

Физика плазмы, 2020, T. 46, № 7, стр. 595-605
Оптическая спектральная диагностика плазмы доломита и кросс-валидация с использованием сканирующей электронной микроскопии с рентгеновской энергодисперсионной спектроскопией и рентгеноспектрального микроанализа
M. Fahad a, *, M. Abrar b, K. H. Shah c, A. Shahzad d,
a Department of Electrical and Computer Engineering, COMSATS University Islamabad (Abbottabad campus)
Abbottabad, Pakistan
b Department of Physics, Hazara University Mansehra
Mansehra, Pakistan
c Department of Chemistry, COMSATS University Islamabad (Abbottabad campus)
Abbottabad, Pakistan
d School of Geosciences and Info-Physics Central South University
Changsha, China
* E-mail: drmuhammadfahad@gmail.com
Поступила в редакцию 26.07.2019
После доработки 11.09.2019
Принята к публикации 18.09.2019
Аннотация
Доломит CaMg(CO3)2 – важный карбонатный минерал, состоящий в основном из карбонатов магния и кальция. Определение доли магния в карбонатной породе является важной задачей при оценке возможности использования породы в тех или иных приложениях. Однако наличие минеральных фаз с различным содержанием магния в образце значительно усложняет задачу. В настоящей работе с помощью лазерно-искровой эмиссионной спектроскопии, рентгеновской дифрактометрии, сканирующей электронной микроскопии и рентгеноспектрального микроанализа качественно и количественно был проанализирован образец доломита. Эмиссионные спектры в области длин волн от 200 до 720 нм содержали спектральные линии Ca, Mg, Al, Sr и Na различной интенсивности. Температура плазмы была найдена из анализа зависимости распределения заселенности возбужденных состояний от их энергии для нейтральных частиц и ионов. Полученная таким образом средняя температура плазмы оказалась 4500 ± 450 K. Плотность электронов, определенная по штарковскому уширению спектральных линий кальция и магния, составила 2.39 × 1017 см–3. Количественное определение состава образца производилось методом безэталонной лазерно-искровой эмиссионной спектроскопии в предположении локального термодинамического равновесия плазмы и ее оптической прозрачности. Содержание основных компонентов доломитового образца – кальция и магния – составило 68.58% и 31.41%, соответственно. Продемонстрирована эффективность и взаимодополняемость методов лазерно-искровой эмиссионной спектроскопии, энергодисперсионной рентгеновской спектроскопии и рентгеноспектрального микроанализа для определения элементного состава карбонатных минералов.
1. ВВЕДЕНИЕ
Лазерно-искровая эмиссионная спектроскопия (ЛИЭС) – метод атомной эмиссионной спектроскопии, получивший широкое распространение среди исследователей, работающих в различных областях количественного и качественного анализа твердых, жидких и газообразных веществ. Для возбуждения и абляции молекул исследуемого вещества в ЛИЭС используются высокоэнергичные лазерные импульсы, которые при взаимодействии с исследуемой поверхностью создают плазму, состоящую из ионизованного вещества образца [1]. При переходе в основное состояние частицы плазмы излучают кванты света, формируя спектр излучения, по которому можно определить химический состав исследуемого материала, а также относительные концентрации составляющих его элементов. Высокая популярность этого метода обусловлена легкостью экспериментальной реализации, возможностью непосредственного (in-situ) проведения химического анализа, хорошим пределом обнаружения и неплохой точностью, портативностью, неразрушающим воздействием на материал и отсутствием необходимости предварительной подготовки исследуемого образца. В последние годы ЛИЭС считается предпочтительным и мощным инструментом для проведения быстрого и точного анализа химического состава вещества на атомном уровне. Количественный анализ вещества по методике ЛИЭС может производиться как с использованием калибровочных зависимостей, так и без предварительной калибровки [2–7]. Традиционный метод использования калибровочных зависимостей предполагает наличие эталонного образца, что не всегда представляется возможным, как, например, при анализе археологических находок и геологических материалов. Для устранения такого рода затруднений был разработан безэталонный метод ЛИЭС (БЭ-ЛИЭС), не имеющий подобного ограничения [2]. Этот метод основывается на модели оптически тонкой плазмы, находящейся в состоянии локального термодинамического равновесия (ЛТР). Необходимым условием для достижения правдоподобных количественных оценок при использовании безэталонного метода ЛИЭС является точное определение таких параметров плазмы, как температура и плотность электронов. Температура плазмы Те может быть определена из относительной заселенности энергетических уровней возбужденных частиц в плазме, а плотность электронов (Ne) – из величины штарковского уширения наблюдаемых спектральных линий.
Доломит – важный минерал класса карбонатов, состоящий из карбоната кальция и магния CaMg(CO3)2. Почти половина всех земных запасов карбонатов находится в виде доломита [8]. Доломит не отличим от кальцита, который, в отличие от доломита, целиком состоит из карбоната кальция CaCO3. Замена Ca2+ в кальците на Mg2+ приводит к возникновению магниевых кальцитов с различным содержанием Mg2+ [9]. Доломит широко используется для раскисления (известкования) почв; в фармакологической промышленности как источник магнезии (MgO); в сталелитейной промышленности как спекающий агент или флюс; как добавка в удобрении, а также при производстве стекла, кирпича и керамики [10–14]. Карбонат кальция с различным содержанием карбоната магния играет важную роль в регуляции атмосферного CO2, карбонатном диагенезе и биоминерализации [15]. Проведенные исследования по содержанию магния в карбонатных породах позволили использовать количество магния в породе как геотермометр [16, 17]. Таким образом, значительный интерес для ученых представляет задача точного определения количества магния в карбонатных породах, которая с учетом наличия в породе нескольких минеральных фаз с различным содержанием магния является достаточно сложной задачей [18].
Сложность и разнообразие минерального и химического состава породы, как известно, зависит от месторождения и глубины залегания. Для определения возможности использования породы для тех или иных промышленных приложений одинаково важно знать как ее элементный состав, так и минералогический. В большом количестве работ метод ЛИЭС использовался для анализа геологических материалов и минералов. Значительная часть работ посвящена спектроскопическому анализу мрамора, однако эти исследования ограничены качественным и количественным анализом материала, давая лишь минимальные сведения о параметрах плазмы, которые являются ключевыми для получения количественных результатов в безэталонном методе ЛИЭС. Это побудило в данной работе использовать возможности метода ЛИЭС для точного элементного анализа и количественного определения содержания Mg в доломите. Полученные результаты сравнивались с традиционными аналитическими методами: рентгеновской дифрактометрией; сканирующей электронной микроскопией (СЭМ), совмещенной с энергодисперсионной рентгеновской спектроскопией (ЭДС); рентгеноспектральным микроанализом (РСМА). Для количественного анализа использовался метод безэталонной ЛИЭС (БЭ-ЛИЭС). Состав образцов, определенный с помощью метода ЛИЭС, находился в хорошем согласии с результатами, полученными посредством РСМА и ЭДС. Приведенные результаты не только дают подробное представление о применимости метода ЛИЭС для точного элементного анализа, но также раскрывают взаимодополняющий характер других применяемых аналитических методов.
2. МАТЕРИАЛЫ И МЕТОДЫ
Для настоящего исследования использовался образец доломитового мрамора. Образец был измельчен и размолот с использованием ступки и пестика, а затем просеян через сито для получения частиц одинакового размера. Одна часть образца была использована для определения фазового состава с помощью метода рентгеновской дифрактометрии, а вторая была спрессована в гранулы толщиной 3–4 мм и 13 мм в диаметре для использования в экспериментах по методу ЛИЭС. Опрессовка проводилась с помощью пресс-формы из нержавеющей стали при давлении 40 МПа в течение 15 мин.
Рентгеновская дифрактометрия – аналитический метод для быстрого и неразрушающего определения фазового состава кристаллического материала, а также типа его кристаллической решетки. Основой рентгеновской дифрактометрии является взаимодействие между рентгеновским излучением определенной длины волны и кристаллической решеткой исследуемого материала. При взаимодействии рентгеновского излучения с образцом создается дифракционная картина, по характеру которой определяется его фазовый состав. Для определения фазового состава доломитового образца в настоящей работе использовался дифрактометр фирмы Брукер (BrukerAXSD8 Advance Discover) с излучением на длине волны 0.154 нм (${{K}_{\alpha }}$-линия меди) и углами сканирования 2θ от 10° до 90° с шагом 0.002° и временем сканирования – 4 с/шаг. Для идентификации фаз образца использовалось программное обеспечение DIFFRAC.SUITEEVA.
Подробное описание экспериментальной установки ЛИЭС приведено в [19–23]. Вкратце, для абляции образца использовался Nd:YAG лазер с модуляцией добротности (Quantel Brilliant, Франция) на основной гармонике (1064 нм) в воздухе атмосферного давления с длительностью импульса 5 нс и частотой следования импульсов 10 Гц. Максимальная энергия импульса на основной гармонике в экспериментальной установке составляла 500 мДж. Энергия импульса в эксперименте варьировалась путем изменения времени задержки модуляции добротности с использованием программного обеспечения OOLIBS. Для направления излучения на поверхность исследуемого образца использовалась кварцевая линза с фокусным расстоянием 20 см. Во избежание пробоя воздуха возле поверхности образца расстояние между поверхностью образца и линзой устанавливалось меньше фокусного расстояния. Для анализа спектрального диапазона от 200 до 720 нм использовался набор из пяти спектрометров (LIBS 2000+, OceanOptics, США), каждый из которых имел ширину щели 5 мкм и спектральное разрешение 0.06 нм. Излучение плазмы собиралось под прямым углом к образцу на расстоянии 0.5 мм от поверхности. Время задержки между лазером и системой диагностики составляло 2 мкс. С помощью программного обеспечения OOLIBS из полученного сигнала излучения вычитался сигнал фона. Перед началом анализа поверхность образца очищалась с помощью нескольких предварительных лазерных импульсов. В настоящей работе производилось усреднение спектра доломитового образца по десяти экспериментам, а анализ спектра излучения проводился с использованием базы данных Национального института стандартов и технологий (NIST) [24].
Сканирующая электронная микроскопия (СЭМ) – один из наиболее широко применяемых методов при исследовании материалов. В этом методе используется узкий пучок ускоренных электронов, который сканирует некоторую область образца для определения его микроструктурных особенностей. СЭМ может оснащаться энергодисперсионным детектором, позволяющим измерять энергию рентгеновских лучей, которые возникают при заполнении вакансий, полученных при выбросе вторичных электронов. Испущенные таким образом рентгеновские лучи формируют эмиссионный спектр, который определяется элементным составом исследуемого образца в данной области. Для анализа структуры и формы поверхности исследуемого образца, а также для количественных оценок его химического состава в данной работе использовался сканирующий электронный микроскоп JEOLJSM 5910 SEM, оснащенный энергодисперсионным датчиком и работающий при напряжении 20 кВ.
Рентгеноспектральный микроанализ (электронно-зондовый микроанализ) – это микро-лучевой аналитический метод, используемый для in-situ неразрушающего анализа химического состава мелких твердых образцов. Основное преимущество данного метода заключается в получении точных количественных значений элементного состава в очень малой области исследуемого образца (1–2 мкм). Метод рентгеноспектрального микроанализа аналогичен методу сканирующей электронной микроскопии с возможностью проведения химического анализа по рентгеновскому спектру излучения, возникающему при бомбардировке поверхности образца пучком электронов. Пространственный масштаб метода, совместно с возможностью получения детальных изображений образца, позволяет исследовать геологические образцы, в нашем случае – доломит, in-situ и определять сложные вариации химического состава образца в пределах одной фазы. Для обнаружения некарбонатных фракций и исследования морфологии поверхности образца в настоящей работе использовался электронный микрозонд Cameca SX-100 в энергодисперсионном режиме, работающий при напряжении 15 кВ и токе 10 нА с постоянной времени 5 с.
Все вышеперечисленные методы, использовавшиеся для характеризации доломита, являются не только дорогостоящими разрушающими методами исследования, но и требуют специальной предварительной подготовки образца перед проведением анализа. С другой стороны, способность метода ЛИЭС проводить измерения в режиме реального времени и выполнять многоэлементный анализ совместно с его неразрушающей способностью делает его наиболее подходящим инструментом исследования для композиционного анализа доломита по сравнению с другими стандартными аналитическими методами. Настоящее исследование направлено на сравнение эффективности метода ЛИЭС с эффективностью других применяемых методов в точном элементном анализе доломита.
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
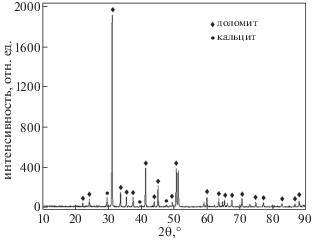
Первоначально была проведена проверка наличия исследуемой фазы в образце с помощью рентгеновской дифрактометрии. Анализ результатов показал, что доломит и кальцит являются основной и второстепенной минеральными фазами в образце (рис. 1). Положения максимумов и их относительная интенсивность на дифрактограмме хорошо согласуются с данными для эталонного образца доломита, что указывает на преобладание доломитовой фазы в образце. Пики при 2θ = 29.448°, 39.461°, 47.685° для расстояний 3.0307 Å, 2.2835 Å и 1.9071 Å соответствовали фазе кальцита в образце и имели малую интенсивность.
Рис. 1.
Зависимость интенсивности рассеянного рентгеновского излучения от угла 2θ для исследованного образца мрамора, в котором основной минеральной фазой является доломит, а второстепенной – кальцит.
3.1. Анализ и описание спектра излучения
Характерные спектры излучения ЛИЭС для различных спектральных областей очищенной поверхности доломита приведены на рис. 2. Спектры были получены в области длин волн от 200 до 720 нм и для лучшего восприятия были разбиты на две спектральные области: от 200 до 500 нм и от 501 до 720 нм. Спектральные линии, наблюдаемые в спектре излучения, обычно соответствуют элементам, которые были выбиты с поверхности образца лазерным излучением. Анализ спектров подтвердил доминирующее присутствие спектральных линий магния и кальция – основных составляющих доломита, а также их ионов. Также наблюдались спектральные линии низкой интенсивности, соответствующие натрию, алюминию и стронцию.
Рис. 2.
Спектр ЛИЭС, полученный с чистой поверхности доломита в различных спектральных областях. а – спектральная область от 250 до 500 нм; б – спектральная область от 501 до 720 нм.
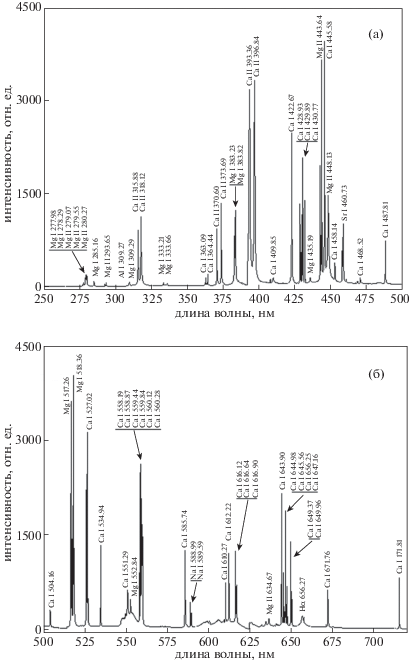
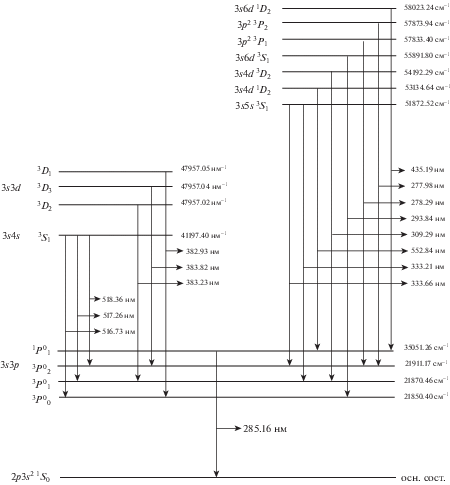
Все определенные энергетические переходы магния с верхних на нижние и основной уровни отмечены на рис. 3. Электронная конфигурация основного состояния магния – 1s22s22p63s2. В полученном спектре возбужденное состояние магния 3s3p формирует энергетический уровень 1P01, для которого разрешен переход только в основное состояние. Для другого возбужденного состояния магния 3s3d возможны 3 различных энергетических уровня – 3D1,2,3. С этих уровней наблюдались переходы в состояния 3s3p3P00,1,2, которые обладают более низкой энергией. Линии кальция в спектре преимущественно наблюдались как в области более длинных, так и более коротких длин волн относительно линий магния. Наиболее интенсивные линии кальция наблюдались на длинах волн 445.58, 558.87, 527.02, 643.90 и 430.25 нм. Самые интенсивные линии в спектре, которые соответствовали переходам в атоме магния, находились на длинах волн 518.36, 517.26 и 383.82 нм. Анализ спектра выявил наличие линий дублета натрия на длинах волн 588.99 и 589.59 нм. Также наблюдались линии атомов стронция и лития на длинах волн 460.73 и 670.79 нм, соответственно. Обнаружение большого числа спектральных линий Mg и Ca соответствует более высокой концентрации этих элементов в исследуемом образце, что соответствует общему химическому составу доломита.
3.2. Определение параметров плазмы
3.2.1. Измерение температуры плазмы Te. Температуру возбуждения можно оценить по относительной интенсивности спектральных линий плазмы при условии, что плазма является оптически тонкой и удовлетворяет условиям локального термодинамического равновесия [25]. Наиболее распространенный способ проверки выполнения условий оптической тонкости плазмы заключается в сравнении отношения интенсивностей двух экспериментально наблюдаемых линий с отношением интенсивности этих же линий, рассчитанных по формуле
(1)
$\frac{{{{I}_{1}}}}{{{{I}_{2}}}} = \frac{{{{A}_{1}}{{g}_{1}}{{\lambda }_{2}}}}{{{{A}_{2}}{{g}_{2}}{{\lambda }_{1}}}}\exp \left( {\frac{{ - \left( {{{E}_{1}} - {{E}_{2}}} \right)}}{{k{{T}_{e}}}}} \right),$Очевидно, что если взять две спектральные линии с общим верхним состоянием, то экспоненциальный член в правой части уравнения (1) станет равным единице, и теоретическое отношение легко может быть рассчитано с использованием спектроскопических констант переходов. Было проведено сравнение отношения интенсивностей спектральных линий кальция на длинах волн 362.41 и 363.09 нм с отношением вероятностей соответствующих переходов. Отношения оказались равными 1.388 и 1.385, что свидетельствует о хорошем согласии между экспериментальным и теоретическим результатами. Для дополнительной проверки использовались спектральные линии иона магния на длинах волн 516.73 и 517.26 нм. Полученное отношение интенсивностей линий в этом случае также находилось в хорошем согласии с теоретическим расчетом. Таким образом, был сделан вывод о выполнении условий оптической прозрачности плазмы.
Температура плазмы определялась из графика заселенности энергетических уровней. Для этого были использованы спектральные линии, для которых выполнялось условие оптической прозрачности плазмы и которые удовлетворяли критерию локального термодинамического равновесия. Наклон линии заселенности определяется уравнением
(2)
$\ln \left[ {\frac{{{{I}_{{k,i}}}{{\lambda }_{{k,i}}}}}{{{{A}_{{k,i}}}{{g}_{{k,i}}}}}} \right] = \ln \frac{{hc{{N}_{0}}}}{{4\pi {{U}_{0}}}} - \frac{{{{E}_{k}}}}{{k{{T}_{e}}}},$Таблица 1.
Спектроскопические параметры линий излучения кальция и магния, выбранных для построения графиков заселенностей возбужденных состояний
| Элемент | Длина волны λ, нм | Энергия возбуждения Ek , см–1 | gk | Вероятность перехода A , с–1 |
|---|---|---|---|---|
| Ca (I) | 336.19 | 45052.37 | 7 | 2.23 × 107 |
| Ca (I) | 364.44 | 42747.38 | 7 | 3.55 × 107 |
| Ca (I) | 428.30 | 38551.55 | 5 | 4.34 × 107 |
| Ca (I) | 429.89 | 38646.80 | 3 | 6.00 × 107 |
| Ca (I) | 430.77 | 38417.54 | 1 | 1.99 × 108 |
| Ca (I) | 431.86 | 38464.80 | 3 | 7.40 × 107 |
| Ca (I) | 610.27 | 31539.49 | 3 | 9.60 × 106 |
| Ca (I) | 643.90 | 35896.88 | 9 | 5.30 × 107 |
| Mg (I) | 333.66 | 51872.52 | 3 | 1.70 ×107 |
| Mg (I) | 382.93 | 47957.05 | 3 | 8.99 × 107 |
| Mg (I) | 435.19 | 58023.24 | 5 | 1.84 × 107 |
| Mg (I) | 517.26 | 41197.40 | 3 | 3.37 × 107 |
| Mg (I) | 518.36 | 41197.40 | 3 | 5.61 × 107 |
| Mg (I) | 552.84 | 43134.64 | 5 | 1.39 × 107 |
| Mg (II) | 279.07 | 71491.06 | 4 | 4.01 × 108 |
| Mg (II) | 279.79 | 71490.19 | 6 | 4.79 × 108 |
| Mg (II) | 292.86 | 69804.95 | 2 | 1.15 × 108 |
| Mg (II) | 293.65 | 69804.95 | 2 | 2.30 × 108 |
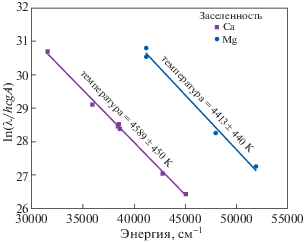
График заселенности энергетических состояний, построенный по спектральным линиям магния и кальция, дан на рис. 4. Температура плазмы, определенная по линиям магния составила 4413 ± 440 K, а по линиям кальция – 4589 ± 450 K. Некоторое расхождение температур может быть объяснено неточностью измерения интенсивностей спектральных линий, площади кривых, а также погрешностью вероятностей переходов.
Рис. 4.
Зависимость заселенности возбужденных уровней от энергии излучения (см–1), определенная из интенсивности спектральных линий атомов кальция и магния, полученных с помощью лазерного пробоя на поверхности исследуемого образца доломита.
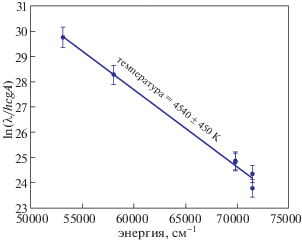
Для увеличения точности расчета температуры плазмы в график заселенности состояний были включены энергетические состояния однократных ионов. В табл. 1 приведены длины волн переходов однократно ионизованного магния, энергии их верхних состояний, вероятности переходов и статистические веса, которые использовались для построения графика заселенности. Температура плазмы, рассчитанная с учетом однократных ионов, составила 4540 ± 450 K (рис. 5). Среднее значение температуры плазмы, известное как кажущаяся температура плазмы, взято равным 4500 ± 450 K и использовалось для количественного расчета элементного состава в безэталонном методе ЛИЭС.
Рис. 5.
Зависимость заселенности возбужденных уровней от энергии излучения (см–1), определенная из интенсивности спектральных линий атома и однократного иона магния, полученных с помощью лазерного пробоя на поверхности исследуемого образца доломита.
3.2.2. Измерение плотности электронов Ne. Одним из наиболее распространенных методов определения плотности электронов является измерение штарковского уширения спектральных линий. В данной работе концентрация электронов оценивалась по штарковскому уширению спектральных линий кальция и магния.
Ширина спектральной линии, уширенной за счет эффекта Штарка, на полувысоте определяется следующим соотношением [1]
(3)
${{N}_{e}}\;({\text{с}}{{{\text{м}}}^{{ - 3}}}) = \left( {\frac{{\Delta {{\lambda }_{{FWHM}}}}}{{2{{\omega }_{S}}\left( {\lambda ,{{T}_{e}}} \right)}}} \right) \times {{N}_{r}}.$В формуле (3) $\Delta {{\lambda }_{{FWHM}}}$ – вклад штарковского уширения в полную ширину спектральной линии, Ne – плотность электронов, ${{\omega }_{S}}$ – штарковский коэффициент уширения, значения которого берутся из литературы [26]. Для определения плотности электронов использовалась спектральная линия кальция на длине волны 445.58 нм, профиль которой представлен на рис. 6. Величина плотности электронов, определенная с помощью соотношения (3), составила (2.39 ± 0.2) × 1017 см–3. Плотность электронов также находилась по спектральной линии иона магния Mg(II) на длине волны 448.13 нм, соответствующей переходу $4{{f}^{2}}F_{{{5 \mathord{\left/ {\vphantom {5 2}} \right. \kern-0em} 2}}}^{0} \to 3{{d}^{2}}{{D}_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}}$. Величина плотности электронов, определенная по этой спектральной линии, составила (2.133 ± 0.2) × × 1017 см–3.
Рис. 6.
Профиль спектральной линии Ca (I) на длине волны 445.58 нм, уширенный эффектом Штарка. Символы – экспериментальные данные, сплошная линия – аппроксимация линии фойгтовским контуром.
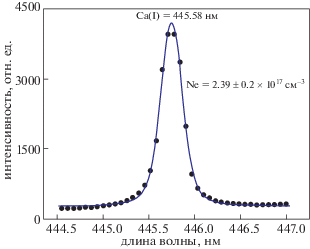
Символы, изображенные на рис. 6, представляют собой экспериментальные данные, а сплошная линия – приближение этих данных фойгтовским контуром. Так как плазма на поверхности образца доломитового мрамора образовывалась при атмосферном давлении воздуха, то в ее спектре также наблюдалась Hα линия водорода. Поскольку Hα линия не подвержена эффекту самопоглощения, то определение по ней плотности электронов является более предпочтительным. Поэтому плотность электронов определялась также и по Hα линии водорода с использованием соотношения [27]
(4)
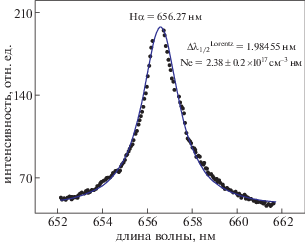
${{N}_{e}}\;({\text{с}}{{{\text{м}}}^{{ - 3}}}) = {{\left[ {\frac{{\Delta {{\lambda }_{{FWHM}}}\;({\text{нм}})}}{{1.098}}} \right]}^{{1.4713}}} \times {{10}^{{17}}}.$Аппроксимация экспериментальных данных фойгтовским профилем спектральной линии Hα приведена на рис. 7. Величина лоренцовской ширины на половине высоты составила 1.98455 нм, а плотность электронов, определенная по этой полуширине, – (2.38 ± 0.2) × 1017 см–3, что совпадает со значением, вычисленным по профилю спектральной линии кальция (рис. 6). Данное обстоятельство позволяет сделать вывод о правомерности оценки плотности электронов, проведенной по профилям спектральных линий атомов и ионов исследуемого образца доломита.
Рис. 7.
Типичный профиль спектральной линии Hα водорода на длине волны 656.27 нм. Сплошная линия, проходящая через экспериментальные данные, – аппроксимация фойгтовским контуром.
Для определения нижней границы плотности электронов, при которой еще выполняются условия локального термодинамического равновесия использовался критерий Маквиртера [28]
(5)
${{N}_{e}}\;({\text{с}}{{{\text{м}}}^{{ - 3}}}) \geqslant \left( {1.6 \times {{{10}}^{{12}}}} \right)\sqrt T {{\left( {\Delta E} \right)}^{3}}.$.Используя для оценки спектральную линию кальция на длине волны 445.58 нм, имеем: $\Delta E$ = = 2.781 эВ, Те = 4500 К и из соотношения (5) Ne = = 2.309 × 1015 см–3. Вычисления по (5) для линии магния 518.36 нм дают величину плотности электронов Ne ≥ 1.467 × 1015 см–3. Полученные граничные величины существенно меньше значений плотностей электронов, которые были получены из обработки спектральных линий кальция, иона магния и Нα линии водорода, что подтверждает наше предположение о выполнении условий локального термодинамического равновесия для плазмы.
3.3. Количественный анализ с использованием безэталонного метода ЛИЭС
Количественное определение основных элементов в доломитовом образце, наблюдавшихся в спектре излучения, проводилось с помощью метода безэталонной ЛИЭС. В основе этого метода лежит прямо пропорциональная зависимость между концентрацией элемента и интегральной интенсивностью его спектральных линий, а также предположения об оптической прозрачности плазмы и о выполнении условий локального термодинамического равновесия. Заселенность возбужденных уровней в плазме определяется законом Больцмана (2), который можно записать в виде линейного соотношения
где $y = \ln \left[ {\frac{{I{{\lambda }_{{ki}}}}}{{hc{{A}_{{ri}}}{{g}_{k}}}}} \right]$; $x = {{E}_{k}}$; $m = \left[ {\frac{1}{{{{k}_{B}}T}}} \right]$; ${{q}_{s}} = $ $ = \ln \left[ {\frac{{F{{C}^{s}}}}{{Z\left( T \right)}}} \right]$.Таким образом, выражение (6) можно переписать в виде
где Сs, U(T) и F – количество излучающего вещества, статистическая сумма состояний данного излучающего соединения и подгоночный параметр, определяемый экспериментально.Как уже упоминалось выше, температуры плазмы, определенные по спектральным линиям магния и кальция, практически совпадают, а образовавшаяся плазма удовлетворяет условиям локального термодинамического равновесия. Таким образом, графики заселенности возбужденных уровней имеют практически одинаковый наклон m с различными свободными членами qs, величины которых определяются логарифмом концентрации вещества. Относительный состав основных компонентов доломитового образца, определенный методом безэталонной ЛИЭС, оказался Ca = 68.58% и Mg = 31.41%. Полученный количественный состав хорошо согласуется с результатами измерений методами рентгеноспектрального микроанализа и сканирующей электронной микроскопии (совмещенной с энергодисперсионной рентгеновской спектроскопией). Данные измерений приведены в табл. 2.
Таблица 2.
Концентрации кальция и магния (в весовых процентах) в образце доломита, определенные с помощью безэталонного метода лазерно-искровой эмиссионной спектроскопии (БЭ-ЛИЭС), сканирующей электронной микроскопии, совмещенной с рентгеновской энергодисперсионной спектроскопией (СЭМ-ЭДС), и рентгеноспектрального микроанализа (РСМА-ЭДС)
| Элементы | БЭ-ЛИЭС | СЭМ-ЭДС | РСМА-ЭДС |
|---|---|---|---|
| Ca, вес.% | 68.58 ± 1.38 | 69.17 ± 1.40 | 68.55 ± 1.37 |
| Mg, вес.% | 31.41 ± 0.65 | 30.83 ± 0.60 | 31.34 ± 0.62 |
3.4. Количественный анализ методами РСМА и СЭМ-ЭДС
В настоящей работе изучалась морфология поверхности образца доломитового мрамора. Состав образца дополнительно определялся с использованием методов сканирующей электронной микроскопии рентгеноспектрального микроанализа. Картина распределения вторичных электронов, которое определяется фазовой структурой исследуемого образца, и рентгеновский спектр, полученный методами энергодисперсионной спектроскопии, представлены на рис. 8.
Рис. 8.
Распределение вторичных электронов в методе СЭМ, иллюстрирующее фазовую структуру поверхности доломитового мрамора, и рентгеновский спектр, полученный с помощью электронов СЭМ и РСМА и записанный методами рентгеновской энергодисперсионной спектроскопии (ЭДС). а – двумерная структура распределения вторичных электронов поверхности доломитового мрамора; б – рентгеновский спектр, полученный с помощью воздействия электронов СЭМ и РСМА на образец доломита.
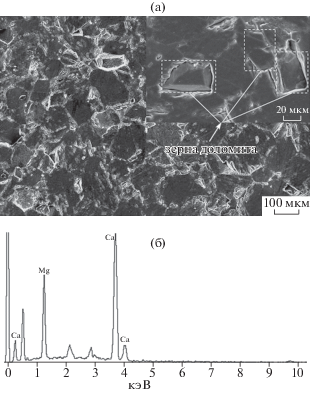
Распределение вторичных электронов, полученное в доломитовом образце (рис. 8а), имеет практически однородную микроструктуру с варьирующимся размером доломитовых и кальцитовых зерен. Аналогичное распределение фаз было также получено с помощью рентгеновской дифрактометрии (рис. 1). При большем увеличении (~20 мкм) можно заметить, что доломитовые зерна погружены в окружающую их кальцитовую массу (рис. 8а).
Полученный энергодисперсионный спектр указывает на то, что основными составляющими элементами исследуемого образца являются кальций и магний (рис. 8б). Величины концентраций Ca и Mg, определенные с помощью энергодисперсионного спектра, составили 69.17 и 30.83 весовых процентов, соответственно. Никаких следов дополнительных элементов с помощью энергодисперсионного анализа выявлено не было. Определение кальция как основного и магния как вторичного элементов, составляющих исследуемый образец, хорошо согласуется с результатами рентгеновской дифрактометрии и лазерно-искровой эмиссионной спектроскопии. Изображение отраженных электронов в методе СЭМ приведено на рис. 9. Можно отметить наличие на полученном изображении карбонатных зерен, а также присутствие трещин/пустот, окружающих доломитовую фазу. Рентгеноспектральный микроанализ использовался как в энергодисперсионном, так и в волнодисперсионном режимах. Размер зерен кальцита и доломита был ~ 100 мкм, а размер пучка во время исследования составлял 5 мкм. Поэтому, чтобы уменьшить повреждения от пучка, измерения в энергодисперсионном режиме также проводились с большим растром для реинтеграции примесей, рассеянных в основной массе образца. По данным этих измерений также не было обнаружено никаких дополнительных элементов.
Рис. 9.
Распределение отраженных электронов, иллюстрирующее трещины/пустоты вдоль границ доломитовых зерен.

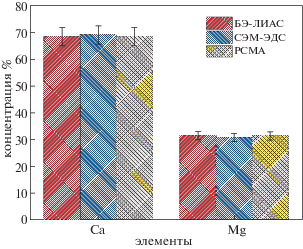
В табл. 2 представлены сравнительные данные элементного анализа по основным элементам, составляющим исследуемый образец доломитового мрамора, полученные методами БЭ-ЛИЭС, СЭМ-ЭДС и РСМА. Видно, что результаты, полученные методом БЭ-ЛИЭС, отличаются от результатов, полученных другими методами, не более чем на 2%. Небольшое отклонение в содержании Mg может быть отнесено к возможному замещению ионов кальция ионами магния в фазе кальцита [29, 30]. На рис. 10 те же результаты анализа даны в виде гистограммы. На рис. 10 не фигурируют стронций, алюминий и натрий, которые были обнаружены в образце в незначительных количествах (<1%), что отражалось в слабой интенсивности их спектральных линий.
4. ВЫВОДЫ
Доломит – карбонатный минерал, состоящий в основном из карбонатов кальция и магния. Он широко применяется в фармацевтической и в сталелитейной промышленности, а также в ряде приложений, касающихся охраны окружающей среды. Содержание магния в доломитах широко используется в качестве геотермометра. При этом точное определение содержания Mg в образце всегда является сложной задачей. В настоящей работе мы использовали метод ЛИЭС совместно с методами рентгеновской дифрактометрии, СЭМ-ЭДС и РСМА для качественного и количественного анализа образца доломита. Анализ полученного спектра излучения выявил присутствие Ca и Mg в качестве основных составляющих образец элементов, в то время как Al, Sr и Na были обнаружены в несущественных количествах. Была проведена оценка температуры плазмы с помощью графиков заселенности возбужденных состояний нейтральных частиц плазмы и ионов. Полученное значение температуры составило 4500 ± 450 K. Концентрация электронов Ne определялась с помощью штарковского уширения спектральной линии кальция и составила 2.39 × × 1017 см-3. С хорошей точностью полученный результат совпал с результатом перекрестной проверки с использованием уширения Нα линии водорода. Был проведен количественный анализ с использованием метода БЭ-ЛИЭС для случая, когда выполняются условия оптической прозрачности плазмы и локального термодинамического равновесия. Количественный состав образца также определялся методами ЭДС и РСМА. Было обнаружено, что результаты, полученные различными методами, находятся в хорошем согласии друг с другом. Сделан вывод о возможности использования метода БЭ-ЛИЭС для точного количественного определения основных и второстепенных элементов, тогда как другие аналитические методы (ЭДС и РСМА) являются взаимодополняющими, а также дополняют метод ЛИЭС в вопросе композиционного анализа геологических образцов.
Коллектив авторов благодарит профессора факультета физики университета Каид-и Азама (Исламабад, Пакистан) Рахиля Али (Raheel Ali) за предоставленное оборудование для проведения экспериментов.
Список литературы
Cremers D.A., Radziemski L.J. Handbook of Laser-induced breakdown Spectroscopy. New York: Wiley, 2006.
Ciucci A., Corsi M., Palleschi V., Rastelli S., Salvetti A., Tognoni E. // Appl. Spectrosc. 1999. V. 53. P. 960.
Wang L., Zhang C., Feng Y. // Chin. Opt. Lett. 2008. V. 6. P. 5.
Tognoni E., Cristoforetti G., Legnaioli S., Palleschi V. // Spectrochim. Acta B. 2010. V. 65. P. 1.
Pandhija S., Rai N.K., Pathak A.K., Rai A.K., Choudhary A.K. // Spectrosc. Lett. 2014. V. 14. P. 579.
Abbass Q., Ahmed N., Ahmed R., Baig M.A. // Plasma Chem. Plasma Proc. 2016. V. 36. P. 1287.
Yang J., Li X., Xu J., Ma X. // Appl. Spectrosc. 2018. V. 72. P. 129.
Zenger D.H., Dunham J.B., Ethington R.L. Concepts and models of dolomitization. Spec. Publ.-SEPM, V. 28 (1980).
Boynton R.S. Chemistry and Technology of Lime and Limestone. New York: Interscience Publishers, 1967.
Yeprem H.A., Turedi E., Karagoz S.A. // Mater. Charact. 2004. V. 52. P. 331.
Gai G.S., Yang Y.F., Fan S.M., Cai Z.F. // Powder Technol. 2005. V. 153. P. 153.
Rabah M., Ewais E.M.M. // Ceram. Int. 2008. V. 35. P. 813.
Iqbal Y., Lii-Cherng L., Fahad M., Ubic R. // JOM. 2013. V. 65. P. 73.
Fahad M., Iqbal Y., Riaz M., Ubic R., Abrar Himal M. // Geol. 2016. V. 37. P. 17.
Bertram M.A., Mankenzie F.T., Bischoff F.C., Bis-choff W.D. // Am. Mineral. 1991. V. 76. P. 1889.
Fahad M., Iqbal Y., Riaz M., Ubic R., Redfern S.A.T. // J. Earth Sci. 2016. V. 27. P. 989.
Fahad M., Sundas S. // Geosci. J. 2018. V. 22. P. 303.
Titschak J., Geotz-Neunhoeffer F., Neubauer J. // Am. Mineral. 2011. V. 96. P. 1028.
Abrar M., Iqbal T., Fahad M., Andleeb M., Farooq Z., Afsheen S. // Laser Phys. 2018. V. 28. 056002.
Fahad M., Abrar M. // Laser Phys. 2018. V. 28. 085701.
Fahad M., Farooq Z., Abrar M., Shah K.H., Iqbal T., Sundas S. // Laser Phys. 2018. V. 28. 125701.
Fahad M., Farooq Z., Abrar M. // Appl. Opt. 2019. V. 58. P. 3501.
Fahad M., Ali S., Iqbal Y. // Plasma Sci. Technol. 2019. V. 21. 085507.
Kramida A., Ralchenko Y., Reader J. and NIST ASD Team. National Institute of Standards and Technology. 2019. http://physics.nist.gov/asd
Griem H.R. Principles of Plasma Spectroscopy. Cambridge: Cambridge University Press, 2005.
Griem H.R. Spectral Line Broadening by Plasma. New York, London: Academic Press, Inc. 1974.
Konjevic N., Ivkovic M., Sakan N. // Spectrochim. Acta B. 2012. V. 76. P. 16.
McWhirter R.W.P. Spectral Intensities Plasma Diagnostics Techniques / R.H. Huddleston and S.L. Leonard, Eds. New York: Academic Press, Inc. 1965.
Barcina L.M., Espina A., Suarez M., Garcia J.R., Rodriguez J. // Thermochim. Acta. 1997. V. 290. P. 181.
Warren J. // Earth-Sci. Rev. 2000. V. 52. P. 1.
Дополнительные материалы отсутствуют.