

Журнал физической химии, 2023, T. 97, № 4, стр. 580-591
Физико-химические свойства лигноцеллюлозных материалов из озонированной древесины
Н. А. Мамлеева a, *, А. Н. Харланов a, М. В. Кузнецова b, Д. С. Косяков b
a Московский государственный университет имени М.В. Ломоносова,
Химический факультет
119992 Москва, Россия
b Северный (Арктический) федеральный университет имени М.В. Ломоносова
163002 Архангельск, Россия
* E-mail: mamleevana@bk.ru
Поступила в редакцию 22.06.2022
После доработки 03.10.2022
Принята к публикации 04.10.2022
- EDN: TGEQDM
- DOI: 10.31857/S0044453723040222
Аннотация
Методами УФ-спектроскопии диффузного отражения (УФДО), спектроскопии комбинационного рассеяния (КР) и флуоресцентной спектроскопии исследованы лигноцеллюлозные материалы (ЛЦМ), полученные после обработки озоном древесины сосны. По данным спектров КР установлено, что при озонировании происходит деструкция лигнина и гемицеллюлоз, уменьшается содержание аморфной целлюлозы. Впервые проведена деконволюция спектров КР озонированных ЛЦМ на индивидуальные компоненты. На основании результатов анализа спектров КР и УФДО сделан вывод о преимущественном разрушении ароматических структур биомассы с сопряженными связями –С=С– и >С=О; делигнификация озоном сопровождается многократным возрастанием интенсивности флуоресценции ЛЦМ. Показано, что спектрально-люминесцентные свойства ЛЦМ контролируются процессом поглощения озона; это позволило выделить области расходов озона, соответствующих преимущественной деструкции лигнина и полисахаридов биоматериала.
В настоящее время особое внимание уделяется разработке инновационных технологий, удовлетворяющих требованиям со стороны экологии. Озон рассматривают как экологически чистый реагент, так как единственным продуктом разложения озона является кислород; реакции с его участием идут при температуре окружающей среды и атмосферном давлении. Прогресс в области синтеза озона, достигнутый в последние годы, привел к существенному снижению стоимости озона и закономерно к возрастанию интереса к озону и расширению области его применения [1]. Озон – сильный окислитель, который активно взаимодействует с ароматическими и непредельными соединениями, что используется для очистки сточных вод ЦБП от фенольных соединений (производных лигнина), а также в ряде других производств [1, 2].
К настоящему времени в мире накоплен значительный экспериментальный материал по взаимодействию растительной биомассы с озоном. Показана перспективность применения озона для удаления лигнина из растительного сырья, отмечена экономическая целесообразность озонирования для последующего получения целлюлозы, моносахаридов и биоэтанола [3, 4]. Среди достоинств озона как делигнифицирующего агента по отношению к биомассе отмечают селективность озона по отношению к лигнину (ЛГ), тогда как целлюлоза (ЦЛ) и гемицеллюлозы (ГЦ) относительно устойчивы к воздействию озона [4–11].
Изучение эффективности делигнификации биомассы при различных условиях проведения озонирования позволило установить оптимальные условия для ее проведения [5–8]. Показано, что для древесины сосны озонирование наиболее эффективно при содержании воды в образце 60–65%, когда, согласно результатам определения содержания ЛГ в озонированных образцах, степень делигнификации древесины может достигать 40% [7, 9]. Методом ВЭЖХ показано, что продуктами озонирования являются алифатические кислоты (муравьиная, щавелевая, глиоксалевая и др.), которые окисляются далее при длительной обработке озоном [7, 9].
ЛЦМ из озонированной древесины сосны исследовали рядом физико-химических методов (ИК-спектроскопия, спектроскопия комбинационного рассеяния (КР), УФ-спектроскопия диффузного отражения (УФДО), рентгеновская диффракция, термический анализ) [6–11]. Показано [7], что процесс делигнификации озоном сопровождается разрушением гемицеллюлоз и аморфной части целлюлозы. Отмечено уменьшение степени полимеризации целлюлозы ЛЦМ при увеличении продолжительности озонирования.
В [11–13] для изучения физико-химических свойств целлюлозосодержащих материалов использовали метод флуоресцентной спектроскопии. В работе [11] впервые показано, что делигнификация лиственной древесины озоном позволяет получить материал, который характеризуется высокой интенсивностью флуоресценции в видимой области. Отмечено, что флуоресцентная спектроскопия может рассматриваться как один из наиболее чувствительных методов контроля озонолитической делигнификации древесины.
Данная работа продолжает цикл исследований физико-химических свойств ЛЦМ из озонированной биомассы. Она посвящена изучению превращений древесины сосны, относящейся к самому распространенному на планете виду древесины. Древесина хвойных пород отличается от лиственной древесины клеточным строением, структурой гемицеллюлоз, а также высокой степенью полимеризации целлюлозы и ее более высоким содержанием. Строение хвойного лигнина также существенно отличается от ЛГ лиственных пород древесины [14, 15]. С практической точки зрения, интерес к изучению превращений хвойной древесины обусловлен и огромным количеством производственных отходов, которые могут служить сырьем для дальнейшей химической переработки [15].
Цель работы – с помощью методов спектроскопии КР, УФДО и флуоресцентной спектроскопии изучить превращения древесины сосны на различных этапах озонирования и оценить возможности этих недеструктивных методов с точки зрения их информативности для анализа динамики деструкции лигнина и других компонентов биомассы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исследуемого материала использовали опилки древесины сосны (Pinus sylvestris) с размером частиц 0.315–0.63 мм. Содержание воды в образце (moisture content –МС) составило 60–65% относительно массы абсолютно-сухой древесины (а.с.д.), МС = (${{m}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}$/mа.с.д) × 100%. Для приготовления образцов к навеске 0.30–0.35 г воздушно сухой древесины (МС = 8%/г а.с.д., определена по методике [16]), добавляли 0.15–0.20 мл дистиллированной воды и выдерживали в течение 5–7 суток при t = 20°С в закрытой емкости для достижения набухания древесины. Конечное содержание воды контролировали гравиметрически.
Проведена серия опытов с различной продолжительностью обработки озоном образцов древесины сосны массой 0.45–0.50 г. Озонирование проводили в проточной установке в термостатированном (25°С) реакторе с неподвижным слоем. Установка состояла из озонатора (Медозон 03/05), фотометра определения концентрации озона (Медозон 254/3) и каталитического патрона для разложения непрореагировавшего озона, описанного в [1]. Озон-кислородную смесь с концентрацией озона 60 ± 2 мг/л пропускали через реактор при скорости потока 1 × 10–3 л/с. Линейная скорость потока 1 см/с. Постоянство объема образцов позволяло проводить эксперимент при постоянном времени контакта реагента (0.8 ± ± 0.05 с) в реакционной зоне.
Количество поглощенного озона Qr(t) в момент времени t рассчитывали из кинетических кривых зависимости концентрации озона, согласно уравнению:
где U – скорость потока газовой смеси (л/с), $C_{t}^{*}$ и ${\text{\;}}~{{C}_{t}}$ – текущие значения концентрации озона (моль/л) на входе и выходе из реактора с исследуемым образцом, соответственно, m – масса а.с.д.; Qr(t) (ммоль/г) рассчитывали на г а.с.д. древесины. Ошибка определения Qr(t) – 10%.Озонированные образцы ЛЦМ промывали дистиллированной водой для удаления водорастворимых продуктов. Воздушно-сухой исходный образец (№ 1) и озонированные ЛЦМ с различной продолжительностью озонирования (О1–О6) исследовали методами спектроскопии КР, УФ-спектроскопии диффузного отражения и флуоресцентной спектроскопии.
Спектры КР регистрировали на приборе Bruker Equinox 55/S, с приставкой FRA 106/S. Длина волны возбуждающего излучения 1064 нм, мощность лазера 850 мВт, размер пятна 0.1 мм. Регистрацию спектра проводили с накоплением по 1024 сканам при разрешении 4 см–1 в интервале 100–3600 см–1. Спектры записывали для пяти случайно выбранных точек образца. Экспериментальные спектры КР нормировали к полосе 1096 см–1, согласно [17], затем определяли средние значения интенсивности некоторых полос в спектре КР. Ошибка определения интенсивности в максимуме 5%.
Деконволюцию спектра исходного образца индивидуальными компонентами осуществляли методом Левенберга–Марквардта с помощью программного обеспечения OPUS 6.0 (Bruker, Германия). Проводили деконволюцию спектров на пять индивидуальных компонент. Выбор количества модельных полос и положение их максимумов осуществляли на основании литературных данных по спектрам КР лигноцеллюлозных материалов [17–20]. Варьирование формы линии компоненты показало, что оптимальной формой линии являются: для компонент ##1–4 – форма 90%Гаусс + 10%Лорентц и для компоненты #5–75%Гаусс + 25%Лорентц. Для озонированных образцов в качестве первичного приближения использовали модельный спектр исходного образца. На первом этапе оптимизировали интенсивности модельных полос, ширину линии и положение максимума фиксировали. На втором этапе оптимизировали ширину линий, значения интенсивности фиксировали. На третьем этапе уточняли положения максимумов и ширину отдельных полос. Среднеквадратичная ошибка отклонения модельного спектра от экспериментального 10%.
УФ-спектры диффузного отражения (УФДО) образцов древесины (100 мг) регистрировали на приборе Specord M-40 (Carl Zeiss Jena, Германия) с интегрирующей сферой в диапазоне 220–820 нм. При регистрации спектров УФДО в качестве эталона использовали образец BaSO4 (его отражение принимается за 100%). Обработку спектров проводили с помощью функции Кубелки–Мунка (F(R)), которая представляет собой отношение коэффициента поглощения к коэффициенту рассеяния среды F(R) = k/s = (1 – R)2/2R, что позволяет оценить поглощение бесконечно толстого слоя образца при данной длине волны. Ошибка определения F(R) составляет 10%.
Спектры возбуждения и эмиссии регистрировали при температуре 25 ± 0.2°С, на флуоресцентном спектрометре Fluorolog-3 (Horiba, Франция), оснащенном двойными монохроматорами в каналах возбуждения и эмиссии и ксеноновой лампой высокого давления мощностью 450 Вт в качестве источника возбуждения. Для регистрации сигнала использовали детектор FL-1073 на основе фотоумножителя R928P, работающий в режиме счета единичных фотонов при комнатной температуре. Перед регистрацией спектров воздушно-сухие образцы прессовали в таблетку массой 150 мг. Образец размещали в держателе под углом 35° по отношению к возбуждающему лучу, чтобы избежать влияния отраженного света. Спектральная ширина щелей монохроматоров возбуждения и эмиссии составляла 2 нм. Спектры флуоресценции образцов древесины получены для λвозб = 360 и 400 нм. Интегрирование спектров проводили с помощью программного обеспечения Opus 6.0 (Bruker, Германия).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Озонирование
На рис. 1 приведены кинетические кривые поглощения озона, соответствующие различной продолжительности обработки. Кривые ложатся на одну кинетическую зависимость, характерную для поглощения озона при данных условиях обработки, которые приняты в работах [7, 8] как оптимальные для делигнификации древесины сосны. На рисунке указаны значения удельного поглощения озона (Qr, ммоль/г), которые для маркированных образцов соответствуют окончанию озонирования. Из рис. 1 видно, что скорость поглощения падает при увеличении продолжительности озонирования. Из компонентов древесины наиболее активно с озоном взаимодействует лигнин, поэтому для начального участка кинетической кривой и образцов О1 (Qr = 0.8 ммоль/г) и О2 (Qr = 1.0 ммоль/г), которые получены в условиях наибольшей скорости поглощения озона (определяемой по наклону кривой), можно ожидать и наиболее эффективной делигнификации. При более высоких значениях Qr скорость поглощения озона уменьшается. Как показано в [7, 9], на этом этапе озонирования в реакциях с озоном участвуют продукты реакции, присутствующие на поверхности.

Спектры КР
На рис. 2 представлены спектры КР исследованных образцов в области 800–1800 см–1 (рис. 2а) и в области валентных колебаний C-H-связей (рис. 2б). Спектры соответствуют известным из литературы спектрам КР хвойной древесины [17–20]. Полоса 1374 см–1 (δС–Н в R3CH в ЦЛ и глюкоманнане [18], 1330 см–1 (деформационные Сар–OH или Сар–O–CH3), 1267 см–1 (Сар–O–CH3 и Сaр–O гваяцильного кольца с С=О-группами [18], 1126 (валентные колебания С–С, С–О в ЦЛ, глюкоманнане), 1096 см–1 (νС–С, νС–О в ГЦ и ЦЛ) [19, 20], 899 см–1 (валентные колебания C–O–С атома С1 и четырех окружающих его атомов в β-гликозидных структурах [21]). В спектре исходного образца (№ 1) присутствуют интенсивные полосы скелетных С–С-колебаний ароматических колец (1600 см–1), а также полосы 1632 см‒1 (С=С-колебания в конифериловом альдегиде), 1656 см–1, (С=С-колебания в конифериловом спирте) [20].
Рис. 2.
Спектры КР ЛЦМ из озонированной древесины сосны в интервале 100–1800 см–1 (а) и 2600–3200 см–1 (б);  1,
1,  О1,
О1,  О2,
О2,  О3,
О3,  О4,
О4,  О5,
О5,  О6; параметры (I/I0) при 1600 (в), 2899, 2940 и 899 см–1 (г), а также содержание ЛГ Класона (в) [9] в ЛЦМ в зависимости от удельного поглощения озона.
О6; параметры (I/I0) при 1600 (в), 2899, 2940 и 899 см–1 (г), а также содержание ЛГ Класона (в) [9] в ЛЦМ в зависимости от удельного поглощения озона.

На рис. 2б представлены спектры КР в области валентных С–Н-колебаний: полоса 3071 см–1 относится к νС–Н ароматического кольца, полоса 2899 см–1 соответствует валентным С–Н-колебаниям целлюлозы, 2940 см–1 – валентные С–Н-колебания в СН3- и ОСН3-группах ЛГ и ГЦ [18, 20]. По сравнению со спектром КР исходного образца (№ 1), в спектрах всех озонированных образцов по мере увеличения Qr уменьшается интенсивность полос 1600, 1632, 1660, 3071, 1267 см‒1, относящихся к ЛГ; уменьшается интенсивность полосы 1126 см–1, относящейся к глюкоманнану. Уменьшается интенсивность полосы 2940 см–1 валентных С–Н-колебаний в метоксигруппах ЛГ и ГЦ (рис. 2).
На рис. 2в приведены значения (I/I0)1600 (интенсивность I в максимуме 1600 см–1 озонированного ЛЦМ, приведенная к значению интенсивности I0 в спектре исходного образца), в зависимости от удельного расхода озона (кривая 1). На рисунке также приведена аналогичная зависимость содержания лигнина Класона (кривая 2), полученная в [7]. Из рис. 2в видно, что (I/I0)1600 падает по мере увеличения Qr, что указывает на деструкцию ароматики. Наиболее заметно уменьшение при Qr ≤ 1.0 ммоль/г. При более высоких значениях Qr величина (I/I0)1600 практически постоянна и составляет 40–42% от исходного значения. Из кривой 2 также следует вывод о деструкции лигнина в озонированных ЛЦМ, содержание ЛГ в исходном образце – 28.5%, а при Qr = = 1.0 ммоль/г содержание ЛГ составляет 14.8%. Из сопоставления с данными спектров КР видно, что эффект разрушения ароматики более заметен из спектров КР. Это неудивительно, так как метод спектроскопии КР дает информацию непосредственно с той части поверхности образцов, которая делигнифицирована при обработке озоном, в отличие от содержания ЛГ на 1 га.с.д, представляющее собой среднее значение ЛГ в объеме образца.
На рис. 2г приведены значения (I/I0)2899 и (I/I0)2940 в зависимости от Qr. Видно, что по мере увеличения Qr интенсивность полос 2899 и 2940 см–1 изменяется симбатно, так как они накладываются друг на друга; контур спектра на рис. 1б у озонированных образцов сохраняется.
Полосу 1096 см–1 (валентные С–С-, С–О-колебания ЦЛ) относят к кристаллической целлюлозе [17, 22]. Интенсивность полосы 899 см–1 асимметричных колебаний глюкопиранозного кольца целлюлозы характеризует относительное содержание аморфной и кристаллической целлюлозы [21, 22]. Как видно из рис. 2г, значение (I/I0)899 уменьшается при расходах озона 1.4–2.4 ммоль/г, т.е. в интервале расходов озона, где деструкция ароматики озоном наименее эффективна (рис. 2в). Уменьшение (I/I0)899 свидетельствует о разрушении аморфной части ЦЛ при озонировании. Следует отметить, что такой же вывод был сделан ранее в работе [7] на основании данных рентгеновской дифракции при изучении ЛЦМ из озонированной древесины сосны.
Деконволюция спектров КР
Для области 1500–1700 см–1 проведена деконволюция спектров КР на пять индивидуальных компонент с максимумами при 1580 и 1599 см–1 (скелетные колебания ароматического кольца) – компоненты #1 и #2, 1620 см–1 (валентные колебания С=О, конъюгированных с ароматическим кольцом) – компонента #3, 1632–1636 см–1 и 1656 см–1 (компоненты #4 и #5, относящиеся к валентным С=С-колебаниям в конифериловом альдегиде и спирте, соответственно. Выбор полос обусловлен присутствием соответствующих им структур в лигнине хвойной древесины [17, 20]. Положение максимумов компонент разложения #1, #2, #3 и #5 для разных образцов постоянно, а для компоненты #4 варьируется от 1632 до 1636 см–1.
На рис. 3а в качестве примера приведены спектры КР образцов № 1, О1, О4, О6 в области 1500–1700 см–1 и разложение их на компоненты. Видно, что проведенное моделирование позволяет достигнуть удовлетворительного соответствия экспериментального (голубой) и модельного (красный) спектров.
Рис. 3.
(а) Деконволюция спектров КР на компоненты (образцы № 1, О1, О3, О6); (б) интегральная интенсивность спектра КР (A/A0) при 1500–1700 см–1 и относительные интенсивности (Ac/$A_{{\text{c}}}^{0}$) компонент разложения с максимумами 1580 (1), 1599 (2), 1620 (3), 1632–1636 (4), 1656 см–1 (5) в зависимости от удельного поглощения озона; (в) вклад компонент разложения (Aс1/A, Aс2/A, Ас3/A, Ас4/A, Ас5/A и Ас3,4,5/A) в интегральную интенсивность спектра при различных значениях удельного поглощения озона. Зависимости Ас2/A (1) и Ас3,4,5/A (2) от удельного поглощения озона (Qr).
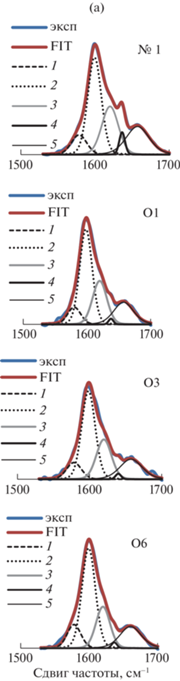
Рис. 3.
Окончание

На рис. 3б представлена зависимость интегральной интенсивности спектра (A), (определяемой как площадь под контуром спектра), нормированной к интегральной интенсивности (A0) спектра исходного образца (кривая 1). По мере увеличения удельного расхода озона значение A/A0 падает в ⁓10 раз вследствие деструкции ароматических систем. На рис. 3б также приведены значения относительной интенсивности компонент разложения (Ac/$A_{{\text{c}}}^{0}$) для компонент разложения 1–5 в зависимости от удельного поглощения озона. Из рис. 3б видно уменьшение интенсивности каждой из компонент разложения с увеличением Qr, причем это наиболее заметно для компоненты 4 с максимумом при 1632–1636 см–1. Следует отметить, что наиболее заметное уменьшение интенсивности всех компонент разложения соответствует области расходов озона ≤1 ммоль/г, что согласуется с данными рис. 1 и 2в.
На рис. 3в приведены значения вклада компонент разложения в интегральный спектр для всех исследованных образцов. Для исходного образца вклад компоненты #1 (Ас1/A0) (1580 см–1) составил 10%, а вклад Ас2/A составляет 40%; ⁓50% приходится на значение Ас3,4,5/A – характеристику спектра, представляющую собой суммарный вклад компонент с максимумами 1620, 1632–1636 и 1656 см–1 ароматических структур, сопряженных с двойными (–С=С–, >С=О) связями. Зависимости Ас2/A0 и Ас3,4,5/A от удельного поглощения озона (Qr) представлены кривыми 1 и 2.
По мере увеличения расхода озона вклад компонент меняется. Так, вклад компоненты с максимумом 1599 см–1 (Ас2/A) в интегральный спектр по мере увеличения Qr постепенно возрастает, и для образца О6 – увеличивается на 9%. Эта тенденция проиллюстрирована с помощью кривой 1. Доля компоненты #1 практически постоянна (9–10%). Суммарный вклад компонент #3, #4, #5 в интегральную интенсивность немного уменьшается; так, значение Ас3,4.5/A компонент с максимумами 1620, 1632–1636, 1656 см–1 при Qr = 2.4 ммоль/г (образец О6) уменьшается на 9% по сравнению с исходным образцом (кривая 2 рис. 3в).
Отмеченные тенденции свидетельствуют о том, что при делигнификации озоном преобладает деструкция ароматики, включенной в сопряжение с С=С-связями, а также разрушение α-карбонильных ароматических структур. Этот вывод следует из того, что доля этих структур в спектре КР озонированных ЛЦМ с ростом удельного расхода озона уменьшается. Согласно результатам деконволюции, образования новых ароматических структур при обработке ЛЦМ озоном не происходит.
Данные спектров КР показывают, что, наряду с ароматическими структурами ЛГ, в озонированных образцах частично разрушены гемицеллюлозы; отмечено уменьшение доли аморфной целлюлозы.
УФ-спектры диффузного отражения
На рис. 4а представлены УФ-спектры диффузного отражения (ДО) для исходного образца и некоторых озонированных образцов. На вставке они приведены в единицах Кубелки–Мунка (F(R)). Исследованные ЛЦМ поглощают в широкой области спектра и характеризуются максимумом при 280 нм, характерным для ароматических соединений с кислородсодержащими заместителями (прежде всего, фенолов). Спектр представляет совокупность многочисленных перекрывающихся полос поглощения, относящихся к разным структурам [23–25]. Несопряженные фенольные (сирингильные, гваяцильные) структуры поглощают при 250, 295–300 нм. Поглощение в области 300–400 нм связывают с наличием в структуре более обширных сопряженных систем, прежде всего включающих гваяцильное ядро, карбонильный кислород и двойную связь в пропановой цепи [24, 25]. Типичные примеры таких структур – феруловая кислота (λmax = 321 нм) и конифериловый альдегид (λmax = 342 нм) [26]. Еще более длинноволновое поглощение характерно для присутствующих в лигнине в незначительных количествах о-хинонных структур и стабильных феноксильных радикалов, а поглощение хинонметидов, образованных стильбеновыми структурами, может простираться до 500 нм и далее в длинноволновую область спектра [24, 26, 27].
Рис. 4.
(а) Спектры УФДО образцов: № 1, О1, О3, О6 ); на вставке – УФДО-спектры в единицах
Кубелки–Мунка; зависимости F(R) (б) и F(R)/F(R)0 (в) при λ = 280, 360 и 400 нм от Qr,  1,
1,  О1,
О1,  О2,
О2,  О3,
О3,  О4,
О4,  О5,
О5,  О6.
О6.

Обработка древесины озоном приводит к снижению поглощения во всей области спектра (рис. 4а), что особенно видно на вставке рис. 4а. С увеличением Qr значения F(R) для длин волн 280, 360 и 400 нм уменьшаются, выходя на плато (рис. 4б), причем это наиболее заметно для F(R)360.
Сравнение значений F(R), нормированных к значению F(R)0 для исходного образца, позволяют сопоставить эффективность озонной обработки по отношению к ароматическим структурам, поглощающим в разной области спектра. Из рис. 4в видно, что, например, при Qr = 2.0 ммоль/г значения F(R)360/F(R)$_{{360}}^{0}$ составляет ∼45% от исходного. Для полос поглощения 280 и 400 нм кривые зависимости F(R)/F(R)0 от Qr близки, при Qr = = 2.0–2.4 ммоль/г значения F(R)280/F(R)$_{{280}}^{0}$ и F(R)400/F(R)$_{{400}}^{0}$, составляют 65–70 и 50–55% относительно исходной величины. Это значит, что хромофоры, поглощающие в указанных областях УФ-спектра, менее эффективно разрушаются озоном. Таким образом, с помощью УФДО-спектров показано преимущественное разрушение озоном хромофоров с поглощением вблизи 360 нм (структуры c двойной α-β-связью и сопряженной карбонильной группой, стильбеновые структуры). Этот вывод соответствует широко известной селективности действия озона на двойные связи и полностью согласуется с данными по деконволюции спектров КР, которые показали, что при озонировании происходит преимущественная деструкция ароматических структур, включенных в сопряжение с С=С- и С=О-связями.
При изучении УФДО-спектров озонированной древесины осины в работе [11] также наблюдали преимущественное разрушение хромофоров с поглощением при λпогл = 360 нм, а структуры с поглощением при 280 и 400 нм составили 30–35 и 40% от исходного значения, соответственно. Если, условно, составить ряд уменьшения эффекта обработки озоном, то для древесины осины получим: 360 → 280 → 400 нм, а для древесины сосны: 360 → 400 → 280 нм. Таким образом, видно, что несопряженные фенольные структуры с λпогл = = 280 нм в древесине сосны более устойчивы к озону по сравнению с несопряженными фенольными структурами в древесине осины. Это различие объясняется различным строением хвойного (гваяцильного) и лиственного (сирингил – гваяцильного) лигнина.
Спектры флуоресценции
Спектры возбуждения флуоресценции (ФЛ) серии образцов древесины сосны (рис. 5а) характеризуются максимумом при 400 нм и идентичны для всех образцов, отличаясь лишь интенсивностью. Для всех образцов наблюдаются плечи при 430, 460 и 520 нм, указывающие на присутствие различных типов фуорофоров. Очевидное отличие спектров рис. 5а от спектров поглощения рис. 4а, с максимумом вблизи 280 нм обусловлено неоднородностью химической структуры лигнина и относительно низким квантовым выходом флуоресценции несопряженных фенольных структур, поглощающих в указанной области спектра. В качестве другой причины несовпадения положения максимумов в спектрах поглощения и возбуждения флуоресценции можно рассматривать так называемый “эффект внутреннего фильтра”, связанный с подавлением флуоресценции за счет высокой оптической плотности образца, которая максимальна в диапазоне 260–300 нм. Наиболее интенсивная флуоресценция характерна для бифенильных групп, некоторых фенилкумарановых и стильбеновых структур [28], а также хинонметидных группировок. Последние поглощают вблизи 500 нм и, по-видимому, отвечают за наличие у спектров возбуждения рис. 4 небольших плеч в области 500–550 нм [27].
Рис. 5.
Спектры возбуждения флуоресценции образцов ЛЦМ (а), спектры флуоресценции образцов
ЛЦМ (λвозб = 360 и 400 нм) из древесины сосны (б) и зависимости интенсивности флуоресценции
Iпр (в) и относительной интенсивности флуоресценции (Iпр/$I_{{{\text{пр}}}}^{0}$) (г) от Qr; λ возб = 360 (1) и 400 нм (2); данные [11]: λвозб = 360 (3) и 400 нм (4),  1,
1,  О1,
О1,  О2,
О2,  О3,
О3,  О4,
О4,  О5,
О5,  О6.
О6.

Спектры флуоресценции образцов ЛЦМ, приведенные на рис. 5б, характеризуются максимумом эмиссии при 452–453 (λвозб = 360 нм) и 470–475 нм (λвозб = 400 нм). Зависимость положения полосы флуоресценции древесины от энергии квантов падающего на ее поверхность излучения характерна для растительных субстратов. Этот эффект был отмечен в работе [29] при изучении структуры хвойной и лиственной древесины, а также при изучении спектров флуоресценции ЛЦМ из озонированной древесины осины [11].
В этом отношении флуоресцентные свойства ЛЦМ отличаются от препаратов лигнина, которые ведут себя как единый флуорофор, в котором энергия электронного возбуждения переносится по механизму Ферстера к структурам – акцепторам энергии с более низко расположенными синглетными уровнями; излучательный переход с этих уровней конкурирует с интеркомбинационной конверсией в триплетные уровни энергии [25–31]. Такой механизм переноса энергии между флуорофорами частично присущ и исследуемому ЛЦМ, что подтверждается весьма значительной разницей в энергиях возбуждения и эмиссии флуоресценции. При этом можно предположить, что максимум ФЛ при ∼450 нм (λвозб = 360 нм) относится к структурам, которые активируются за счет вторичного поглощения излучения, испускаемого флуорофорами с λвозб = 360 нм.
Как видно из рис. 5б, спектры ФЛ озонированных образцов характеризуются более высокой интенсивностью, причем увеличение интенсивности не сопровождается изменением положения максимума или контура спектра. Это согласуется с результатами спектров КР, которые показали, что новые ароматические структуры при озонировании ЛЦМ не возникают.
Интенсивность ФЛ зависит от интенсивности поглощения возбуждающего излучения, поэтому далее рассматриваются приведенные значения (Iпр) в зависимости от удельного поглощения озона (Iпр – интенсивность ФЛ, отнесенная к величине (F(R) при λвозб = 360 нм (Iпр = I/(F(R)360) и λвозб = 400 нм (Iпр = I/(F(R)400)). Зависимости Iпр от Qr представлены на рис. 5в. Сравнение кривых 1 и 2 показывает, что значения Iпр для λвозб = 400 нм выше для всей области Qr, что согласуется со спектрами возбуждения, которые показали максимум при λпогл = 400 нм.
Значения Iпр/$I_{{{\text{пр}}}}^{0}$ ($I_{{{\text{пр}}}}^{0}$ – интенсивность ФЛ исходного образца) позволяют оценить влияние обработки озоном на интенсивность ФЛ ЛЦМ. Как показывает рис. 5г, с увеличением Qr значение Iпр/$I_{{{\text{пр}}}}^{0}$ по сравнению с исходным образцом возрастает в 3–4 раза для λвозб = 400 нм, и в 6–7 раз для λвозб = 360 нм, постепенно выходя на насыщение. Более заметное возрастание (Iпр/$I_{{{\text{пр}}}}^{0}$)360 закономерно, объясняются данными УФ-спектров, которые показали, что структуры ЛЦМ с поглощением при 360 нм в наибольшей степени подверглись деструкции озоном.
Из рис. 5в и 5г видно, что для расходов озона ≤1 ммоль/г интенсивность флуоресценции заметно возрастает. Эта область расходов озона соответствует линейному участку кривой поглощения озона на рис. 1. Скорость поглощения озона на этом участке кривой максимальна, так как озон реагирует преимущественно с лигнином, и этой области Qr соответствует наиболее эффективное уменьшение содержания ЛГ в ЛЦМ (рис. 2в). В области Qr = 1.4–2.4 ммоль/г, где содержание ароматических структур практически не меняется (рис. 2в), а идет частичная деструкция полисахаридов (рис. 2г) интенсивность ФЛ практически постоянна (рис. 5в, 5г). На этом участке кривой рис. 1 скорость поглощения озона также уменьшается. Отмеченные корреляции между зависимостью интенсивности ФЛ и кинетической кривой поглощения озона свидетельствуют о возможности регулировать люминесцентные свойства ЛЦМ, варьируя условия обработки ЛЦМ озоном.
Многократное увеличение интенсивности ФЛ образца ЛЦМ после обработки озоном было отмечено нами в работе [11] при изучении озонированной древесины осины, представленное на рис. 5г (кривые 3 и 4). Учитывая, что флуоресценция обусловлена присутствием лигнина – компонента, наиболее эффективно разрушающегося озоном, а образования новых ароматических структур, устойчивых к действию O3, не отмечено, то в качестве объяснения усиления флуоресценции было предложено изменение свойств лигнинсодержащего материала, образовавшегося после обработки озоном.
Рассматривая в этом ракурсе данные по озонированию древесины сосны, следует отметить, что происходящее при озонировании разрушение лигнина и лигнин-углеводных ковалентных связей, а также внутри- и межмолекулярных водородных связей, отмеченное в [7, 9, 10], препятствует безызлучательным механизмам потери энергии в форме колебаний и способствует усилению эмиссии электромагнитного излучения. Интенсивность флуоресценции конкретного флуорофора определяется соотношением вероятностей излучательной и безызлучательной диссипации энергии электронного возбуждения [29, 30], поэтому важным фактором является окружение флуорофора. Методом ИКС показано [7, 9], что делигнификация древесины озоном сопровождается образованием многочисленных алифатических карбоксильных соединений. Связанное с этим понижение рН среды может играть важную роль в изменении интенсивности эмиссии, вследствие смещения протолитических равновесий в направлении образования неионизированных форм фенольных структур лигнина.
Кроме того, рост интенсивности ФЛ у озонированных ЛЦМ может быть связан со снижением уже упомянутого эффекта внутреннего фильтра по мере удаления лигнина, способного активно поглощать испускаемое излучение и тушить флуоресценцию по аналогии с концентрированными растворами флуорофоров. В пользу этой версии свидетельствуют данные работ [12, 13], где отмечено возрастание интенсивности ФЛ при уменьшении содержания лигнина в ЛЦМ.
Возможно, имеет значение и уменьшение концентрации структур с низким выходом флуоресценции вследствие их деструкции озоном, что может способствовать возрастанию интенсивности ФЛ. На роль последних могут претендовать С=О-группы, сопряженные с ароматическим кольцом, так как в работах [13, 30] отмечено, что уменьшение содержания арил-карбонильных групп в препаратах лигнина, приводит к усилению флуоресценции. Это соображение подкрепляется и данными деконволюции КР-спектров, которые указали на их деструкцию озоном. Очевидно, что надежное установление роли отмеченных механизмов изменения интенсивности ФЛ в наблюдаемом эффекте при обработке озоном требует более глубокого изучения с применением современных физических и физико-химических методов исследования структуры лигнинов в ЛЦМ и находится за рамками настоящей работы.
На рис. 6 представлена зависимость интенсивности ФЛ (Iпр/$I_{{{\text{пр}}}}^{0}$) от интенсивности (I/I0)1600 полосы 1600 см–1 в спектре КР ЛЦМ. Так как значение (I/I0)1600 характеризует содержание ЛГ, то данные рис. 6 характеризуют зависимость интенсивности ФЛ от содержания ЛГ в ЛЦМ. Для исходного образца (I/I0)1600 = 1и (Iпр/$I_{{{\text{пр}}}}^{0}$) = 1, для озонированных ЛЦМ по мере уменьшения (I/I0)1600 интенсивность ФЛ возрастает в несколько раз, причем для λвозб = 360 нм это возрастание вдвое больше, чем для λвозб = 400 нм (кривые 1 и 2).
Рис. 6.
Зависимости относительной интенсивности флуоресценции (Iпр/$I_{{{\text{пр}}}}^{0}$) ЛЦМ от (I/I0)1600 полосы 1600 см–1 в спектрах КР; λвозб = 360 (1) и 400 нм (2); кривые 3 и 4 – данные [11], λвозб = 360 (3) и 400 нм (4).

Из рис. 6 также следует, что в сравнении с ЛЦМ из озонированной древесины осины (кривые 3, 4 из работы [11]) для одной и той же области значений (I/I0)1600 возрастание флуоресценции ЛЦМ из озонированной сосны вдвое меньше. С формальной точки зрения, эти различия достаточно просто объяснить рассмотренными выше изменениями УФ-спектров ЛЦМ из озонированной древесины осины и сосны.
Согласно данным [9], содержание ЛГ в древесине сосны на ~5% выше, чем в древесине осины. Приблизительно такое же различие в содержании ЛГ сохраняется и у озонированных образцов. Более высокое содержание ЛГ у озонированных образцов древесины сосны способствует проявлению отмеченного выше “эффекта внутреннего фильтра” и может служить объяснением более низкой ФЛ ЛЦМ из древесины сосны на рис. 6.
Озонолитическая делигнификация древесины
Изучение закономерностей поглощения озона различными видами биомассы показало [6–10], что реакции озона осуществляются на поверхности пористой структуры биоматериала, доступной молекулам озона. В данной работе обработка озоном проводится в условиях, оптимальных для делигнификации биомассы, когда с лигнином преимущественно взаимодействует молекулярный озон, растворенный в воде, присутствующей в пористой структуре биомассы [5, 6, 8]. Молекулярный озон вступает в реакции электрофильного циклоприсоединения с последующим раскрытием ароматического кольца и образованием продуктов – алифатических кислот [9–11]. С молекулярным озоном взаимодействуют преимущественно функциональные группы с высокой электронной плотностью [32]. Сопоставление активности органических соединений в реакциях с озоном показало, что наиболее активны непредельные структуры и стильбены; в ряду активности далее идут сирингильные, – гваяцильные структуры, – карбонилсодержащие и полиароматические соединения [33, 34]. Относительно медленно окисляются озоном алифатические связи в углеводородах и углеводах [32, 33].
Исследованная серия озонированных образцов различается значениями Qr, достигнутыми при различной продолжительности озонирования, что позволило рассмотреть последовательно деструкцию биоматериала. Взаимодействие озона с древесиной протекает в аморфных областях лигноуглеводного комплекса, в основном, состоящего из полисахаридов и лигнина [7]. Доступные реагенту ароматические структуры – наиболее реакционноспособная часть структурной организации, поэтому разрушаются в первую очередь, что подтверждено данными спектров КР, УФ и флуоресценции. Показана преимущественная деструкция сопряженных ароматических структур, что свидетельствуют об избирательном действии озона на функциональные группы лигнина в древесине.
Сопоставление этих результатов с данными работы [11] по изучению спектрально-люминесцентных свойств ЛЦМ из озонированной древесины осины также указывает на избирательное действие озона по отношению к разным ароматическим структурам ЛГ. Лигнин лиственной древесины содержит гваяцильные и сирингильные структурные субъединицы (соотношение (1 : 1)), лигнин хвойной древесины на ~90% состоит из гваяцильных структур [35]. Известно, что для гваякола и 2,6-диметоксифенола, моделирующих, соответственно, гваяцильные и сирингильные остатки, константы скорости реакции с озоном различаются на несколько порядков (105 и >107 л/(моль с) – для гваякола и сирингола соответственно) [34]. Таким образом, более эффективное воздействие озона на лиственную древесину и модифицирование физико-химических свойств ЛЦМ предопределено структурой нативного ЛГ.
Преобладание гваяцильных структурных единиц и, как следствие, более низкое содержание метоксильных групп, а также бόльшая склонность к конденсации за счет свободного положения 5 в ароматическом кольце гваяцилпропанового звена играют существенную роль при радикальных процессах с участием феноксильных радикалов [24, 25]. Феноксильные радикалы возникают и в реакциях озона с фенольными соединениями [32]. В рассматриваемом случае озонирования хвойной древесины, проведенного в условиях, благоприятных для делигнификации с участием молекулярного озона, роль радикальных процессов минимизирована [7–9].
В работе использованы методы УФ-, КР- и флуоресцентной спектроскопии, с помощью которых получена информация о свойствах поверхности ЛЦМ, часть которой модифицирована озоном. Это позволило наблюдать деструкцию ароматических структур лигнина и отметить избирательный характер этого процесса под воздействием сильного окислителя – озона. Делигнификация сопровождается значительным возрастанием интенсивности флуоресценции материала. Разрушение ЛГ озоном приводит и к удалению из ЛЦМ глюкоманнана – одной из структур гемицелллюлоз хвойной древесины, ковалентно связанных с ЛГ.
При более высоких значениях Qr озон расходуется не только на деструкцию доступной ароматики, но и на реакции с продуктами делигнификации и деструкцию полисахаридов. Наблюдается окисление аморфной ЦЛ. Отмеченные закономерности изменения физико-химических свойств ЛЦМ соответствуют активности озона в ряду органических соединений.
Таким образом, с помощью недеструктивных методов исследования в работе рассмотрены закономерности декомпозиции структуры биомассы при озонолитической делигнификации. Показано, что спектрально-люминесцентные свойства ЛЦМ контролируются процессом поглощения озона, который определяет области расходов озона, соответствующих преимущественной деструкции ЛГ и полисахаридов биоматериала. Отмечено, что особенности модифицирования физико-химических свойств ЛЦМ при озонировании предопределяются, главным образом, структурой нативного лигнина.
Можно предположить, что использованная в работе комбинация методов изучения превращений древесины под воздействием озона перспективна и для исследования делигнификации растительной биомассы другими способами.
Работа выполнена с использованием оборудования ЦКП “Нанохимия и наноматериалы” при Химическом факультете МГУ имени М.В. Ломоносова при финансовой поддержке Минобрнауки РФ в рамках госбюджетной темы: “Физикохимия поверхности, адсорбция и катализ” ААА-А21-121011990019-4 (Озонолитическая делигнификация ЛЦМ, УФ, спектроскопия КР) и Центра коллективного пользования научным оборудованием “Арктика” Северного (Арктического) федерального университета имени М.В. Ломоносова в рамках проекта государственного задания № FSRU-2021-0009 (Изучение люминесцентных характеристик компонентов древесины).
Авторы благодарят профессора Алтайского государственного университета Н.Г. Базарнову за предоставленные образцы древесины.
Список литературы
Самойлович В.Г., Ткаченко С.Н., Ткаченко И.С., Лунин В.В. / Теория и практика получения и применения озона. Ред. В.В. Лунин. М.: Изд-во Моск. ун-та, 2016. 432 с.
Мамлеева Н.А., Бенько Е.М., Лунин В.В. / Методы обезвреживания сточных вод, газовых выбросов и отходов производства и потребления. Ред. В.В. Лунин. М.: Изд-во Моск. ун-та, 2019. 359 с.
Perrone O.M., Colombari F., Rossi J. et al. // Bioresour. Technol. 2016. V. 218. P. 69.
García-Cubero M.T., Palacín L.G., González-Benito G. et al. // Ibid. 2012. V. 107. P. 229.
Li C., Wang L., Chen Z., Li Y. et al. // Ibid. 2015. V. 183. P. 240.
Benko E.V., Chukhchin D.G., Lunin V.V. // Holzforschung, 2020. V. 74. № 12. P. 1157.
Мамлеева Н.А., Харланов А.Н., Чухчин Д.Г. и др. // Химия растительного сырья. 2019. № 1. С. 85.
Мамлеева Н.А., Харланов А.Н., Купреенко С.Ю., Чухчин Д.Г. // Журн. физ. химии, 2021. Т. 95. № 11. С. 1658.
Мамлеева Н.А., Бенько Е.М., Шумянцев А.В. и др. // Там же. 2021. Т. 95. № 3. С. 577.
Мамлеева Н.А., Харланов А.Н., Лунин В.В. // Там же. 2019. Т. 93. № 12. С. 1901
Мамлеева Н.А., Харланов А.Н., Кузнецова М.В., Косяков Д.С. https://istina.msu.ru/workers/418035/// Там же. 2022. Т. 96. № 9. С. 2043.
Billa E., Koutsoula E., Koukios E.G. //Biores. Technol. 67 (1999). C. 25.
Заказов А.Н., Чупка Э.И. // Химия древесины. 1983. № 2. С. 52.
Papadopoulos A.N., Hill C.A.S., Gkaraveli A. // Holz als Roh- und Werlag. 2003. V. 61. P. 453.
Азаров В.И. / Химия древесины и синтетических полимеров. СПб., 1999. 629 с.
Оболенская А.В., Ельницкая З.П., Леонович А.А. / Лабораторные работы по химии древесины и целлюлозы. М.: Экология, 1991. 320 с.
Agarwal U.P. // Frontiers in Plant Science. 2014. V. 5. Article 490. https://doi.org/10.3389/fpls.2014.00490
Zhe Ji, Jianfeng Ma, and Feng Xu // Microsc. Microanal. 2014. V. 20. P. 566.
Kihara M., Takayama M., Wariishi H., Tanaka H. // Spectrochim. Acta. Part A. 2002. V. 58. P. 2211.
Lupoi J.S., Singh S., Parthasarathi R. et al. // Renewable and Sustainable Energy Reviews. 2015. V. 49. P. 871.
Ciolacu D., Ciolacu F., Popa V. // Cellulose Chem. Technol. 2011. V. 45. № 1–2. P. 13.
Molina-Guerrero C.E., de la Rosa G., Castillo-Michel H. et al. // Chem. Eng. Technol. 2018. V. 41. Is. 7. P. 1350.
Физическая химия лигнина / Под ред. К.Г. Боголицына, В.В. Лунина. Архангельск: Арханг. гос. технич. ун-т, 2009. 489 с.
Sadeghifar H., Ragauskas A. // Polymers.2020. V. 12. P. 1134. https://doi.org/10.3390/polym12051134
Paulsson M., Parkås J. // BioResources. 2012. V. 7 (4). P. 5995.
Косяков Д.С., Горбова Н.С., Боголицын К.Г., Гусаков Л.В. // Журн. физ. химии. 2007. Т. 81. № 7. С. 1227.
Чупка Э.И., Бурлаков В.М. // Химия древесины. 1984. № 2. С. 31.
Albinsson B., Li S., Lundquist K., Stomberg R. // J. Mol. Struct. 1999. V. 508. P. 19.
Donaldson L. / International Association of Wood Anatomists (IAWA), 2013. Published by Koninklijke Brill NV, Leiden https://doi.org/10.1163/2294193200000002
Panfilova M.V., Kosyakov D.S., Bogoltsin K.G. / Europe Workshop on Lignocelulosics and Pulp. EWLP. P. 627, 2014. June 24–27. 2014. Seville. Spain.
Кузнецова М.В., Косяков Д.С., Горбова Н.С., Боголицын К.Г. // Журн. физ. химии. 2020. Т. 94. № 8. С. 1185.
Разумовский С.Д., Заиков Г.Е. / Озон и его реакции с органическими соединениями. М.: Наука, 1974. С. 219.
Olkkonen C., Tylli Y., Forsskåhl I. et al. // Holzforschung. 2000. V. 54. P. 397.
Kaneko H., Hosoya S., Iiyama K., Nakano J. // J. Wood Chem. Technol. 1983. V. 3. P. 399.
Holladay J.E., Bozell J.J., White J.F., Johnson D / Top Value-Added Chemicals from Biomass. V. II. 2007. USA. http://www.ntis.gov/ordering.htm
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии