

Генетика, 2019, T. 55, № 2, стр. 214-222
Молекулярно-генетическое исследование поясно-конечностной мышечной дистрофии 2D в выборках больных различными формами прогрессирующих мышечных дистрофий
М. В. Булах 1, *, Н. М. Галеева 1, Д. А. Полякова 1, О. П. Рыжкова 1, Е. Л. Дадали 1, А. В. Поляков 1, **
1 Медико-генетический научный центр
115522 Москва, Россия
* E-mail: mariya.bulakh@gmail.com
** E-mail: apol@dnalab.com
Поступила в редакцию 15.03.2018
После доработки 19.06.2018
Принята к публикации 06.06.2018
Аннотация
Поясно-конечностная мышечная дистрофия 2D (ПКМД 2D) является самой частой в мире формой саркогликанопатий – аутосомно-рецессивных ПКМД, обусловленных различными молекулярными дефектами в генах, кодирующих белки саркогликанового семейства (α, β, γ и δ). ПКМД 2D возникает при наличии мутаций в гене SGCA, кодирующем α-саркогликан, и в целом по своей клинической картине сложно отличима как от других псевдогипертрофических форм ПКМД, так и от мышечной дистрофии Дюшенна/Беккера (МДД/Б). В настоящей работе проведено исследование ПКМД 2D на основании анализа встречаемости повторяющихся вариантов гена SGCA (c.229C>T, c.271G>A и c.850C>T) в трех выборках больных прогрессирующими мышечными дистрофиями, манифестирующими после периода нормального моторного развития. В выборке больных с направляющим диагнозом “мышечная дистрофия Эмери–Дрейфуса” не было выявлено ни одного больного ПКМД 2D. В выборке больных с направляющим диагнозом “ПКМД” доля ПКМД 2D составила не менее 1.9%, а в выборке больных с направляющим диагнозом “МДД/Б” – не менее 0.6%. Примечательно, что в этих двух выборках на долю самого частого в мире варианта гена SGCA – c.229C>T и варианта c.271G>A, зарегистрированного всего в одной итальянской семье, приходится практически одинаковое число больных хромосом. Следовательно, больные ПКМД 2D из РФ отличаются от больных во всем мире наличием двух мажорных вариантов этого гена (c.229C>T и c.271G>A). Информативность тест-системы для поиска трех вариантов гена SGCA (способность выявить хотя бы один мутантный аллель) оказалась в среднем довольно высокой (70% в группе больных ПКМД и 50% в группе больных МДД/Б), следовательно, данная тест-система может быть рекомендована в качестве первичного этапа молекулярной диагностики ПКМД 2D у больных с соответствующей клинической картиной.
Еще на рубеже XXI в. зарубежными исследователями в группе поясно-конечностных мышечных дистрофий (ПКМД) была обособлена подгруппа саркогликанопатий. К данной подгруппе заболеваний относятся четыре аутосомно-рецессивные формы, вызываемые молекулярными дефектами в генах, кодирующих белки саркогликаны (α, β, γ и δ). Эти четыре белка представляют собой субъединицы, формирующие тетрамерный трансмембранный саркогликановый подкомплекс, который является частью большого дистрофин-гликопротеинового комплекса, участвующего в соединении цитоскелета мышечных волокон с окружающим их внеклеточным матриксом [1]. Наиболее частая саркогликанопатия во всем мире – α-саркогликанопатия, по номенклатуре, принятой на международном семинаре экспертов в 1995 г., именуемая ПКМД 2D. Доля ПКМД 2D среди всех ПКМД, включая аутосомно-доминантные формы, составляет от 4% (США) до 26.9% (Индия) [2, 3], в Чехии на эту форму приходится 2.8% случаев ПКМД с аутосомно-рецессивным типом наследования [4]. При этом в подгруппе саркогликанопатий доля ПКМД 2D составляет от 34 до 55% [5–7]. Первые упоминания о ПКМД 2D в научной литературе появились в 1994 г., когда двумя независимыми авторами в семьях с тяжелой “дюшенноподобной” детской аутосомно-рецессивной мышечной дистрофией была установлена связь заболевания с локусом 17q21, идентифицирован ген SGCA и его белковый продукт, названный адгалином (α-саркогликан) [8, 9]. На сегодняшний день по информации, предоставляемой непрерывно пополняемой базой данных HGMD (Human Gene Mutation Database Professional 2017.4), зарегистрировано 98 патогенных и вероятно патогенных вариантов гена SGCA (NM_000023), большинство из которых приходятся на однонуклеотидные миссенс- и нонсенс-замены; реже встречаются нуклеотидные варианты, приводящие к сдвигу рамки считывания (18%) либо затрагивающие донорные или акцепторные сайты сплайсинга (8%). Для гена SGCA выявлена общая гено-фенотипическая корреляция типа молекулярного дефекта и тяжести клинической картины. Наиболее тяжелый – “дюшенноподобный” – вариант течения мышечной дистрофии с манифестацией в раннем возрасте развивается при наличии так называемых null-мутаций (Loss-of-function вариантов, приводящих к сдвигу рамки считывания или образованию преждевременного терминирующего кодона), обусловливающих резкое снижение или тотальное отсутствие α-саркогликана в мышечной ткани [10]. Миссенс-варианты меньше влияют на экспрессию гена, приводя к снижению биосинтеза белка в той или иной степени, вследствие чего у больных формируется более мягкий фенотип [11]. Основные клинические симптомы ПКМД 2D типичны для всей группы ПКМД, выражаются прогрессирующей слабостью и атрофией скелетной мускулатуры верхнего и нижнего поясов конечностей, а также проксимальных мышечных групп, снижением и исчезновением рефлексов, постепенным формированием контрактур крупных суставов и сколиоза. К особенностям как данной формы, так и группы саркогликанопатий в целом относятся псевдогипертрофии икроножных мышц; у некоторых больных развивается кардиомиопатия. Как и при большинстве первично мышечных заболеваний, в периферической крови больных наблюдается повышение сывороточной креатинфосфокиназы (КФК) (в 5–100 раз). Возраст манифестации заболевания варьирует от 2 до 30 лет, инвалидизация наступает, как правило, после 10–20 лет от начала заболевания. Интеллект больных при этом заболевании остается сохранным. Таким образом, по совокупности клинических и лабораторных признаков ПКМД 2D зачастую сложно дифференцировать не только от других аутосомных форм ПКМД, но и от мышечной дистрофии Дюшенна/Беккера (МДД/Б), особенно в изолированных случаях. Некоторое время назад в РФ было проведено первичное молекулярно-генетическое исследование саркогликанопатий на выборке больных женского пола, которым в связи с тяжестью клинических проявлений и несмотря на половую принадлежность был установлен диагноз “МДД/Б”. В результате исследования патогенные варианты идентифицированы у 13.2% пробандов, при этом большинство патологических аллелей пришлись всего на два варианта: с.229С>T (p.Arg77Cys) и с.271G>A (p.Gly91Ser) [12]. Согласно литературным данным вариант с.229С>T является самой распространенной в мире мутацией этого гена, причем в Финляндии она выявляется практически в 100% случаев ПКМД 2D [13–15]. Вариант нуклеотидной последовательности с.271G>A (p.Gly91Ser) описан единожды в компаунд-гетерозиготном состоянии у двоих сибсов в итальянской семье [5]. Таким образом, поскольку ранее у российских больных уже были выявлены повторяющиеся варианты гена SGCA, очевидна целесообразность создания простой в техническом исполнении диагностической системы для их детекции на основании уже существующих рутинных лабораторных методик. Такая система могла бы найти применение как первичный этап для диагностики ПКМД 2D у больных с подходящей клинической картиной.
МАТЕРИАЛЫ И МЕТОДЫ
Материалом для исследования послужили образцы ДНК 516 больных с входящим диагнозом “ПКМД” (303 женщины и 213 мужчин), 1355 больных мужского пола с входящим диагнозом “МДД/Б” и 104 больных (76 мужчин и 28 женщин) с входящим диагнозом “мышечная дистрофия Эмери–Дрейфуса” (МДЭД). Диагностические критерии для включения пациентов в исследование различались в зависимости от выборки:
• Выборка больных ПКМД: 1) преимущественное поражение мышц тазового и плечевого поясов; 2) повышение уровня креатинфосфокиназы; 3) первично-мышечный характер поражения на ЭМГ; 4) отсутствие вертикального типа наследования (применялся только в семейных случаях).
• Выборка больных МДД/Б: 1) преимущественное поражение мышц тазового и плечевого поясов; 2) мужской пол; 3) резкое повышение уровня креатинфосфокиназы.
• Выборка больных МДЭД: 1) преимущественное поражение тазово-перонеальных и плече-лопаточных мышц; 2) контрактуры крупных суставов, тугоподвижность позвоночника; 3) наличие признаков аритмогенной кардиомиопатии у пробанда или его родственников; 4) умеренно повышенный уровень КФК; 5) первично-мышечный характер поражения на ЭМГ.
Контрольную выборку составили 528 образцов ДНК биобанка лаборатории ДНК-диагностики ФГБНУ “МГНЦ”, полученных от необследованных неродственных жителей различных регионов РФ.
Забор биологического материала проводили в лабораторных кабинетах медико-генетических консультаций и лечебно-профилактических учреждений разных регионов РФ и стран СНГ. От всех пациентов получено информированное согласие на проведение исследования, а в случае несовершеннолетних пациентов – от их родителей или законных представителей.
Выделение ДНК осуществляли из образцов цельной венозной крови или сухих пятен крови с помощью наборов реактивов для выделения Wizard® Genomic DNA Purification Kit (Promega, США) и DLAtomтм DNA Prep100 (ООО “Изоген”, Россия) по стандартным протоколам производителей. Поиск повторяющихся вариантов гена SGCA выполнен методом мультиплексной проба-зависимой лигазной реакции с последующей амплификацией (Multiplex Ligation-dependent Probe Amplification – MLPA). Дизайн олигонуклеотидных проб проведен в лаборатории ДНК-диагностики ФГБНУ “МГНЦ” таким образом, чтобы в “одной пробирке” детектировать три варианта гена SGCA (табл. 1). MLPA-реакция осуществлялась на программируемом термоциклере МС2 фирмы “ДНК-технология” (Россия) в течение двух часов в реакционной смеси следующего состава: 1) 0–50 нг геномной ДНК, 2) 7–10 фМ/мл каждого оригинального олигонуклеотида, 3) 0.4 ед. активности Pfu-DNA-лигазы, 4) буфер для лигирования (20 мM трис-HCl с pH 7.5, 20 мM KCl, 10 мM MgCl2, 0.1% Igepal, 0.01 мM rATP, 1 мM DTT), 5) 20–30 мл минерального масла. На втором этапе проводили стандартную полимеразную цепную реакцию (ПЦР) с олигопраймерами (универсальными), комплементарными участкам последовательностей, специально для данной реакции синтезированным в олигонуклеотидных пробах. В смесь, в которой предварительно провели лигазную реакцию, добавляли реакционную смесь для ПЦР следующего состава: 1) по 0.25 мМ каждого оригинального олигопраймера, 2) по 200 мМ каждого нуклеозидтрифосфата, 3) 1.0 ед. активности ДНК-полимеразы Biotaq (БиоМастер, Россия), 4) буфер для ПЦР (67 мM трис-HCl, 16.6 мM (NH4)2SO4, 0.01% твин-20, рН 8.8). Амплификацию проводили со следующими параметрами: I этап – предварительная денатурация при 95°С – 5 мин; лигирование олигонуклеотидных проб при температуре 62°С – 1 ч; II этап – предварительная денатурация при 95°С – 5 мин, 94°С – 2 с, температура отжига праймера – 2 с, элонгация при 72°С – 2 с, 30–32 цикла, с последующей окончательной достройкой при 72°С в течение 5 мин.
Таблица 1.
Олигонуклеотидные пробы и праймеры, применявшиеся для детекции искомых вариантов гена SGCA
| Детектируемый вариант | Последовательность олигонуклеотидов (5' → 3') | Длина фрагмента, пн |
|---|---|---|
| c.229C>T, экзон 3 |
FN: GTTCGTACGTGAATCGCGGTACGACCTGCCCCGGTGGCTCC | 85 – С
(норма), 88 – T (c.229C>T) |
| FM: GTTCGTACGTGAATCGCGGTACTCAGACCTGCCCCGGTGGCTCT | ||
| RN: GCTACACCCAGCGCAGCCTTTCGATGCGATCCGATGCCTTCATG | ||
| c.271G>A, экзон 3 |
FN: GTTCGTACGTGAATCGCGGTACCACCCTGGCTTCCTCTACG | 98 – G
(норма), 102 – T (c.271G>A) |
| FM: GTTCGTACGTGAATCGCGGTACTCACCACCCTGGCTTCCTCTACA | ||
| RN: GCTCTGCCACCCCAGAAGATCTTTATTGTTATTTC GATGCGATCCGATGCCTTCATG | ||
| c.850С>T, экзон 7 |
FN: GTTCGTACGTGAATCGCGGTACCCACTGAGGCCCCAGACC | 124 – С
(норма), 128 – T (c.850С>T) |
| FM: GTTCGTACGTGAATCGCGGTACTCACCCACTGAGGCCCCAGACT | ||
| LINKER: GTGACTTCTTGGTGGATGCTCTGGTCAC | ||
| RN: CCTCCTGGTGCCCCTGCTGTTTATTGTTATTTTCGATGCGATCCGATGCCTTCATG | ||
| Универсальные MLPA-праймеры | F: GTTCGTACGTGAATCGCGGTAC R: CATGAAGGCATCGGATCGCATC |
– |
Визуализацию результатов амплификации проводили методом электрофореза в 8%-ном полиакриламидном геле (ПААГ) 20 × 20 см с массовым соотношением акриламида (АА) и бисакриламида (БА) 19 : 1. В качестве электрофорезного буфера использовали 1× TBE. Результаты электрофореза визуализировались после окрашивания геля раствором бромистого этидия с помощью документирующей системы GelDoc фирмы Bio-Rad (США) в УФ-излучении с длиной волны 312 нм.
Поиск второго патологического аллеля у пациентов с частым вариантом гена SGCA в гетерозиготном состоянии осуществлялся прямым секвенированием фрагментов нуклеотидных последовательностей всех кодирующих и некодирующего экзонов этого гена по методу Сенгера на приборе ABI Prism 3100 (Applied Biosystems, США) согласно протоколу фирмы-производителя. Матрицы для секвенирования получали методом стандартной ПЦР с использованием ДНК-полимеразы Biotaq (Биомастер) в объеме 25 мкл реакционной смеси состава, аналогичного смеси для этапа II в реакции MLPA, но с добавлением MgCl2 в указанной в табл. 2 концентрации. Праймеры и условия амплификации, используемые в данной работе, приведены в табл. 2.
Таблица 2.
Олигонуклеотидные праймеры и условия амплификации, применявшиеся для анализа последовательности гена SGCA
| Фрагмент | Последовательность праймеров, 5' → 3' | Размер, пн | Температура отжига* | MgCl2, мМ |
|---|---|---|---|---|
| SGCA Экзон 1 |
F: GGGAGGCTGACGCTGGGAC R: CCACACCTCCGTCTTACCACC |
195 | 65°С | 4 |
| SGCA Экзоны 2–3 |
F: GCCCAGGACTGAGGCTGGC R: CTCCTAGGAGGCCACTCTGGC |
475 | 68°С | 2 |
| SGCA Экзоны 4–5 |
F: CACGTTTCTCCCCTAACCCAC R: GCCACAAGCTGCATCCTCCTC |
730 | 68°С | 2 |
| SGCA Экзон 6 |
F: CCCCTGGCAGAGTGGTGCC R: GAGACGGAGATAGGAAGCAGC |
300 | 68°С | 2 |
| SGCA Экзоны 7–8 |
F: GTGTCCAGCCAGCCACTTCC R: CAACAACTGTCTAGGTGGGCTC |
620 | 68°С | 2 |
| SGCA Экзоны 9–10 |
F: GGGCAGCTGACTCCCAGAGC R: GAATCAGCAGGTGGGATGTGC |
590 | 58°С | 2 |
Для поиска протяженных делеций/дупликаций гена SGCA использовался набор SALSA MLPA P116 SGC probemix (MRC-Holland, Нидерланды); реакцию проводили по протоколам фирмы-производителя. Для точного количественного анализа данные обрабатывали с помощью программного обеспечения Coffalyser V8, предоставляемого компанией-разработчиком MLPA [16].
Биоинформатический анализ клинической значимости выявленных изменений нуклеотидной последовательности проводили с помощью интернет-ресурсов SIFT, Provean, UMD-Predictor, FATHMM, PolyPhen-2 и NetGene2 [17–21]. Для статистической обработки данных применялись пакеты программы STATISTICA 6.0.
РЕЗУЛЬТАТЫ
Используя полученные в предыдущем исследовании данные [12], была разработана диагностическая MLPA-система для поиска повторяющихся вариантов с.229С>T (p.Arg77Cys) и с.271G>A (p.Gly91Ser), в которую включили еще один патогенный вариант гена SGCA – c.850C>T (p.Val247Met), отобранный из базы данных Leiden Open (source) Variation Database на основании высокой частоты встречаемости среди европейских больных (рис. 1). С помощью этой системы протестированы три выборки больных мышечными дистрофиями, манифестирующими после периода нормального моторного развития, сформированные на основании направляющего клинического диагноза.
Рис. 1.
Электрофореграмма результатов тест-системы для детекции трех вариантов гена SGCA методом MLPA. 1 – пробанд с вариантом c.271G>A в гомозиготном состоянии; 2 – пробанд с вариантами c.229C>T и c.271G>A в компаунд-гетерозиготном состоянии; 3 – пробанд с вариантами c.229C>T и с.850С>T в компаунд-гетерозиготном состоянии; 4 – пробанд с вариантом c.229C>T в гетерозиготном состоянии; 5 – пробанд без мутаций.
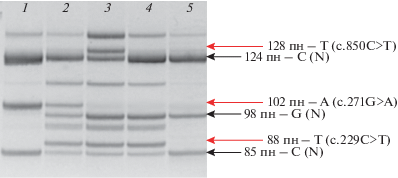
В выборке больных ПКМД анализ проведен 371 больному; для 145 пациентов молекулярно-генетический диагноз был подтвержден ранее, при этом мутации выявлялись в широком спектре генов, ответственных за развитие различных форм мышечных дистрофий с разными типами наследования, а также спинальной мышечной атрофии: CAPN3, FKRP, DMD, SMNt, LMNA, ANO5, ZNF9, POMT1, LAMA2 [22]. В данной выборке “частые” варианты гена SGCA выявлены у девяти пациентов (1.7%) на 14 хромосомах (1.4%). Варианты с.229С>T и с.271G>A встретились как в гомозиготном, так и в гетерозиготном состоянии, причем на каждый из них приходится одинаковое число хромосом больных – 6; вариант c.850C>T встретился на двух хромосомах в гетерозиготном состоянии с другими вариантами. Для четырех больных с повторяющимся вариантом в гетерозиготном состоянии с целью поиска второго патологического аллеля проведено секвенирование всей кодирующей последовательности гена SGCA по методу Сенгера. В результате идентифицированы нонсенс-варианты с.402C>G (Tyr134Ter) и c.574C>T (p.Arg192Ter), миссенс-вариант c.308T>C (p.Ile103Thr) и прежде не описанный в литературе вариант c.223delT (p.Trp75Glyfs*136), приводящий к сдвигу рамки считывания и образованию преждевременного терминирующего кодона. Таким образом, у всех девяти больных выявлены два патологических аллеля (табл. 3). Исходя из уравнения Харди–Вайнберга, на основании наблюдаемого числа гомо- и гетерозигот повторяющихся вариантов в исследованной выборке рассчитано ожидаемое число больных (Nож, “идеальный” вариант событий, при котором все 100% патологических аллелей у 100% больных обнаружены), в данном случае Nож = 10. Соответственно, доля ПКМД 2D в этой выборке составила не менее 1.9%, а суммарная аллельная частота трех повторяющихся вариантов (из уравнения Харди–Вайнберга) составляет 71.4%. Информативность MLPA-системы для поиска повторяющихся вариантов гена SGCA, под которой понимается способность с помощью настоящей тест-системы выявить хотя бы один тестируемый мутантный аллель, у больных в группе с ПКМД достигает 70%.
Таблица 3.
Генотипы больных, идентифицированные в исследованных выборках
| Пациент | Пол | Входящий диагноз | Генотип: нуклеотидный вариант |
Генотип: аминокислотная замена |
|---|---|---|---|---|
| C306 | Ж | ПКМД | c.229C>T/c.229C>T | p.Arg77Cys/p.Arg77Cys |
| F100 | Ж | ПКМД | c.271G>A/c.271G>A | p.Gly91Ser/p.Gly91Ser |
| L468 | Ж | ПКМД | c.271G>A/c.271G>A | p.Gly91Ser/p.Gly91Ser |
| F4.1 | М | ПКМД | c.271G>A/c.229C>T | p.Gly91Ser/p.Arg77Cys |
| C140 | Ж | ПКМД | c.850C>T/c.229C>T | p.Val247Met/p.Arg77Cys |
| C70.1 | Ж | ПКМД | c.850C>T/с.402C>G | p.Val247Met/p.Tyr134Ter |
| АА9085 | М | ПКМД | c.271G>A/c.308T>C | p.Gly91Ser/p.Ile103Thr |
| FKRP121 | Ж | ПКМД | c.229С>T/c.223delT | p.Arg77Cys/p.Trp75Glyfs*136 |
| LGMD 486 | М | ПКМД | с.229С>T/c.574C>T | p.Arg77Cys/p.Arg192Ter |
| 1330 | М | МДД/Б | c.229C>T/c.229C>T | p.Arg77Cys/p.Arg77Cys |
| 2080 | М | МДД/Б | c.271G>A/c.271G>A | p.Gly91Ser/p.Gly91Ser |
| 2380 | М | МДД/Б | c.271G>A/c.271G>A | p.Gly91Ser/p.Gly91Ser |
| 2435* | М | МДД/Б | c.229C>T/N | p.Arg77Cys/N |
В выборке 1355 больных МДД/Б анализ проведен 548 больным, поскольку для 608 из них ранее был установлен молекулярно-генетический диагноз на основании анализа мутаций гена DMD (делеции/дупликации одного или нескольких экзонов) и “частых” в РФ вариантов генов CAPN3 (c.550delA, c.598_612del15), FKRP (c.826C>A, c.229C>T) и SMNt (делеция экзонов 7–8); материал 199 пациентов оказался непригоден для исследования используемой методикой. Искомые варианты гена SGCA выявлены у четырех пациентов (0.3%) на семи хромосомах (0.3%) (табл. 3). У больного с вариантом с.229С>T в гетерозиготном состоянии дополнительно проведено секвенирование всей кодирующей последовательности и областей экзон-интронных соединений гена SGCA, а также произведен поиск протяженных делеций/инсерций этого гена, однако второй патологический аллель выявить не удалось.
Как известно, вариант с.229С>T является самым частым патогенным вариантом гена SGCA в мире (частота аллеля T = 0.0004562, данные базы-агрегатора gnomAD). В настоящей работе выявлено, что в контрольной выборке (n = 528) вариант с.229С>T выявлен у одного человека в гетерозиготном состоянии. Следовательно, при довольно грубой оценке каждый 528-й житель РФ является популяционным носителем этой мутации, в то время как в группе больных также выявлен один предположительно носитель варианта с.229С>T. Применение точного критерия Фишера для двух независимых групп с целью определения статистической значимости в данном случае невозможно вследствие большого объема выборок. Поэтому статистическая значимость оценивалась путем расчета показателя отношения шансов (OR) популяционного носительства (фактор) в выборке больных и в контрольной группе (исходы), при этом получили значение OR = 0.96 (95%-ный доверительный интервал от 0.06 до 15.44). При отношении шансов, близком к единице, шансы обнаружить фактор риска в сравниваемых группах приблизительно одинаков; при этом доверительный интервал включает в себя 1, что свидетельствует об отсутствии статистической значимости связи между фактором и исходом при уровне значимости p > 0.05. Из этого следует, что больной с вариантом с.229С>T в гетерозиготном состоянии вероятнее всего – его популяционный носитель и выявленный вариант не имеет отношения к развитию заболевания.
Поскольку в выборке больных МДД/Б не встретилось пробандов с генотипами, образованными комбинацией повторяющегося и любого другого патогенного варианта гена SGCA, расчет ожидаемого числа больных ПКМД 2D производился на основании значений, полученных из уравнения Харди–Вайнберга для выборки больных ПКМД. Вычислив Nож = 6 для выборки больных МДД/Б, которым удалось провести анализ MLPA-системой (n = 548), был осуществлен перерасчет этого значения на число всех больных без мутаций в других исследованных генах (n = 747, Nож = 8). Таким образом рассчитали долю ПКМД 2D в данной выборке, которая составила не менее 0.6%. При этом информативность MLPA-системы в группе больных МДД/Б оказалась ниже, чем в первой выборке, и составила 50%.
В выборке больных МДЭД MPLA-системой протестировано 49 пациентов, у которых ранее не было выявлено мутаций в генах, ответственных за три формы МДЭД (LMNA, EMD, FHL1), а также частых в РФ мутаций в генах CAPN3 и FKRP [23]. Ни у одного пациента искомые варианты не обнаружены.
ОБСУЖДЕНИЕ
ПКМД 2D (α-саркогликанопатия) является самой частой в мире формой в группе саркогликанопатий, на которые в общей сложности в зависимости от популяции приходится от 2 до 53.8% всех ПКМД [24]. В настоящей работе продемонстрировано, что в выборке больных ПКМД из РФ на ПКМД 2D приходится не менее 1.9% случаев всех аутосомно-рецессивных ПКМД, в то время как среди больных МДД/Б мужчин доля этой формы составляет не менее 0.6%, что в 3 раза ниже по сравнению с первой выборкой. Данный результат может быть объяснен в рамках особенностей клинической картины МДД/Б, которые изначально подразумевают более тяжелое, злокачественное течение с обязательным вовлечением миокарда и более ранним возрастом манифестации заболевания. В то время как ПКМД 2D характеризуется более вариабельной по тяжести клинической картиной и характером течения, а кардиомиопатия развивается гораздо реже. Следовательно, большинство болеющих ПКМД 2D мужчин уже на этапе клинического обследования попадали в выборку больных ПКМД, а наличие больных ПКМД 2D в выборке больных МДД/Б обусловлено формированием у них тяжелого “дюшенноподобного” фенотипа, клинически неотличимого от МДД/Б. Свойство мутаций гена SGCA приводить к тяжелой “дюшенноподобной” мышечной дистрофии также объясняет значительно более высокую долю ПКМД 2D среди женщин с направляющим диагнозом “МДД/Б” (13.2%, результаты исследования [12]), по сравнению с двумя исследованными в этой работе выборками. Столь явное различие значений доли ПКМД 2D в настоящем и предыдущем исследованиях очевидно обусловлено тем, что выборка больных женщин с направляющим диагнозом “МДД/Б” являлась более подходящим объектом исследования для определения вклада саркогликанопатий в спектр ПКМД по большинству критериев (женский пол, с высокой долей вероятности исключающий диагноз “МДД/Б”, тяжелая клиническая картина проксимальной миопатии, ранняя манифестация заболевания), нежели исследуемые в этой работе. Следовательно, с высокой долей вероятности можно утверждать, что доля ПКМД 2D в РФ ни в одной из выборок больных ПКМД (за исключением тех, для которых доступны результаты иммуногистохимического исследования биоптатов мышечной ткани) не может превышать 13.2%, что и было подтверждено в настоящей работе.
Спектр мутаций гена SGCA и их относительная аллельная частота различались в зависимости от выборки. В группе больных МДД/Б выявлены только два “частых” варианта c.229C>T и c.271G>А, на которые приходится соответственно 33 и 66% выявленных вариантов. Тот факт, что в этой группе не встретился вариант c.850C>T, можно считать закономерным, поскольку по литературным источникам больные, имеющие в генотипе данный вариант в гомо- или компаунд-гетерозиготном состоянии, отличаются более мягкой клинической картиной ПКМД и относительно доброкачественным течением заболевания с поздней инвалидизацией [7, 14, 25].
Спектр мутаций гена SGCA в группе больных ПКМД шире, чем в выборке больных МДД/Б. На долю трех искомых вариантов гена SGCA (c.229C>T, c.271G>A и c.850C>T) в этой группе больных приходится 78% всех выявленных вариантов. При этом доля вариантов c.229C>T и c.271G>A среди больных хромосом оказалась одинакова (33%). Сравнение долей этих вариантов, полученных в данной работе для двух выборок (ПКМД и МДД/Б), со значениями, полученными в предыдущей работе (50% для варианта c.229C>T и 30% для варианта c.271G>A [12]), не позволяет выявить наличие статистически значимых различий между сопоставляемыми значениями в этих трех выборках вследствие малого числа выявленных хромосом с данными вариантами [12]. Отдельно стоит отметить, что в исследованных выборках больных доля варианта c.271G>A, который по литературным источникам встретился всего в одной итальянской семье, сравнима с долей патогенного варианта c.229C>T. Следовательно, настоящая работа в совокупности с результатами предыдущего исследования подтверждает, что больные ПКМД 2D в РФ, в отличие от больных в остальных популяциях мира, имеют две превалирующие (мажорные) мутации гена SGCA: c.229C>T и c.271G>A. Вариант c.850C>T встретился в группе больных ПКМД дважды и только в гетерозиготном состоянии с другими вариантами. Для больного с генотипом c.850C>T(;)229C>T были доступны анамнез и минимальное описание клинических особенностей. Пробанд 27 лет с дебютом заболевания в 12 лет, на момент осмотра отмечались мышечные боли и признаки мышечной слабости в проксимальных отделах, гипотрофия мышц бедер и умеренные псевдогипертрофии икроножных мышц, данных за кардиомиопатию нет. При этом пробанд оставался амбулаторным, но передвигался с поддержкой. Этот случай подтверждает данные литературных источников, что вариант c.850C>T является “мягким” и даже в компаунд-гетерозиготном состоянии приводит к развитию сравнительно мягкого фенотипа ПКМД.
Помимо трех искомых вариантов гена SGCA в выборке больных ПКМД выявлены четыре неповторяющихся варианта нуклеотидной последовательности, три из них зарегистрированы в литературных источниках и внесены в базу данных HGMD: c.574C>T (p.Arg192Ter), с.402C>G (p.Tyr134Ter) и c.308T>C (p.Ile103Thr). Нонсенс-вариант c.574C>T (p.Arg192Ter) описан в одной публикации в гомозиготном состоянии у двух албанских сибсов, имеющих клиническую картину ПКМД 2D и высокий уровень сывороточной креатинфосфокиназы в крови; при иммуногистохимическом исследовании у обоих сибсов обнаружили снижение α- и δ-саркогликана в образцах мышечной биопсии [26]. Нонсенс-вариант с.402C>G (p.Tyr134Ter) прежде был упомянут в двух публикациях, причем только в одной из них указано, что он выявлен у больного с клинической картиной МДД/Б [27, 28]. Миссенс-вариант c.308T>C (p.Ile103Thr) описан также в одной публикации в компаунд-гетерозиготном состоянии у больного из Европы с “дюшенноподобным” фенотипом и ранней инвалидизацией (12 лет). Поскольку каждый из трех ранее описанных вариантов в экспериментальных работах упомянут единожды, как и один из тестируемых MLPA-системой вариантов (c.271G>A), для них проведено определение клинической значимости согласно существующим в РФ критериям интерпретации вариантов, представленным в “Руководстве по интерпретации данных, полученных методами массового параллельного секвенирования (MPS)” [29]. Та же процедура проведена для ранее не описанного в литературе варианта c.223delT (p.Trp75Glyfs*136). Результаты представлены в табл. 4.
Таблица 4.
Результаты интерпретации выявленных вариантов гена SGCA
| Нуклеотидный вариант | Аминокислотная замена | Критерии патогенности | Клиническая значимость |
|---|---|---|---|
| c.271G>A | p.Gly91Ser | PM2, 5 + PP1, 3 | Вероятно патогенный |
| c.223delT | p.Trp75Glyfs*136 | РVS1 + PM2 | Вероятно патогенный |
| c.308T>C | p.Ile103Thr | PM2, 5 + PP3 | Неопределенного клинического значения |
| с.402C>G | p.Tyr134Ter | РVS1 + PM2 | Вероятно патогенный |
| c.574C>T | p.Arg192Ter | РVS1 + PM2 + PP1 | Патогенный |
Необходимо отметить, что при интерпретации вариантов не был задействован критерий категории “средней тяжести” PM3, поскольку материал родителей пробандов с идентифицированными вариантами не был доступен и проверка цис/транс-положения этих вариантов не проводилась. Следовательно, клиническая значимость проанализированных вариантов занижена по объективным причинам. Из всех проанализированных вариантов только один оказался вариантом неопределенного клинического значения (c.308T>C). При этом постановка молекулярно-генетического диагноза возможна на основании наличия в генотипе больного либо двух патогенных вариантов, либо комбинации патогенного и вероятно патогенного вариантов или же двух вероятно патогенных вариантов, в последнем случае – при наличии четко очерченной и специфической клинической картины заболевания. Это означает, что для установления молекулярно-генетического диагноза у пробанда, имеющего в генотипе этот вариант (c.271G>A(;)308T>C), требуется как минимум либо исследование цис/транс-положения выявленных вариантов, либо накопление сегрегационных данных для варианта с неопределенным клиническим значением, что позволило бы перевести его в категорию “вероятно патогенный”.
В случае с обследованной нами выборкой больных МДЭД следует отметить, что пациенты с этой клинической картиной довольно хорошо распознаются опытными врачами-генетиками или неврологами уже на клиническом уровне. Однако нужно помнить, что МДЭД, как и остальные исследуемые в данной работе заболевания, характеризуется прогрессирующим течением. Следовательно, существует вероятность, что отличительные для МДЭД признаки (отсутствие гипертрофий, ранние контрактуры крупных суставов, ретракция ахилловых сухожилий) могли отсутствовать или быть незначительно выраженными на момент обследования, либо паттерн вовлеченных в патологический процесс мышечных групп не позволял однозначно заподозрить ту или иную форму миопатии. Исходя из этих соображений, в группе больных МДЭД также проведен поиск повторяющихся вариантов гена SGCA, который оказался неэффективен. Данный результат в очередной раз демонстрирует существование очевидных различий клинических проявлений МДЭД и ПКМД 2D уже на ранних этапах развития этих заболеваний.
Подводя итоги работы, мы получаем возможность усовершенствовать существующий алгоритм молекулярно-генетической диагностики ПКМД 2D. На группе больных ПКМД было продемонстрировано, что эффективность разработанной MLPA-системы для детекции повторяющихся вариантов гена SGCA оказалась довольно высокой (70%). Следовательно, данная система рекомендуется как первичный этап молекулярной диагностики в первую очередь для больных с клинической картиной ПКМД и наличием псевдогипертрофий, а также для женщин с тяжелой “дюшенноподобной” формой проксимальной мышечной дистрофии.
Список литературы
Davies K.E., Nowak K.J. Molecular mechanisms of muscular dystrophies: old and new players // Nat. Rev. Mol. Cell Biol. 2006. V. 7. № 10. P. 762–773. doi 10.1038/nrm2024
Moore S.A., Shilling C.J., Westra S. et al. Limb-girdle muscular dystrophy in the United States // J. Neuropathol. Exp. Neurol. 2006. V. 65. № 10. P. 995–1003. doi 10.1097/01.jnen.0000235854.77716.6c
Meena A.K., Sreenivas D., Sundaram C. et al. Sarcoglycanopathies: a clinico-pathological study // Neurol. India. 2007. V. 55. № 2. P. 117–121. doi 10.4103/0028-3886.32781
Ginjaar H.B., van der Kooi A.J., Ceelie H. et al. Sarcoglycanopathies in Dutch patients with autosomal recessive limb girdle muscular dystrophy // J. Neurol. 2000. V. 247. № 7. P. 524–529.
Guglieri M., Magri F., D’Angelo M.G. et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients // Hum. Mutat. 2008. V. 29. № 2. P. 258–266. doi 10.1002/humu.20642
Duggan D.J., Gorospe J.R., Fanin M. et al. Mutations in the sarcoglycan genes in patients with myopathy // N. Engl. J. Med. 1997. V. 336. № 9. P. 618–624. doi 10.1056/nejm199702273360904
Trabelsi M., Kavian N., Daoud F. et al. Revised spectrum of mutations in sarcoglycanopathies // Eur. J. Hum. Genet. 2008. V. 16. № 7. P. 793–803. doi 10.1038/ejhg.2008.9
Roberds S.L., Leturcq F., Allamand V. et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy // Cell. 1994. V. 78. № 4. P. 625–633.
McNally E.M., Yoshida M., Mizuno Y. et al. Human adhalin is alternatively spliced and the gene is located on chromosome 17q21 // Proc. Natl Acad. Sci. USA. 1994. V. 91. № 21. P. 9690–9694.
Campbell K.P. Adhalin gene mutations and autosomal recessive limb-girdle muscular dystrophy // Ann. Neurol. 1995. Sep. V. 38. № 3. P. 353–354. doi 10.1002/ ana.410380302
Eymard B., Romero N.B., Leturcq F. et al. Primary adhalinopathy (alpha-sarcoglycanopathy): clinical, pathologic, and genetic correlation in 20 patients with autosomal recessive muscular dystrophy // Neurology. 1997. V. 48. № 5. P. 1227–1234.
Щагина О.А., Полякова Д.А., Рыжкова О.П. и др. Причины мышечной дистрофии у женщин с направляющим диагнозом “мышечная дистрофия Дюшена/Беккера” // Мед. генетика. 2017. T. 16. № 11. C. 17–22.
Moreira E.S., Vainzof M., Suzuki O.T. et al. Genotype–phenotype correlations in 35 Brazilian families with sarcoglycanopathies including the description of three novel mutations // J. Med. Genet. 2003. V. 40(2):E12. doi 10.1136/jmg.40.2.e12
Hackman P., Juvonen V., Sarparanta J. et al. Enrichment of the R77C alpha-sarcoglycan gene mutation in Finnish LGMD2D patients // Muscle Nerve. 2005. V. 31. № 2. P. 199–204. doi 10.1002/mus.20267
Carrie A., Piccolo F., Leturcq F. et al. Mutational diversity and hot spots in the alpha-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D) // J. Med. Genet. 1997. V. 34. № 6. P. 470–475.
http://mrc-holland.com.
http://provean.jcvi.org/index.php.
http://umd-predictor.eu/.
http://www.cbs.dtu.dk/services/NetGene2/.
http://genetics.bwh.harvard.edu/pph2/.
http://fathmm.biocompute.org.uk/.
Рыжкова О.П. Клинико-молекулярно-генетический анализ изолированных поясно-конечностных мышечных дистрофий, являющихся ферментопатиями: Автореф. дис. … канд. мед. наук, 2011. 27 с.
Адян Т.А. Клинико-генетическое разнообразие и молекулярная диагностика мышечной дистрофии Эмери–Дрейфуса: Автореф. дис. … канд. мед. наук, 2015. 24 с.
Mahmood O.A., Jiang X.M. Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review) // Mol. Med. Rep. 2014. V. 9. № 5. P. 1515–1532. doi 10.3892/mmr.2014.2048
Klinge L., Dekomien G., Aboumousa A. et al. Sarcoglycanopathies: can muscle immunoanalysis predict the genotype? // Neuromuscul. Disord. 2008. V. 18. № 12. P. 934–941.
Babameto-Laku A., Tabaku M., Tashko V. et al. The first case of primary alpha-sarcoglycanopathy identified in Albania, in two siblings with homozygous alpha-sarcoglycan mutation // Genet. Couns. 2011. V. 22. № 4. P. 377–383.
Schara U., Gencik M., Mortier J. et al. Alpha-sarcoglycanopathy previously misdiagnosed as Duchenne muscular dystrophy: implications for current diagnostics and patient care // Eur. J. Pediatr. 2001. V. 160. № 7. P. 452–453.
Xiong H.Y., Alipanahi B., Lee L.J. et al. The human splicing code reveals new insights into the genetic determinants of disease // Science. 2015. V. 347. № 6218:1254806. Epub. 2014. Dec. 18. doi 10.1126/science.1254806
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных, полученных методами массового параллельного секвенирования (MPS) // Мед. генетика. 2017. T. 16. № 7. C. 4–17.
Дополнительные материалы отсутствуют.