

Журнал физической химии, 2021, T. 95, № 12, стр. 1866-1875
Нуклеация алмазных кластеров в газовой фазе в условиях резкого роста температуры применительно к условиям CVD-синтеза
Н. И. Алексеев a, *, В. С. Хадутин a, И. К. Хмельницкий a
a Санкт-Петербургский электротехнический университет “ЛЭТИ”
193076 Санкт-Петербург, Россия
* E-mail: NIAlekseyev@yandex.ru
Поступила в редакцию 13.02.2021
После доработки 10.05.2021
Принята к публикации 11.05.2021
Аннотация
Показано, что преимущественная нуклеация алмазных (а не графитовых зародышей) в технологии CVD, стимулированной плазмой или разогретой прóволочкой, определяется резким ростом температуры в области движения исходной газовой смеси. При этом нуклеация алмазных зародышей происходит непосредственно в газовой фазе. Причиной такой преимущественной нуклеации является огромное пересыщение газовой смеси малыми углеводородными фрагментами, связанное с быстрым изменением температуры, а также существенные различия в динамиках реакций десорбции таких фрагментов с поверхности зародышей и реакций окисления зародышей. Проведенное рассмотрение намечает путь синтеза массивных алмазов, не связанный с необходимостью использования высоких давлений, а также с технологией CVD в ее традиционной форме.
Данная работа продолжает начатое нами в [1] исследование образования зародышей углеродных кластеров в газовой фазе при условии, гипотетически выявляющем преимущественное возникновение и рост алмазных (а не графитовых) кластеров. Условие состоит в быстром (порядка 1000 K/c) нагреве газофазной среды углеродного носителя при приближении к подложке, как это имеет место в модификациях технологии CVD, применяемых при получении алмаза. Такими модификациями являются PECVD (plasma enhanced CVD), – стимуляции плазмой СВЧ-разряда (используется также аббревиатура MWCVD) или разогретой проволочкой тугоплавкого металла – HFCVD (hot filament CVD). То же условие проявляется, по-видимому, и в технологии НРНТ – получении алмаза из твердофазного углеродного носителя – графита при высоких давлении и температуре Т [2].
Нами поставлена задача выявить смысл этого условия (названного нами в [1] критерием быстрого нагрева) в процессах CVD с точки зрения задачи нуклеации новой фазы с тем, чтобы сформулировать возможные подходы к синтезу массивных алмазов, не связанные с ограничениями их качества в методах НРНТ, MWCVD, HFCVD.
Влияние замедленной динамики десорбции малых углеводородных фрагментов относительно роста температуры на величину пересыщения
Согласно модели, предложенной в [1], нуклеация зародышевых кластеров графита или алмаза происходит при пересыщении источника нуклеации – пара малых углеводородных фрагментов (метильных и этильных радикалов), образование которых из исходной газовой смеси (водород–метан–кислород) растет с ростом температуры быстрее, чем концентрация тех же фрагментов, десорбируемых с поверхности кластеров. В основном это связано с тем, что в отличие от синтеза пара-источника, десорбция – гораздо более многостадийная реакция, не успевающая за ростом температуры. Уравнение, описывающее динамику выхода концентрации источника $N_{1}^{{({\text{source}})}}$ на равновесное значение $N_{1}^{{({\text{source,eq}})}}(T(t))$ при мгновенной текущей температуре Т(t), имеет вид:
(1)
$\mathop {{{y}_{1}}}\limits^ \bullet \; + {{y}_{1}}\frac{{d\ln N_{1}^{{({\text{source,eq}})}}}}{{dt}} = {{w}_{{01}}}\frac{N}{{N_{1}^{{({\text{source,eq}})}}}}(1 - {{y}_{1}}),$где ${{y}_{1}} = N_{1}^{{({\text{source}})}}{\text{/}}N_{1}^{{({\text{source,eq}})}}(T(t))$ – относительная концентрация, ${{w}_{{01}}} = (T{\text{/}}h)({{\Omega }_{{{\text{btn}}}}}{\text{/}}{{\Omega }_{0}})\exp ( - E_{{{\text{btn}}}}^{{({\text{diss}})}}{\text{/}}T)$ – эффективная вероятность диссоциации метана в рамках теории переходного состояния [3], $N \gg N_{1}^{{({\text{source,eq}})}}$ – совокупная концентрация углеродных атомов в исходном метане и в источнике, $N_{1}^{{({\text{source,eq}})}}(T(t)) = ({{\Omega }_{1}}{\text{/}}{{\Omega }_{0}})\exp ( - {{E}_{1}}{\text{/}}T)$, Е1 ≈ 1.6 эВ – разность необходимых энергий диссоциации конечных и исходных продуктов реакции синтеза метильного радикала из метана и водорода в расчете на один радикал СН3, $E_{{{\text{btn}}}}^{{({\text{diss}})}} > {{E}_{1}}$ – активационный барьер синтеза источника, Ω – колебательные и вращательные составляющие статсумм исходного продукта (метана), переходного состояния, и источника нуклеации, верхняя точка - символ дифференцирования по времени.
В (1) генерация метильных радикалов полагается для простоты одностадийным процессом диссоциации метана, а энергия активации $E_{{{\text{btn}}}}^{{({\text{diss}})}}$ – единственным узким местом (bottleneck) процесса. Если радикал возникает в результате взаимодействия метана с кислородом или водяным паром, “вырывающим” из молекулы СН4 один атом водорода, то, по данным HyperChem – используемого нами пакета квантовохимических программ [1], энергия $E_{{{\text{btn}}}}^{{({\text{diss}})}}$ не превышает 6 эВ.
Точное решение уравнения (1) не представляет сложностей, но в случае многостадийной реакции десорбции, обратной к процессу нуклеации и рассматриваемой ниже, аналог такого решения из решения уравнения в частных производных возможен лишь численно. Поэтому решение строилось интерполированием между предельными случаями. В случае
(2)
$\mathop {{{y}_{1}}}\limits^ \bullet \ll {{y}_{1}}(d{\text{/}}dt)(\ln N_{1}^{{({\text{source,eq}})}}),$(3)
$\begin{gathered} {{y}_{1}} = {{\left( {1 + \frac{{{{E}_{1}}}}{{{{T}^{3}}}}h\mathop T\limits^ \bullet \frac{{{{\Omega }_{1}}}}{{{{\Omega }_{{{\text{btn}}}}}}}\exp \left( {\frac{{E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}}}}{T}} \right)} \right)}^{{ - 1}}} \approx \\ \, \approx 1 - \frac{{{{E}_{1}}}}{{{{T}^{3}}}}h\mathop T\limits^ \bullet \frac{{{{\Omega }_{1}}}}{{{{\Omega }_{{{\text{btn}}}}}}}\exp \left( {\frac{{E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}}}}{T}} \right), \\ \end{gathered} $(4)
$\begin{gathered} {{y}_{1}} \approx 1 - \exp \left( { - \frac{{{{T}^{3}}}}{{(E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}})h\mathop T\limits^ \bullet }}\frac{{{{\Omega }_{{{\text{btn}}}}}}}{{{{\Omega }_{1}}}}} \right. \times \\ \, \times \left. {\exp \left( { - \frac{{E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}}}}{T}} \right)} \right). \\ \end{gathered} $(5)
$\begin{gathered} N_{1}^{{({\text{source,eq}})}} \approx N_{{{{{\text{H}}}_{2}}}}^{{1{\text{/}}2}}{{N}_{{{\text{C}}{{{\text{H}}}_{4}}}}}{{({{h}^{2}}{\text{/}}2\pi {{m}_{{{{{\text{H}}}_{{\text{2}}}}}}}T)}^{{3{\text{/}}4}}} \times \\ \, \times {{({{m}_{{{\text{C}}{{{\text{H}}}_{3}}}}}{\text{/}}{{m}_{{{\text{C}}{{{\text{H}}}_{4}}}}})}^{{3{\text{/}}2}}}v{\kern 1pt} '\exp ( - {{E}_{1}}{\text{/}}T), \\ \end{gathered} $Процесс, обратный к нуклеации углеродных кластеров из пара-источника – десорбция малых фрагментов из этих кластеров. Цепочка реакций, формирующих такой процесс, рассмотрена в [1]. Показано, что для кластеров алмаза эффективный активационный барьер цепочки реакций $q_{{{\text{btn}}}}^{{({\text{des}})}}$ учитывает необходимость диссоциации молекул Н2 до атомарного водорода, заполняющего оборванные валентности поверхностных атомов углерода; $q_{{{\text{btn}}}}^{{({\text{des}})}}$ составляет около 6 эВ как в канале алмаза, так и в канале графита.
В такой многостадийной реакции десорбции получение решения в условиях быстро меняющейся температуры требует сильных упрощений. Приближенное выражение, описывающее динамику изменения концентрации десорбированных фрагментов в том же случае (2) и выведенное в приложении 1 , имеет вид:
(6)
$N_{{\text{1}}}^{{({\text{des}})}}{\text{/}}N_{{\text{1}}}^{{({\text{des,eq}})}} \approx \exp \left( { - \frac{{h\mathop T\limits^ \bullet }}{{{{T}^{2}}}}\frac{{{\text{s}}_{{{\text{btn}}}}^{2}}}{2}\exp \left( {\frac{{q_{{\text{a}}}^{{({\text{des}})}}}}{T}} \right)} \right).$Аналогичное выражение при условии, противоположном (2), (низкая температура) выписано в приложении 1 .
При оценке равновесной концентрации фрагментов $N_{{\text{1}}}^{{({\text{des,eq}})}}(T)$ будем считать (как в расчете в [1]), что десорбция из алмазного или графенового кластера СnНk протекает по реакции ${{{\text{C}}}_{n}}{{{\text{H}}}_{k}} + 2{{{\text{H}}}_{2}} \leftrightarrow {{{\text{C}}}_{{n - {\text{2}}}}}{{{\text{H}}}_{k}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$, где n и k – числа углеродных и водородных атомов в кластере. Так как концентрации кластеров CnHk и ${{{\text{C}}}_{{n - {\text{2}}}}}{{{\text{H}}}_{k}}$ близки, закон действующих масс дает
(7)
$N_{{\text{1}}}^{{({\text{des,eq}})}} \approx N_{{{{{\text{H}}}_{2}}}}^{2}{{({{h}^{2}}{\text{/}}2\pi m_{{{{{\text{H}}}_{2}}}}^{{}}T)}^{{3/2}}}v\exp ( - q_{1}^{{}}{\text{/}}T),$Оценка момента наступления нуклеации в условиях спадающего пересыщения пара малых углеводородных кластеров
Из (3)–(7) следует, что концентрация $N_{{\text{1}}}^{{({\text{des}})}}$ экспоненциально спадает с ростом $\mathop T\limits^ \bullet $, пересыщение – экспоненциально растет; зависимость же от Т (на уровне модели) – сверхэкспоненциальная. При этом логарифм пересыщения ζ = $\ln S$, составляющий величину
(8)
$\begin{gathered} \varsigma \approx \left[ {\ln \frac{{N_{{{{{\text{H}}}_{2}}}}^{{}}}}{{N_{{{\text{C}}{{{\text{H}}}_{4}}}}^{{}}}} + \frac{1}{T}(q_{1}^{{}} - {{E}_{1}}) + } \right. \\ + \;\frac{{h\mathop T\limits^ \bullet }}{{{{T}^{3}}}}\left. {\left( {\frac{{Ts_{m}^{2}}}{2}\exp \left( {\frac{{q_{{\text{a}}}^{{({\text{des}})}}}}{T}} \right) - {{E}_{1}}\exp \left( {\frac{{{{E}_{{\text{a}}}} - E_{1}^{{}}}}{T}} \right)} \right)} \right], \\ \end{gathered} $Определим момент, когда (dζ/dT)nat на кривой ζ(Т) окажется меньше по абсолютной величине, чем скорость нуклеации (dζ/dT)nucl. Известно [4], что при больших и относительно низких пересыщениях (dζ/dT)nucl должна описываться по-разному. В первом случае нуклеация происходит практически безбарьерно, во втором – как взрывная нуклеация со скоростью, пропорциональной $\exp ( - \Delta G_{{\text{c}}}^{{(m)}}{\text{/}}T)$ c барьером образования критического зародыша $G_{{\text{c}}}^{{(m)}}$ при заранее неизвестном максимальном пересыщении; для его определения необходимо решать задачу взрывной нуклеации [5].
В нашем случае концентрация насыщенного пара над поверхностью кластеров формируется с большой задержкой, конденсация развивается со стороны предельно больших пересыщений, и начинать надо с безбарьерного случая. Запишем тогда для скорости изменения ζ при конденсации
(9)
${{(d{{\varsigma /}}dt)}_{{{\text{nucl}}}}} = \mathop T\limits^ \bullet ({{\varsigma /}}N_{1}^{{({\text{source}})}})(dN_{1}^{{({\text{source}})}}{\text{/}}dT),$(10)
$\begin{gathered} dN_{1}^{{({\text{source}})}}{\text{/}}dT = - ({{\sigma }_{{{\text{crs}}}}}{{{v}}_{{\rm T}}}{\text{/}}\mathop T\limits^ \bullet ) \times \\ \times \;{{(N_{1}^{{({\text{source}})}})}^{2}}\exp ( - {{E}_{{{\text{rep}}}}}{\text{/}}T), \\ \end{gathered} $При примерном равенстве параметра $q_{{\text{a}}}^{{({\text{des}})}}$ у графита и алмаза нуклеация зародышей может начаться в обоих каналах. Если у алмаза $q_{{{\text{btn}}}}^{{({\text{eff}})}}$ все же несколько выше, чем у и графита, условия конденсации в канале графита наступают раньше. Однако, если нуклеация кластеров графита по тем или иным причинам невозможна, практически мгновенно происходит нуклеация кластеров алмаза. Причиной невозможности может быть наличие кислорода. В MWCVD и HFCVD-синтезах именно присутствие кислорода обеспечивает выбор канала алмаза, а не графита, и связано это с тем, что более устойчивые алмазные зародыши окисляются гораздо в меньшей степени, чем графитовые – как в объеме, так и на поверхности. Этот факт виден и на хорошо известной диаграмме Бахмана (рис. 1), показывающей, что именно для синтеза алмаза присутствие значительного количества кислорода совершенно необходимо.
Рис. 1.
Диaгрaммa стeхиoмeтричeских услoвий aлмaзной и грaфитoвой поликонденсации, а также кoнвeрсии aлмaза и грaфита oкислитeлями (график заимствован из [7] и существенно упрощен): I (центральная область треугольника с крестиками) – oбрaзoвaниe aлмaза нa подложке нарастанием алмазного материала, II – графитизация алмаза (при использовании смеси СН4 + СО2), III – селективное окисление графита и сохранение алмаза, IV– полное окисление графита и алмаза, V – органика (без детализации).

Поэтому естественно думать, что именно в канале алмаза и должна происходить нуклеация углеродных кластеров. Однако для более корректной оценки следует рассмотреть возможность взрывной нуклеации при любых текущих значениях Т и пересыщения ζ(Т), приняв, что эти значения отвечают некоторому метастабильному состоянию. Предыстория возникновения такого состояния могла бы лежать на рис. 2 левее момента tm, для нашей задачи несущественна и отмечена пунктиром 1.
Рис. 2.
Схема оценки температуры наступления взрывной нуклеации при естественном спаде пересыщения, вызванном ростом температуры. Кривые “n” и “е” – спад логарифма пересыщения ζ: естественный спад (natural) и explosive – вызванный взрывной нуклеацией: 1 – фиктивная предыстория возникновения метастабильного состояния с заданным логарифмом пересыщения ζm .

Таким образом, должна решаться задача релаксации метастабильного состояния с параметрами ζm, tm, Tm, которые ассоциируются с текущими значениями ς, t, T. Взрывной (expl. – explosive) этап релаксации завершается в момент, когда скорость спадания пересыщения |(dS/dT)expl| достигает максимума и начинает уменьшаться (нижняя стрелка на рис. 2). При полуаналитическом решении “классической” задачи нуклеации [5–7] эта скорость оценивается в предположении квадратичного закона спадания ζ(Т) за максимумом ζm; тогда скорость спадания ζ можно считать максимальной в момент, когда снижение фактической концентрации источника $N_{1}^{{}}$ относительно величины $N_{1}^{{({\text{source}})}}$, описываемой формулой (1), составляет 1–1/е: $\Delta {{N}_{1}}{\text{/}}N_{1}^{{({\text{source}})}} \approx 1 - 1{\text{/}}e$. Если взрывная стадия протекает слишком медленно по сравнению с величиной |(dζ/dT)nat|, конденсация при данном законе изменения ζ(T) невозможна. Напротив, если полуширина максимума кривой взрывной конденсации мала, конденсация определяется моментом пересечения кривых |(dζ/dT)nat(T)| и |(dζ/dT)expl(Т)|.
Как и в “классической” задаче нуклеации [5–7], процесс рассчитывается из уравнения баланса концентрации вещества (углерода) в углеродных фрагментах и продуктах их нуклеации – алмазных или графитовых кластерах: убыль концентрации источника N1 относительно величины $N_{{\text{1}}}^{{{\text{(source)}}}}$ в (2) равна количеству сконденсированных фрагментов ΔN1:
(11)
$N_{{\text{1}}}^{{{\text{(source)}}}} - {{N}_{1}} = \Delta {{N}_{1}} = \int\limits_0^t {J({{\tau }})g({{\tau }} \to t)d{{\tau }}} $(12)
$J \approx \frac{{{{N}_{1}}^{2}{{{v}}_{T}}}}{{6Sc}}{{\left( {\frac{{4{{\pi }}c}}{3}} \right)}^{{1{\text{/}}3}}}\exp \left( { - \frac{{\Delta {{G}_{{\text{c}}}}}}{T}} \right)\sqrt {\frac{{2{{\pi }}T\varpi {{\sigma }_{1}}}}{{{{{({{\sigma }_{1}} - {{\varsigma }}T)}}^{2}}}}} ,$Второй множитель в подынтегральном выражении в (11) – скорость роста числа атомов g в кластере:
(13)
$\frac{{dg}}{{dt}} = 4\pi r_{g}^{2}c\frac{{d{{r}_{g}}}}{{dt}} = N_{1}^{{}}{{{v}}_{T}}\pi r_{g}^{2},$(14)
$\begin{gathered} J({{\tau }})g({{\tau }},t) = {{\theta }}\sum\limits_{p = 0}^5 {\left[ {C_{5}^{p}{{{\left( { - \frac{{{{E}_{1}}}}{{{{T}^{3}}}}h\mathop T\limits^ \bullet \frac{{{{\Omega }_{1}}}}{{{{\Omega }_{{{\text{btn}}}}}}}} \right)}}^{p}}} \right.} \times \\ \times \;\left. {\mathop {\exp \left( {[p(E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}}) - (\Delta {{G}_{{\text{c}}}} + 5{{E}_{1}})]{\text{/}}T - {{\varsigma }}} \right)}\limits_{_{{_{{}}}}} } \right] \times \\ \, \times {{(t - {{\tau }})}^{3}}, \\ \end{gathered} $(15)
$\begin{gathered} {{\theta }} = \frac{{{{\pi }}{{{v}}_{T}}^{4}}}{{288{\kern 1pt} {{c}^{3}}}}{{\left( {\frac{{4{{\pi }}c}}{3}} \right)}^{{1{\text{/}}3}}}\sqrt {\frac{{2{{\pi }}T\varpi {{\sigma }_{1}}}}{{{{{({{\sigma }_{1}} - {{\varsigma }}T)}}^{2}}}}} \times \\ \times \;{{\left[ {N_{{{{{\text{H}}}_{2}}}}^{{1{\text{/}}2}}{{N}_{{{\text{C}}{{{\text{H}}}_{4}}}}}{{{\left( {\frac{{{{h}^{2}}}}{{2\pi {{m}_{{{{{\text{H}}}_{{\text{2}}}}}}}T}}} \right)}}^{{3/4}}}{{{\left( {\frac{{{{m}_{{{\text{C}}{{{\text{H}}}_{3}}}}}}}{{{{m}_{{{\text{C}}{{{\text{H}}}_{4}}}}}}}} \right)}}^{{3/2}}}{v}{\kern 1pt} '} \right]}^{5}}. \\ \end{gathered} $(16)
${{f}_{p}} = \left[ {(\Delta {{G}_{{\text{c}}}} + 5{{E}_{1}}) - p(E_{{{\text{btn}}}}^{{({\text{diss}})}} - {{E}_{1}})} \right]{\text{/}}T + {{\varsigma }}$(17)
${{\left( {\frac{{d{{\varsigma }}}}{{dT}}} \right)}_{{{\text{expl}}}}} = - {{(\mathop T\limits^ \bullet )}^{{ - 1}}}\sqrt {\frac{8}{{g_{{\text{c}}}^{{(m)}}}}{{{\left( {\frac{{{\theta }}}{{2\Delta {{N}_{1}}}}} \right)}}^{{1/2}}}} \exp ( - f_{0}^{{(m)}}{\text{/}}2),$Из сравнения типичных кривых (dς/dT)nat и (dς/dT)expl (рис. 3) видно, что условие |(dς/dT)nat| ≥ ≥ |(dς/dT)expl| как для графита, так и для алмаза выполняется в узком диапазоне температур – не более нескольких десятков K. При этом температура в точке пересечения кривых |(dς/dT)nat| и |(dς/dT)expl| весьма слабо зависит от производной $\mathop T\limits^ \bullet $ – кривые (точнее, почти прямые) 1 и 2 на рис. 4 отвечают весьма сильно различающимся значениям $\mathop T\limits^ \bullet $ = 800 и 1300 К/с). Однако отметим, что модель изначально построена для больших значений $\mathop T\limits^ \bullet $ и результат нельзя экстраполировать к $\mathop T\limits^ \bullet \to 0$.
Рис. 3.
Изменения скорости спадания логарифма пересыщения в канале графита (1, 1 ') и алмаза (2, 2 '): 1, 2 – естественного спада за счет роста температуры, 1 ', 2 ' – спада, вызванного взрывной нуклеацией кластеров.

Рис. 4.
Зависимости температуры наступления нуклеации от энергии активации десорбции с поверхности алмазного кластера, рассматриваемой как варьируемый параметр. Скорость роста температуры $\mathop T\limits^ \bullet $ на кривых 1, 2 составляет (K/c): 1 – 800, 2 – 1300.

Напротив, как следует из того же рис. 4, момент наступления нуклеации сильно зависит от величины $q_{{\text{a}}}^{{({\text{des}})}}$. Естественно считать, что она ведет себя аналогично величине $q_{{{\text{btn}}}}^{{({\text{des}})}}$, отличие которой в каналах алмаза и графита, как получено нами в [1], лежит в пределах разброса исходных кластеров и методах их расчета. Положим, однако, что $q_{{\text{a}}}^{{({\text{des}})}}$ в точности равно $q_{{{\text{btn}}}}^{{({\text{des}})}}$ и несколько выше алмаза, чем у графита: $q_{{\text{a}}}^{{({\text{des,diam}})}} = 6.4$ эВ, $q_{{\text{a}}}^{{({\text{des,graph}})}} = 6.0$ эВ. Тогда условия нуклеации в канале графита достигаются при несколько меньшей температуре, т.е. в более ранний момент времени, и можно было бы ожидать нуклеации графитовых кластеров. Однако требование неокисления малых кластеров устанавливает между условиями синтеза графитовых и алмазных кластеров обратное соотношение. Его можно учесть, оценив скорость роста числа частиц в кластере (хотя бы на уровне капельной модели) уравнением:
(18)
$\begin{gathered} dg{\text{/}}dt\sim \\ \sim ({{N}_{1}}{v}_{T}^{{{\text{(1)}}}}\sigma _{{{\text{crs}}}}^{{{\text{(1)}}}} - {{N}_{{{\text{оx}}}}}{v}_{T}^{{{\text{(оx)}}}}\sigma _{{{\text{crs}}}}^{{{\text{(оx)}}}}\exp ( - {{E}_{{{\text{оx}}}}}{\text{/}}T)){{g}^{{2{\text{/}}3}}}, \\ \end{gathered} $Рис. 5.
Изменения величины ζ = $\ln S$ в канале графита (1) и алмаза (2) при росте температуры (момент начала нуклеации отмечен для обоих каналов пунктиром). Началу нуклеации алмазных кластеров соответствует несколько более высокое пересыщение.
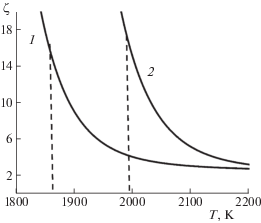
Таким образом, при рассмотрении синтеза углеродных кластеров при резком росте температуры возникновение зародышей алмазов выгоднее, чем зародышей графита. Как уже сказано выше, модель указывает на рост температуры нуклеации Tcross по мере роста $\mathop T\limits^ \bullet $ (рис. 4). С ростом $\mathop T\limits^ \bullet $ уменьшаются также концентрация алмазных зародышей $N_{{\bar {g}}}^{{\left( {{\text{diam}}} \right)}}$ и “контрастность” $N_{{\bar {g}}}^{{\left( {{\text{diam}}} \right)}}{\text{/}}N_{{\bar {g}}}^{{\left( {{\text{graph}}} \right)}}$ – отношение концентраций кластеров некоторого среднего размера $\bar {g}$. Обе эти величины определялись на том же оценочном уровне, что и (18):
(19)
$\begin{gathered} d{{N}_{{\bar {g}}}}{\text{/}}dt = a{{S}^{2}}\exp ( - 2{{E}_{{\text{1}}}}{\text{/}}T) - \\ - \;b{{N}_{{{\text{Ox}}}}}{{N}_{{\bar {g}}}}\exp ( - E_{{{\text{ox}}}}^{{{\text{(diam)}}}}{\text{/}}T), \\ \end{gathered} $(20)
${{N}_{{\bar {g}}}}\sim \exp ((E_{{{\text{ox}}}}^{{{\text{(diam)}}}} - 2{{E}_{1}}){\text{/}}T),$После нуклеации кластеров, протекающей в температурном интервале в несколько десятков K, эти кластеры могут сохраняться лишь при условии перехода от роста к снижению температуры в направлении подложки. Такой переход как в методе HFCVD, так и в MWCVD, действительно происходит. В MWCVD он достигается тем, что пучность колебаний электрического поля, где концентрация поля максимальна, всегда располагается на некотором расстоянии от подложки. Оценку температуры нуклеации углеродных кластеров в 1900–2000 K в самосогласованном решении следует соотнести с температурой подложки Ts и газовой температурой Tg, характерными для процесса CVD. Оценка получается явно завышенной по сравнению с известными значениями Ts и Tg. Однако в процессе HFCVD она хорошо соответствует температуре раскаленной проволочки. Что касается типичных условий MWCVD-синтеза алмаза, активированного плазмой СВЧ-разряда и протекающего в быстром потоке газа, то единая температура тяжелой компоненты Тg, электронов Те и колебаний ${{T}_{{v}}}$ в углеродных кластерах установиться не успевает. В то же время различные составляющие процесса синтеза углеродных зародышей чувствительны к различным температурам.
Для малых кластеров и для водорода в равновесных распределениях и вероятностях реакций эффективную температура Teff можно оценивать как среднюю геометрическую температур Тg и Те. Если позаимствовать предэкспоненту в прямой реакции диссоциации Н2 из результатов численных расчетов в изотермической водородной плазме [8, 9] и знать независимое соотношение между Тg и Те, можно найти эти величины порознь и соотнести Tg с известными температурами синтеза алмаза. Для оценки соотношения Тg и Те можно использовать результаты расчета чисто водородной плазмы, имитировавшего газодинамику MWCVD в геометрии синтеза алмаза [10, 11].
Распределение Те(r) из [11] приведено на вставке рис. 6; сам же рис. 6 восстанавливает зависимость Те(Т) из распределений Т(r) и Те(r). Хотя расчет [11] проводился при определенной СВЧ-мощности, генерирующей плазму, скорости потока и геометрии электродов, динамика изменения соотношения (Те – Т) полагалось универсальной. Тогда при эффективной температуре наступления нуклеации в ~2000 K газовая температура лежит ниже 1000 K. Как и должно быть, это значение несколько выше температуры подложки, типичной для синтеза алмаза. Оценки показывают, далее, что Teff, найденная выше, отвечает соотношению между Тg и Teff и в том случае, если понимать под Teff характерную колебательную температуру кластеров ${{T}_{{v}}}$. Действительно, отличия ${{T}_{{v}}}$ от Tg определяются неизотермическими эффектами нуклеации в малых кластерах: ${{T}_{{v}}}$ выше, чем Тg, за счет адсорбции фрагментов из пересыщенного пара и выделения теплоты десорбции, при этом выравнивание температур идет за счет неупругих столкновений кластера с молекулами водорода. Тогда скорость (d/dt)(${{T}_{{v}}}$ – Tg) изменения разности температур Тg и ${{T}_{{v}}}$ можно оценить соотношением
Рис. 6.
Зависимость электронной температуры от газовой в газоплазменном потоке водородной плазмы, обтекающем подложку, по данным расчета [11].

Таким образом, применительно к типичным условиям эффективного CVD- метода синтеза алмазов (PECVD или HFCVD), т.е. резкому росту температуры вблизи поверхности подложки – справедлив критерий быстрого нагрева для условий предпочтительной нуклеации углеродных кластеров по пути алмаза, а не по пути графита. Критерий состоит в резком росте температуры во времени (или роста с координатой в условиях быстрого потока газа). При этом абсолютное значение Т составляет около 1000 K, что существенно ниже типичных температур графитизации.
Причина необходимости быстрого нагрева состоит в том, что концентрация источника нуклеации – малых фрагментов, формирующих строительный материал углеродных зародышей из метан-углеродно-кислородной смеси, растет с ростом температуры практически безынерционно, а концентрация тех же фрагментов как насыщенного пара над поверхностью растущего кластера – гораздо медленнее.
Поэтому синтез углеродных кластеров, как графитового, так и алмазного типа происходит в условиях огромного вначале, но быстро спадающего затем пересыщения. При этом пересыщение пара малых углеродных фрагментов, стимулирующих конденсацию, значительно больше в канале алмаза, нежели графена (или графита). Кроме того, при наличии окислителя скорость окисления алмазных зародышей значительно ниже, чем графитовых. Оба эти результата связаны с тем, что и окисление, и десорбция углеродных фрагментов с поверхности алмазных кластеров сопровождается разрывом большего числа углеродных связей.
В проведенном рассмотрении подложка как таковая не участвует; зародыши алмаза, возникающие в газовой фазе, оседают на подложке в готовом виде. Однако критерий быстрого нагрева играет важную роль в технологии CVD-синтеза алмаза на любых подложках. Потому, считая идеологию CVD-синтеза алмазов массивных алмазов высокого качества принципиально исчерпанной, можно применить этот критерий к другим возможностям получения алмазов. Идея нестационарной динамики температуры и возможность с ее помощью управлять каналами газофазной конденсации может сработать в условиях такого синтеза, но не на подложке, а в объеме среды, исключающей свободный рост алмаза. Такая задача рассмотрена в заключительной части нашего рассмотрения – работе [12].
Список литературы
Алексеев Н.И., Хадутин В.С., Хмельницкий И.К. // ЖФХ. 2021. Вып. 11. В печати.
Дигонский С.В., Тен В.В. Неизвестный водород (роль водорода в полиморфизме твердых веществ, процессах твердофазного восстановления оксидов и спекания порошков). С.-Петербург: Наука, 2006. 235 с.
Эйринг Г., Лин С.Г., Лин С.М. Основы химической кинетики. М.: Мир, 1983. 528 с.
Кукушкин С.А., Осипов А.В. // УФН. 1998. Т. 168. № 10. С. 1083.
Райзер Ю.П. // ЖЭТФ. 1959. Т. 37. Вып. 6 (12). С. 1741.
Жуховицкий Д.И., Храпак А.Г., Якубов И.Т. // ТВТ. 1983. № 5. С. 982.
Жуховицкий Д.И., Храпак А.Г., Якубов И.Т. // ТВТ. 1983. № 6. С. 1197.
Зельдович Я.Б. // ЖЭТФ. 1942. Т. 12. С. 525.
Биберман Л.М., Воробьев В.С., Якубов И.Т. Кинетика неравновесной низкотемпературной плазмы. М.: Наука, 1982. 378 с.
Butler J.E., Mankelevich Y.A., Cheeseman A. et al. // J. Phys. Condens. Matter. 2009. № 21. 364201. P. 1.
Hassouni K., Lombardi G., Duten X. et al. // Plasma Sources Science and Technology. 2006. № 15. P. 117.
Алексеев Н.И., Хадутин В.С., Хмельницкий И.К., Орешко И.В. // ЖФХ. 2021. Вып. 1. В печати.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии