

Журнал физической химии, 2020, T. 94, № 7, стр. 1093-1098
Сорбция U(VI) из водных растворов химически модифицированными волокнами Luffa cylindrica
А. Н. Туранов a, *, В. К. Карандашев b, c, Г. А. Емельченко a, Ш. Су, S. Su d, К. Лью, Q. Liu d, Д. Ванг, J. Wang d
a Российская академия наук, Институт физики твердого тела
Черноголовка, Россия
b Российская академия наук, Институт проблем технологии микроэлектроники и особо чистых материалов
Черноголовка, Россия
c Национальный исследовательский технологический университет “МИСиС”
Москва, Россия
d Key Laboratory of Superlight Materials and Surface Technology, Ministry of Education, Harbin Engineering University
Harbin, P.R. China (Харбинский инженерный университет, Харбин, 150001, КНР)
* E-mail: turanov@issp.ac.ru
Поступила в редакцию 27.06.2019
После доработки 13.12.2019
Принята к публикации 16.12.2019
Аннотация
Волокна Luffa cylindrica, модифицированные лимонной кислотой, исследованы как новый биосорбент для извлечения урана(VI) из водных растворов. Изучено влияние рН водного раствора, времени контакта фаз и температуры на эффективность извлечения U(VI) в фазу сорбента. Кинетические зависимости в процессе сорбции урана(VI) описываются уравнением псевдо-второго порядка реакции. Изотерма адсорбции U(VI) описывается уравнением Ленгмюра, максимальная сорбционная емкость сорбента по урану составляет 204.3 мг/г. Показана возможность повторного использования сорбента в нескольких циклах сорбция – десорбция.
Сорбционные методы широко используются для извлечения урана(VI) из водных растворов [1]. В качестве адсорбентов исследованы полимерные смолы, мезапористые силикагели, углеродные материалы с функциональными группами различной природы, получаемые путем химической модификации поверхности сорбента [2–17]. Однако широкое применение таких материалов ограничено их достаточно высокой стоимостью. В последнее время возрос интерес к использованию недорогих растительных материалов, таких как побочные продукты сельского хозяйства и деревообрабатывающей промышленности, в качестве адсорбентов для удаления ионов металлов из промышленных сточных вод [18]. Биосорбенты обладают рядом достоинств, таких как дешевизна, доступность, биоразлагаемость в окружающей среде. Они содержат гидроксильные, фенольные, карбонильные, карбоксильные и другие функциональные группы, которые могут участвовать в комплексообразовании с ионами металлов в процессе адсорбции. Однако, адсорбционная емкость таких материалов достаточно низкая. Для ее повышения проводят химическую модификацию исходных материалов различными реагентами [19]. Поскольку карбоксильные функциональные группы обладают высокой комплексообразующей способностью по отношению к ионам тяжелых металлов, введение –С(О)ОН-групп в матрицу целлюлозных материалов повышает их сорбционную способность. Доступным реагентом для проведения такой модификации является лимонная кислота (ЛК). При нагревании ЛК образуется реакционно-способный ангидрид, который реагирует с гидроксильными группами полисахаридных фрагментов сорбента [20]. Увеличение числа карбоксильных групп на поверхности биосорбента приводит к увеличению его сорбционной способности по отношению к ионам тяжелых металлов и катионных красителей [21, 22].
Показано, что перспективным биосорбентом является губка из плодов Luffa cylindrica (LС) [23]. Она представляет собой лигноцеллюлозный материал и ее волокна содержат 60% целлюлозы, 30% гемицеллюлозы и 10% лигнина [24, 25]. Структура этих волокон представляет собой микроклеточную архитектуру с непрерывными полыми микроканалами (макропоры диаметром 10–20 мкм), которые образуют сосудистые пучки и дают многомодальную иерархическую систему пор [26].
Возможность использования LС в качестве биосорбента для удаления тяжелых металлов и красителей из водных растворов показана в [25, 27–29]. Для повышения сорбционной способности LС по отношению к U(VI) исследована возможность химической модификации LC этилендиаминтетрауксусной кислотой, полиакриловой кислотой и полиэтиленимином [30–34]. Актуальной задачей является развитие других более доступных методов модификации поверхности LC.
Цель настоящей работы – получение нового дешевого биосорбента из волокон Luffa cylindrica путем термохимической модифицикации лимонной кислотой и исследование сорбционных свойств этого материала по отношению к урану(VI).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные образцы губки Luffa cylindrica были получены из Handan, China. Используемые в работе реактивы – лимонная кислота, NaOH, HNO3, UO2(NO3)2 · 6H2O и Pb(NO3)2 соответствовали марке “х.ч.” или “ч.д.а.”. Для приготовления растворов и для промывки сорбента использовали бидистиллированную воду.
Получение биосорбента. Исходную губку Luffa cylindrical (LС) сушили при 50°C в течение 12 ч, измельчали и фракцию 20–40 меш использовали для модификации. Модификацию исходного материала лимонной кислотой проводили согласно методике [35]. Волокна LС обрабатывали 0.1 М водным раствором NaOH при комнатной температуре и промывали водой до нейтральной реакции. Полученный материал перемешивали с 0.6 М водным раствором ЛК в течение 1 ч при комнатной температуре, отделяли от водной фазы фильтрацией и сушили при 50°C в течение 12 ч. Термохимическую этерификацию проводили при 120°C в течение 3 ч. После охлаждения сорбент промывали горячей водой для удаления избытка ЛК (до отсутствия помутнения фильтрата при добавлении 0.1 M раствора Pb(NO3)2). Затем полученный продукт перемешивали с 0.1 M раствором NaOH при комнатной температуре в течение 2 ч и промывали водой до нейтральной реакции. После этого сорбент (LC-C) высушивали при 50°C до постоянного веса.
Морфологические характеристики исходного материала и полученного биосорбента LC-C исследовали методом сканирующей электронной микроскопии с использованием прибора INC250, Japan Electronic. Удельную поверхность сорбентов определяли методом низкотемпературной адсорбции азота с использованием прибора Quantachrome QuadraSorb SI. Образцы для измерения ИК-спектров готовили в таблетках из KBr. ИК-спектры образцов записывали в области 500–4000 см–1 на Фурье-спектрофотометре AVATAR 360 FT-IR.
Распределение урана(VI) в сорбционных системах изучали в статических условиях при соотношении объема водного раствора и массы сорбента V/m в диапазоне 100–1000 мл/г. Исходный раствор U(VI) с концентрацией 1000 мг/л готовили растворением UO2(NO3)2 · 6H2O в воде. Рабочие растворы U(VI) с концентрацией 1–600 мг/л готовили разбавлением исходного раствора. Требуемые значения рН водной фазы устанавливали добавлением растворов NaOH или HNO3. Контакт фаз осуществляли на роторном аппарате для перемешивания со скоростью вращения 60 об./мин в течение 3 ч. Предварительно установлено, что этого времени достаточно для установления равновесия в системе. Концентрацию урана(VI) в исходных и равновесных водных растворах определяли методом масс-спектрометрии с ионизацией пробы в индуктивно связанной плазме с использованием масс-спектрометра X-7 (Thermo Electron, США). Концентрацию U(VI) в фазе сорбента, qe (мг/г), определяли по уравнению материально баланса
где C0 и Ce (мг/л) – исходная и равновесная концентрация U(VI) в водной фазе, V (мл) – объем водной фазы и m (г) – масса сорбента. Коэффициент распределения U(VI) (Kd, мл/г) рассчитывали как Погрешность определения Kd не превышала 5%.ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Характеристики сорбента
Результаты сканирующей электронной микроскопии показали, что поверхность образцов LС и LС-С является шероховатой и состоит из нитевидных волокон, которые обеспечивают ей высокую прочность и гибкость. Удельная поверхность исходного материала LС составляет 409 м2/г и снижается до 6.74 м2/г для LС-С. Подобное снижение величины удельной поверхности, наблюдавшееся при модификации других растительных материалов лимонной кислотой, было объяснено заполнением части пор продуктами термохимической реакции [22].
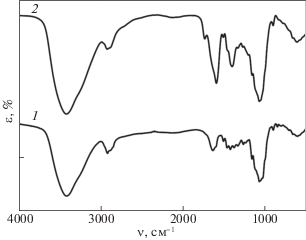
Инфракрасные спектры образцов LС и LС-C представлены на рис. 1. Интенсивные полосы около 3400 и 1051 см–1 свидетельствовали о наличии гидроксильных групп в полисахаридной фракции LС. Полосы вблизи 2914 и 2853 см–1 обусловлены валентными колебаниями групп –CH3 и –CH2–. Пики при 1736 и 1595 см–1 относятся к валентным колебаниям связей C=O, что указывает на привитые карбоксильные группы в структуре LС-C.
Адсорбция ионов урана(VI) на LC-C
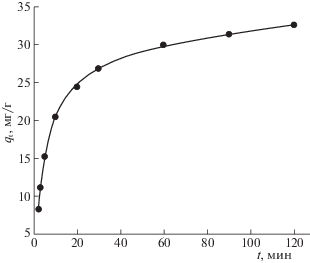
Рассмотрение зависимости сорбции U(VI) от времени контакта фаз (рис. 2) показало, что извлечение U(VI) в фазу сорбента быстро возрастает в течение первых 20 мин, а затем постепенно приближается к равновесию в течение 3 ч. Время, в течение которого исходная концентрация U(VI) снижается в 2 раза (t1/2) составляет 7 мин. Приблизительно 90% максимальной сорбционной емкости сорбента достигается в течение 60 мин.
Рис. 2.
Влияние времени контакта фаз на адсорбцию U(VI) сорбентом LС-C. pH 5.0; V/m = 1000 мл/г; исходная концентрация U(VI) 47.6 мг/л.
Для описания кинетических зависимостей в процессе адсорбции U(VI) использовали уравнения (3) и (4) [6]
(4)
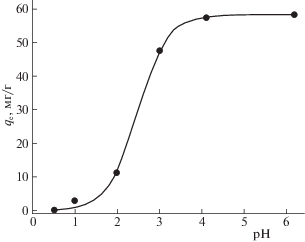
$t{\text{/}}{{q}_{{\text{t}}}} = 1{\text{/}}{{k}_{2}}q_{{\text{e}}}^{2} + t{\text{/}}{{q}_{{\text{e}}}}$Рассмотрение влияния кислотности водной фазы на адсорбцию U(VI) сорбентом LС-C показало, что адсорбция U(VI) возрастает с ростом рН (рис. 3). Такой же характер зависимости qe–рН наблюдался при адсорбции U(VI) другими сорбентами, содержащими функциональные группы кислотного характера [6, 7]. Подавление кислотной диссоциации таких групп с увеличением кислотности водной фазы препятствует их взаимодействию с катионами ${\text{UO}}_{2}^{{2 + }}$. Замедление роста адсорбции U(VI) при увеличении рН выше 4 (рис. 3) может быть связано с образованием частично гидролизованных форм U(VI) в водной фазе [36] и снижением содержания свободных карбоксильных групп в фазе сорбента по мере насыщения его ионами металла.
Рис. 3.
Влияние рН на адсорбцию U(VI) сорбентом LС-C, V/m = 1000 мл/г; исходная концентрация U(VI) 71.4 мг/л; время контакта фаз 3 ч.
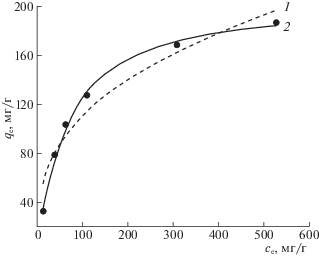
Распределение ионов U(VI) между водным раствором и сорбентом изучено при постоянной температуре в интервале исходной концентрации U(VI) от 1 до 600 мг/л при постоянном рН (рис. 4). Для описания изотермы адсорбции U(VI) использовали уравнение Фрейндлиха
(где KF и n – константы) и уравнение Ленгмюра(5)
${{q}_{{\text{e}}}} = {{K}_{{\text{L}}}}{{q}_{{\text{m}}}}{{C}_{{\text{e}}}}{{(1 + {{b}_{{\text{L}}}}{{C}_{{\text{e}}}})}^{{--1}}},$Рис. 4.
Распределение U(VI) между равновесной водной фазой и сорбентом LС-C. V/m = 1000 мл/г; pH 6.0; температура 21°C. Описание уравнениями Фрейндлиха (1) и Ленгмюра (2); точки – эксперимент.
Проведена оценка влияния посторонних ионов – компонентов реальных природных вод – на эффективность адсорбции U(VI) сорбентом LС-С. Отмечено, что при сорбции микроколичеств U(VI) (1 мг/л) из модельного раствора морской воды, содержащего NaCl (22 г/л), MgCl2 (9.7 г/л), CaCl2 (1.0 г/л), KCl (0.7 г/л), NaHCO3 (0.2 г/л) и H3BO3 (0.02 г/л), степень извлечения U(VI) составляет 83%, тогда как в отсутствие посторонних ионов в фазу сорбента переходит 94.7% U(VI).
Рассмотрение влияния температуры на адсорбцию U(VI) сорбентом LС-C показало, что эффективность адсорбции U(VI) возрастает с ростом температуры (рис. 5), т.е. процесс адсорбции является эндотермическим. Изменение стандартной дифференциальной молярной энергии сорбции Гиббса (ΔG°, кДж/моль), стандартные молярные изменения энтальпии (ΔH°, кДж/моль) и энтропии (ΔS°, Дж/(моль K)) при переходе U(VI) из водного раствора в фазу сорбента были вычислены с использованием уравнений
где T – абсолютная температура, R – универсальная газовая постоянная, и приведены в табл. 1. Положительное значение ΔH° отражает эндотермическую природу адсорбции U (VI) на LC-C и характерно для процесса сорбции U(VI) на других сорбентах, содержащих карбоксильные функциональные группы [37, 38]. Отрицательное значение ΔG° свидетельствует о смещении равновесия в сторону перехода ионов U(VI) из водного раствора в фазу сорбента. Положительное значение ΔS° означает нерегулярное увеличение хаотичности на границе раздела сорбент – водный раствор в процессе адсорбции U(VI).Рис. 5.
Температурная зависимость коэффициентов распределения U(VI) при его сорбции на LС-C. pH = 6.0; V/m = 500 мл/г; исходная концентрация U(VI) 2.38 мг/л; время контакта фаз 3 ч.
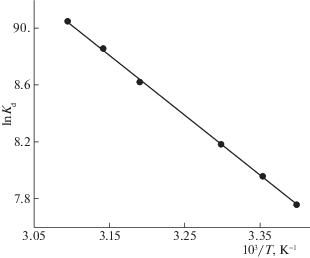
Таблица 1.
Термодинамические параметры адсорбции U(VI) из водных растворов на LС-C
| Т, K | –ΔG°, кДж/моль | ΔH°, кДж/моль | ΔS°, Дж/(моль K) |
|---|---|---|---|
| 294 | 18.98 | 34.10 | 180.5 |
| 298 | 19.73 | ||
| 303 | 20.61 | ||
| 313 | 22.44 | ||
| 318 | 23.43 | ||
| 323 | 24.31 |
Экспериментально установлено, что практически полная (>99%) десорбция U(VI) из фазы сорбента достигается с помощью 0.25 М раствора HNO3. Для повторного использования сорбента его промывали водой после десорбции и высушивали. Результаты по испытанию сорбента LС-C в шести циклах сорбция–десорбция показали, что его сорбционная способность снижается лишь незначительно (табл. 2). В условиях этих испытаний на каждой стадии достигается 10-кратное концентрирование U(VI).
Таблица 2.
Адсорбция U(VI) из водных растворов на LС-C (А) и его десорбция раствором 0.25 M HNO3 (D)
| Стадия | А, % | D, % |
|---|---|---|
| 1 | 94.7 | 99.9 |
| 2 | 95.2 | 99.9 |
| 3 | 94.9 | 99.8 |
| 4 | 94.3 | 99.8 |
| 5 | 93.9 | 99.9 |
| 6 | 93.7 | 99.7 |
Представленные данные показали, что биосорбент на основе волокон Luffa cylindrica, модифицированных лимонной кислотой, обладает высокой адсорбционной способностью по отношению к урану(VI) и может использоваться для его извлечения из водных растворов.
Работа выполнена в рамках Государственного задания 2019 г. ИФТТ РАН и ИПТМ РАН при частичной поддержке Министерства образования и науки РФ по программе повышения конкурентоспособности НИТУ “МИСиС” среди ведущих мировых научно-образовательных центров на 2013−2020 г. (№ К2-2016-070).
Список литературы
Johnson B.E., Santschi P.H., Shane Addleman R. et al. // Appl. Radiat. Isot. 2011. V. 69. P. 205.
Johnson B.E., Santschi P.H., Chuang C.-Y., Otosaka S. // Environ. Sci. Technol. 2012. V. 46. P. 11251.
Saghatchi H., Ansari R., Mousavi H.Z. // Nucl. Eng. Technol. 2018. V. 50. P. 1112.
Zhang W., Ye G., Chen J. // RSC Adv. 2016. V. 6. P. 1210.
Chen X.T., He L.F., Liu R.Z. et al. // RSC Adv. 2015. V. 5. P. 56658.
Walcarius A., Mercier L. // J. Mater. Chem. 2010. V. 20. P. 4478.
Deb A.K.S., Ilaiyaraja P., Ponraju D., Venkatraman B. // J. Radioanal. Nucl. Chem. 2012. V. 291. P. 877.
Carboni M., Abney C.W., Liu C.B., Lin W.B. // Chem. Sci. 2013. V. 4. P. 2396.
Zhao Y., Li J., Zhang S., Wang X. // RSC Adv. 2014. V. 4. P. 32710.
Budnyak T.M., Strizhak A.V., Gladysz-Plaska A. et al. // J. Hazard. Mater. 2016. V. 314. P. 326.
Wang H., Ma L., Cao K. et al. // J. Hazard. Mater. 2012. V. 229. P. 321.
Gorka J., Mayer R.T., Baggetto L. et al. // J. Mater. Chem. A 2013. V. 1. P. 3016.
Zhao Y., Li J., Zhang S. et al. // RSC Adv. 2013. V. 3. P. 18952.
Hadjittofi L., Pashalidis I. // J. Radioanal. Nucl. Chem. 2015. V. 304. P. 897.
Tian K., Wu J.L., Wang J.L. // Radiochim. Acta. 2018. V. 106. P. 719.
Liatsou I., Michail G., Demetriou M., Pashalidis I. // J. Radioanal. Nucl. Chem. 2017. V. 311. P. 871.
Bykov G.L., Ershov B.G. // Radiochemystry. 2009. V. 51. P. 292.
Bailey S.E., Olin T.J., Brica R.M., Adrin D.D. // Water Res. 1999. V. 33. P. 2469.
Wing R.E. // Ind. Crop. Prod. 1996. V. 5. P. 301.
Marshal W.E., Wartelle L.H., Boler D.E. et al. // Bioresour. Technol. 1999. V. 69. P. 263.
Wartelle L.H., Marshal W.E. // Adv. Environ. Res. 2000. V. 4. P. 1.
Zhu B., Fan T., Zhang D. // J. Hazard. Mater. 2008. V. 153. P. 300.
Oboh I.O., Aluyor E.O. // Afr. J. Agric. Res. 2009. V. 4. P. 684.
Mazali I.O., Alves O.L. // An. Acad. Bras. Cienc. 2005. V. 77 P. 25.
Siqueira G., Bras J., Dufresne A. // Bioresources. 2010. V. 5. P. 727.
Altinişik A., Gür E., Seki Y. // J. Hazard. Mater. 2010. V. 179. P. 658.
Hassan L.M. // J. Appl. Polym. Sci. 2006. V. 101. P. 2495.
Demir H., Top A., Balköse D., Ülkü S. // J. Hazard. Mater. 2008. V. 153. P. 389.
Tang X., Zhang Q., Liu Z. et al. // J. Mol. Liq. 2014. V. 199. P. 401.
Gupta V.K., Agarwal S., Singh P., Pathania D. // Carbohydr. Polym. 2013. V. 98. P. 1214.
Liu C., Yan C., Luo W. et al. // Mater. Lett. 2015. V. 157. P. 303.
Su S., Li Q., Liu J. et al. // Sci. Rep. 2017. V. 7. P. 44156.
Su S., Liu Q., Liu J. et al. // J. Colloid Interface Sci. 2018. V. 530. P. 538.
Su S., Chen R., Liu Q. et al. // Chem. Eng. J. 2018. V. 345. P. 526.
Vaughan T., Seo C.W., Marshal W.E. // Bioresour. Technol. 2001. V. 78. P. 133.
Choppin G.R. // J. Radioanal. Nucl. Chem. 2007. V. 273. P. 695.
Song Q., Ma L., Liu, J. et al. // J. Colloid Interface Sci. 2012. V. 386. P. 291.
Cai H., Lin X., Qin Y., Luo X. // J. Radioanal. Nucl. Chem. 2017. V. 311. P. 695.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии