

Генетика, 2019, T. 55, № 12, стр. 1410-1416
Таргетное секвенирование для исследования хозяйственно полезных признаков и филогенетического разнообразия древних овец
А. А. Кечин 1, 2, 3, *, **, М. А. Дымова 1, А. А. Тишкин 4, С. П. Грушин 4, П. К. Дашковский 5, М. Л. Филипенко 1, 3, 4
1 Институт химической биологии и фундаментальной медицины
Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
2 Новосибирский государственный университет,
кафедра молекулярной биологии и биотехнологии
630090 Новосибирск, Россия
3 Новосибирский государственный университет, кафедра клинической биохимии
630090 Новосибирск, Россия
4 Алтайский государственный университет, кафедра археологии, этнографии и музеологии
656049 Барнаул, Россия
5 Алтайский государственный университет, кафедра политической истории,
национальных и государственно-конфессиональных отношений
656049 Барнаул, Россия
* E-mail: aa_kechin@niboch.nsc.ru
** E-mail: a.a.kechin@gmail.com
Поступила в редакцию 31.01.2019
После доработки 18.04.2019
Принята к публикации 23.04.2019
Аннотация
Овца считается одним из первых одомашненных животных. История ее доместикации и распространения насчитывает около 10 тыс. лет, в течение которых популяции овец изменялись как внешне, так и на генетическом уровне. Авторами разработана система из 49 олигонуклеотидных праймеров для исследования с помощью таргетного секвенировавния локусов, используемых в филогенетическом анализе или ассоциированных с хозяйственно полезными признаками. Всего были приготовлены и секвенированы на приборе MiSeq (Illumina) NGS-библиотеки для 48 образцов, для 40 из которых удалось определить филогенетические линии: 28 относились к материнской линии B, 10 – к линии A и по одному образцу – к линиям C и D. Исследование генов, ассоциированных с хозяйственно полезными признаками, выявило образцы с нуклеотидными заменами в гене MC1R, приводящими к черному окрасу шерсти: два образца – c.218T>A, один – c.361G>A и два – обе замены одновременно; а также один образец с заменой в гене GDF8, связанной с мышечной гипертрофией и один – с заменой в гене TYRP1, ассоциированной с коричневым окрасом шерсти. Полученные данные подтверждают высокое генетическое разнообразие овец юга Западной Сибири в конце III и начале II тыс. до н.э. и применимость таргетного секвенирования для исследования образцов архивной ДНК.
Одомашнивание животных стало одним из ключевых этапов развития человечества и привело к появлению новых форм хозяйства [1]. Последовательный и продолжительный отбор животных по предпочтительным признакам позволял получать менее агрессивных, но более продуктивных, выносливых и/или быстрых особей. Другими полезными свойствами одомашненных животных могли стать плодовитость, окрас, приспособленность к особым условиям содержания.
Овцы (Ovis aries) были одомашнены одними из первых животных около 9–11 тыс. лет назад. Вероятнее всего, это произошло в области, получившей название “плодородный полумесяц” (территория от северного Загроса до юго-восточной Анатолии) [2]. Считается, что первоначально отбор овец проводился по продуктивности мяса и молока и лишь позже – по качеству шерсти [3]. Определение скрытых для того времени генетических детерминант, по которым осуществлялся отбор этих животных, имеет не только фундаментальное значение, раскрывая подробности эволюции человечества, но и практическое. Понимание механизмов исторического развития хозяйственно полезных признаков у современных пород дает основу для последующего улучшения их свойств.
История возникновения и формирования современных пород может быть частично восстановлена путем исследования митохондриальной ДНК (мтДНК), отражающей наследование по материнской линии. На сегодняшний день выделяют пять материнских линий или гаплогрупп – A, B, C, D и E, из которых две последние являются наиболее редкими [4].
Основным методом определения аллелей как гаплогрупп мтДНК, так и локусов, ассоциированных с хозяйственно полезными признаками, является секвенирование по Сэнгеру. Реже используется подход полногеномного секвенирования, основанный на массовом параллельном секвенировании (MPS или NGS). Последний подход, с одной стороны, определяет аллели всех необходимых локусов, предоставляя исследователю максимально полную генетическую информацию об объекте, а, с другой стороны, большáя часть производительности этого метода оказывается занята “мусорной” информацией, наличие которой не позволяет сделать никаких дополнительных выводов.
В данной работе использовался подход, который получил широкое распространение в клинике: таргетное секвенирование. При его реализации происходит обогащение только целевых последовательностей генома исследуемого объекта [5]. Обогащение может производиться несколькими путями: с помощью гибридизации и амплификацией. Это и нашло отражение в данном исследовании. В статье показана применимость таргетного секвенирования при изучении образцов архивной ДНК (аДНК) для получения последовательностей маркеров филогенетического развития популяций древних овец и определения генетических детерминант хозяйственно полезных признаков.
МАТЕРИАЛЫ И МЕТОДЫ
Образцы костей и выделение из них ДНК
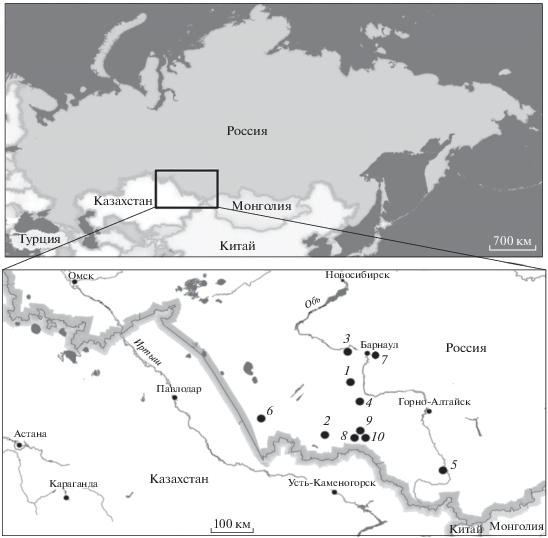
Подробное описание образцов, привлеченных к работе (всего 48 экз.) приведено в табл. 1 Приложения. Карта-схема расположения поселений и могильников представлена на рис. 1. Выделение аДНК из 0.5 г перетертой кости проводилось по описанному ранее методу [6]. Параллельно был поставлен отрицательный контроль выделения.
Выбор целевых локусов и конструирование праймеров
Локусы для амплификации и последующего секвенирования были отобраны на основании данных литературы (табл. 2 Приложения). Праймеры для амплификационного обогащения целевых последовательностей автоматически сконструированы модифицированной нами программой hi-plex [7]. Все сконструированные праймеры проверяли на нецелевую гибридизацию с участками генома (сборка Oar_v4.0) с помощью программы BWA и Python-скриптов.
Приготовление NGS-библиотек и секвенирование
Приготовление библиотек осуществляли по методике, описанной ранее [5] и включающей две стадии амплификации: наработка целевых последовательностей и включение адаптерных и индексирующих последовательностей. Секвенирование проводили на приборе MiSeq Illumina с использованием реагентов “MiSeq reagent kit v3” (150 циклов) в соответствии с инструкцией производителя.
Анализ данных NGS
Из полученных после секвенирования прочтений программой Trimmomatic [8] были удалены последовательности адаптеров. Прочтения короче 50 нуклеотидов не учитывали, поскольку ожидаемая длина ПЦР-продуктов вместе с праймерами составляла около 95 пн. Оставшиеся прочтения картировали программой BWA [9] на извлеченные из генома фрагменты, соответствующие амплифицируемым областям. При этом близко расположенные фрагменты (до 1000 пн) были объединены в один. Получившиеся SAM-файлы конвертировали в BAM-файлы, сортировали в них прочтения и добавляли информацию о группах прочтений программой picard (http:// broadinstitute.github.io/picard/). Далее из прочтений в BAM-файлах удаляли последовательности праймеров программой cutPrimers [10]. Определение генотипов по интересующим локусам проводили программой samtools, консенсус получали с помощью программы Pisces (https://github.com/ Illumina/Pisces) и собственных Python-скриптов. Для определения принадлежности исследуемых образцов к той или иной филогенетической линии (A, B, C, D и E) использовались референсные последовательности, применявшиеся ранее [11].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Общая статистика таргетного NGS
Для амплификации выбранных фрагментов были сконструированы 49 пар праймеров (табл. 3 Приложения). Всего оказалось получено 22.5 млн пар прочтений, из которых 19 млн пар относились к исследуемым 48 образцам. Медианно на каждый образец пришлось по 386 860 (от 19 до 1 478 630) пар прочтений. Медианный процент прочтений, которые были картированы на целевые последовательности, составил 2.31% (от 0.08 до 57.97%). Столь низкий процент связан с повреждениями в аДНК, из-за чего гибридизация праймеров между собой происходит эффективнее, чем с ДНК-матрицей. Это подтверждает и тот факт, что два наибольших значения (9.30 и 57.97%) отмечены у образцов монгольского и современного времени соответственно.
Филогенетический анализ последовательностей D-петли мтДНК
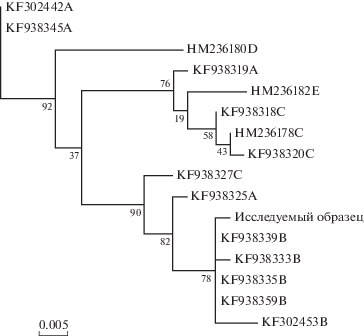
Для определения принадлежности исследуемых образцов к той или иной филогенетической линии (A, B, C, D и E) были составлены консенсусы фрагментов последовательности D-петли (МТ: 14652–16501). Каждый полученный консенсус выравнивали с референсными последовательностями, выравнивания обрезались по самому короткому общему участку и строили филогенетическое дерево методом максимального правдоподобия программой MEGA (версия 6.06) [12]. Пример такого дерева представлен на рис. 2. Для восьми образцов вышеуказанный фрагмент D-петли не был покрыт вовсе. Наиболее распространенной линией оказалась линия B (28 образцов, 70%), следующей – линия A (10–25%). По одному образцу было найдено линий C (2.5%) и D (2.5%).
Рис. 2.
Пример филогенетического дерева, по которому определялась принадлежность к филогенетической линии (последняя буква в названиях референсных образцов обозначает линию; числа рядом с узлами отражают уровень поддержки соответствующего узла).
Распределение филогенетических линий по географическому расположению образцов костей и датировке представлены на рис. 3 и 4. Так же, как и в случае с общей статистикой, большинство образцов, найденных на поселении Березовая Лука и погребально-поминальном комплексе Телеутский Взвоз-I (памятники с наибольшим числом образцов), были отнесены к линиям A и B. Для памятника Березовая Лука 83% образцов относились к линии B, а для Телеутского Взвоза-I – 54%. По датировке наиболее представленной была группа образцов из археологических объектов, определяемых последней четвертью III–первой четвертью II тыс. до н.э. В ней 72% образцов относились к линии B, 24% – к линии A и 3% – к линии C. Таким образом, в данной работе было зафиксировано то, что линия B являлась преобладающей среди всех представленных на сегодняшний день гаплогрупп, как в отдельно взятых районах, так и в разные хронологические отрезки.
Рис. 3.
Распределение образцов по филогенетическим линиям в местах их обнаружения. 1 – Березовая Лука; 2 – Телеутский Взвоз-I; 3 – Чинета-II; 4 – Колыванское-I; 5 – Ханкаринский Дол; 6 – Фирсово-XIV; 7 – Яломан-II; 8 – Мышиный Лог-I; 9 – Рублево-VI; 10 – Центральная Монголия.
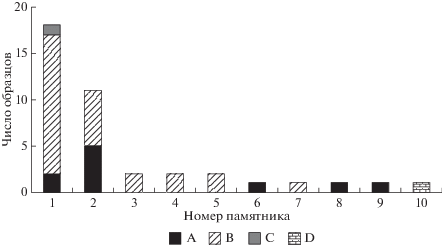
Рис. 4.
Распределение образцов по филогенетическим линиям и датировкам: 1 – последняя четверть III–первая четверть II тыс. до н.э., ранний бронзовый век; 2 – V–III вв. до н.э., ранний железный век; 3 – X–VIII вв. до н.э., поздний бронзовый век; 4 – 2-я половина IV–1-я половина V вв. н.э. (жужанское время), ранний железный век; 5 – IX–X вв. н.э., раннее средневековье; 6 – XIII–XIV вв. н.э., монгольское время; 7 – XXI в. н.э., современный период.

Генетические локусы, ассоциированные с хозяйственно полезными признаками
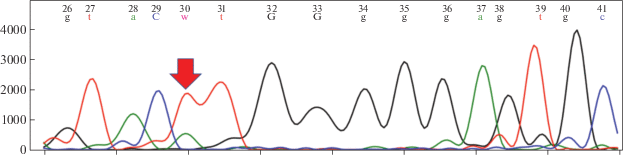
При секвенировании библиотек, полученных с помощью разработанной системы олигонуклеотидных праймеров, были прочитаны 78% целевых районов. По большинству локусов образцы оказались гомозиготны по аллелям дикого типа. Пять образцов из поселения периода ранней бронзы Березовая Лука были гетерозиготны по одной или нескольким однонуклеотидным заменам: два – по MC1R c.218T>A, один – по MC1R c.361G>A и два – по обоим локусам. Данный ген ассоциирован с черным окрасом шерсти с аутосомно-доминантным типом наследования признака. Один образец из памятника Телеутский Взвоз-I был гетерозиготен по локусу: GDF8 g.118146642G>A, один образец из поселения Березовая Лука – по TYRP1 c.869G>T. Первый локус ассоциирован с мышечной гипертрофией (увеличением мышечной массы) [13], второй – с коричневым окрасом шерсти, причем для второго показано аутосомно-доминантное наследование признака [14]. Выявленные варианты были подтверждены секвенированием по Сэнгеру (рис. 5).
Рис. 5.
Пример секвенограммы для подтверждения наличия вариации в исследуемых образцах. Стрелкой отмечена гетерозиготная замена A>T, соответствующая замене c.218T>A в гене MC1R.
Таким образом, среди исследованных 48 образцов костных останков овец были найдены пять овец с генетическими детерминантами черного окраса шерсти, одна оказалась коричневого цвета и еще одна – с мышечной гипертрофией.
Исследование образцов ДНК древних останков животных имеет колоссальное значение для понимания процессов эволюции и одомашнивания. Одним из наиболее распространенных, а также простых и удобных методов генетического анализа архивной ДНК является полногеномное секвенирование. В то же время, его существенным недостатком отмечается нецелевое использование мощностей массового параллельного секвенирования на области генома, прочтение которых не несет никакой полезной для исследования информации. В связи с этим, мы разработали амплификационную панель для таргетного секвенирования локусов генома овец, ассоциированных с их филогенетическим развитием и хозяйственно полезными признаками (табл. 2 Приложения). Ранее уже предпринимались попытки представления таких систем для образцов архивной ДНК [15]. Однако широкое применение они получили только в клинических исследованиях образцов ДНК, выделенных из парафинированных гистологических блоков и плазмы крови [16], что связано со сложностью проведения мультиплексной амплификации с архивной ДНК. В то же время, таргетное секвенирование позволяет получить последовательности многих целевых районов за один запуск, что значительно сокращает расходы столь ценного генетического материала.
Используя разработанную NGS-панель, удалось получить достаточно высокое покрытие большинства выбранных локусов (78%). Для остальных локусов специфических прочтений выявлено не было. Вероятно, это связано со структурой соответствующих праймеров, для которых формирование вторичных структур было термодинамически более выгодным. При этом процент прочтений, которые были картированы на целевые последовательности, был значительно больше для более современных образцов (58.0 и 9.3% против медианного 2.3% для остальных).
Одним из основных результатов проделанной работы стало определение принадлежности образцов к филогенетическим линиям по нуклеотидным последовательностям D-петли митохондриального генома. Большинство образцов были отнесены к линиям B (70% образцов) и A (25%). Линии C и D составили лишь по 2.5%. При этом последняя гаплогруппа была выявлена только для образца овцы современной породы. Полученное распределение филогенетических линий соответствует описанным в литературе [17]. Возможно, что преобладание линий B и A в исследованных районах связано с их первоочередным появлением здесь по сравнению с остальными линиями [18]. Однако возможны и другие объяснения. Также неясным остается, почему на данной территории закрепились в первую очередь линии A и B, а гаплогруппа C стала минорной.
Нами выявлены образцы, которые несли генетические детерминанты черного и коричневого окраса шерсти. При этом важным является тот факт, что наряду с образцами гетерозиготными по одному из локусов обнаружен образец гетерозиготный по двум локусам гена MC1R (c.218T>A и c.361G>A), из чего следует, что в какое-то время в одной популяции сосуществовали особи, несущие по одной гомозиготной или гетерозиготной замене в каждом из этих локусов. Все это говорит о высоком генетическом разнообразии овец юга Западной Сибири в конце III и начале II тыс. до н.э.
Таким образом, с целью повышения эффективности применения NGS при исследовании аДНК был разработан метод приготовления таргетной NGS-библиотеки, который позволяет проводить как филогенетический анализ, так и получать последовательности генома овец, ассоциированные с хозяйственно полезными признаками. Метод может быть использован как для исследования этих же локусов в других образцах, так и в качестве основы для разработки новых таргетных NGS‑панелей, с помощью которых можно изучать другие интересующие локусы овец и других животных. Основным его преимуществом по сравнению с полногеномным секвенированием является более высокое покрытие целевых районов, а по сравнению с секвенированием по Сэнгеру – меньшие затраты извлекаемого генетического материала. Кроме того, результаты применения описанного подхода расширяют наши знания об эволюции и распространении одомашненных овец по территории Западной Сибири.
Список литературы
Vigne J.D. The origins of animal domestication and husbandry: A major change in the history of humanity and the biosphere // C. R. Biol. 2011. V. 334. № 3. P. 171–181. https://doi.org/10.1016/j.crvi.2010.12.009
Zeder M.A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact // PNAS USA. 2008. V. 105. № 33. P. 11597–11604. https://doi.org/10.1073/pnas.0801317105
Ryder M. Sheep & Man. Duckworth, 1983. 846 p.
Meadows J.R.S., Hiendleder S., Kijas J.W. Haplogroup relationships between domestic and wild sheep resolved using a mitogenome panel // Heredity (Edinb.). 2011. V. 106. № 4. P. 700–706. https://doi.org/10.1038/hdy.2010.122
Ermolenko N.A., Boyarskikh U.A., Kechin A.A. et al. Massive parallel sequencing for diagnostic genetic testing of brca genes – a single center experience // Asian Pac. J. Cancer Prev. 2015. V. 16. № 17. P. 7935–7941. https://doi.org/10.7314/apjcp.2015.16.17.7935
Pääbo S., Gifford J.A., Wilson A.C. Mitochondrial DNA sequences from a 7000-year old brain // Nucl. Ac. Res. 1988. V. 16. № 20. P. 9775. https://doi.org/10.1093/nar/16.20.9775
Nguyen-Dumont T., Pope B.J., Hammet F. et al. A high-plex PCR approach for massively parallel sequencing // Biotechniques. 2013. V. 55. № 2. P. 69–74. https://doi.org/10.2144/000114052
Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data // Bioinformatics. 2014. V. 30. № 15. P. 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform // Bioinformatics. 2009. V. 25. № 14. P. 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Kechin A., Boyarskikh U., Kel A., Filipenko M. cutPrimers: A new tool for accurate cutting of primers from reads of targeted next generation sequencing // J. Comput. Biol. 2017. V. 24. № 11. P. 1138–1143. https://doi.org/10.1089/cmb.2017.0096
Dymova M.A., Zadorozhny A.V., Mishukova O.V. et al. Mitochondrial DNA analysis of ancient sheep from Altai // Anim. Genet. 2017. V. 48. № 5. P. 615–618. https://doi.org/10.1111/age.12569
Tamura K., Stecher G., Peterson D. et al. MEGA6: Molecular evolutionary genetics analysis Version 6.0 // Mol. Biol. Evol. 2013. V. 30. № 12. P. 2725–2729. https://doi.org/10.1093/molbev/mst197
Clop A., Marcq F., Takeda H. et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep // Nat. Genet. 2006. V. 38. № 7. P. 813–818. https://doi.org/10.1038/ng1810
Hinten G.N., Hale M.C., Gratten J. et al. SNP-SCALE: SNP scoring by colour and length exclusion // Mol. Ecol. Notes. 2007. V. 7. № 3. P. 377–388. https://doi.org/10.1111/j.1471-8286.2006.01648.x
Stiller M., Knapp M., Stenzel U. et al. Direct multiplex sequencing (DMPS) – a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA // Genome Res. 2009. V. 19. № 10. P. 1843–1848. https://doi.org/10.1101/gr.095760.109
Kechin A., Khrapov E., Boyarskikh U. et al. BRCA-analyzer: Automatic workflow for processing NGS reads of BRCA1 and BRCA2 genes // Comput. Biol. Chem. 2018. V. 77. P. 297–306. https://doi.org/10.1016/j.compbiolchem.2018.10.012
Tapio M., Marzanov N., Ozerov M. et al. Sheep mitochondrial DNA variation in european, Caucasian, and Central Asian areas // Mol. Biol. Evol. 2006. V. 23. № 9. P. 1776–1783. https://doi.org/10.1093/molbev/msl043
Lv F.-H., Peng W.-F., Yang J. et al. Mitogenomic meta-analysis identifies two phases of migration in the History of Eastern Eurasian sheep // Mol. Biol. Evol. 2015. V. 32. № 10. P. 2515–2533. https://doi.org/10.1093/molbev/msv139
Дополнительные материалы отсутствуют.