J. Chem. Phys. 1962. V. 36. № 3. P. 580.
Lednev I.K., Ye N.-Q., Matousek P. et al. // Chem. Phys. Lett. 1998. № 290. P. 68.
Стрейтвизер Э. Теория молекулярных орбит для химиков-органиков. М.: Мир, 1965. С. 217.
Nägele T., Hohe R., Zinth W., Wachtveitl J. // Chem. Phys. Lett. 1997. V. 272. № 5, 6. P. 489.
Хюккель В. Химическая связь. Критическое рассмотрение систематики, способов выражения и изображения в формулах. М.: Изд-во иностр. лит., 1960. С. 77.
Теренин А.Н. Фотоника молекул красителей и родственных органических соединений. Л.: Наука, 1967. 616 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии